
Abstract
In this study, randomly amplified polymorphic DNA-PCR (RAPD-PCR) was employed to investigate the genetic diversity of 308 wild isolates of Listeria monocytogenes. These isolates were taken from 1288 samples isolated from six food sources obtained within the Baoding and Shijiazhuang areas of Hebei province, China. Genetic distant coefficient analysis of the band profiles was carried out using unweighted pair-group method analysis (UPGMA) and cluster analysis. The oligomer RP1 was used as a primer to type 308 isolates of foodborne L. monocytogenes and 228 different RAPD-PCR fingerprinting patterns were obtained. The clustering analysis showed that the 308 isolates of L. monocytogenes could be grouped into six genetic clusters. Among them, Clusters I and II were the dominant species from this region. Also, many differences between genetic clusters from different foods, sampling times, and regions were found. The differences in RAPD-PCR fingerprinting patterns obtained from the six food types sampled indicate that the L. monocytogenes subtypes isolated were genetically diverse.
Similar content being viewed by others


Introduction
Listeria monocytogenes is an important foodborne pathogen due to its widespread distribution (Ryser and Marth 2007). Consumption of food contaminated with this pathogen can lead to listeriosis. This condition has high rates of morbidity and mortality (25–30 % overall) (EFSA 2010; Scallan et al. 2011). The organism can endure adverse conditions such as freezing, drying, mild heat, and anaerobic conditions. It can also grow at temperatures used in refrigeration (from −1.5 to 4 °C) (Gandhi and Chikindas 2007). Because of these characteristics, L. monocytogenes is a great concern for food safety.
Frequent reports of foodborne disease associated with L. monocytogenes are reported globally each year (Mead et al. 2006). L. monocytogenes contributes significantly to foodborne illness and death in the U.S., with approximately 2500 cases of foodborne listeriosis in 2010, with an estimated cost of about $2.04 billion (Scallan et al. 2011; Schraff 2012). Prepared food plays an important role in foodborne disease (Aurora et al. 2009). These products have gained widespread acceptance due their convenience and are popular in products such as meat, cheeses, milk, and instant noodles. The threat of L. monocytogenes to human health becomes more severe with an increase in the consumption of prepared food. Because the incubation period of L. monocytogenes is long (McLauchlin et al. 2004), contaminated food has usually been discarded leaving few contaminated samples for examination once infection has been established. Thus, it is very difficult to trace sources of L. monocytogenes contamination. This makes the investigation of the genetic background of L. monocytogenes important for molecular epidemiological analysis, detection, control, and tracking of this pathogen.
To trace the vehicles of infection and carry out an epidemiological investigation, various methods have been applied to type L. monocytogenes strains, including serotyping, phage typing, and molecular typing. Randomly amplified polymorphic DNA-PCR (RAPD-PCR) is another molecular typing method that can be used to characterize L. monocytogenes strains. This technique detects DNA polymorphisms and is based on the common technique of PCR (Welsh and McClelland 1990). RAPD-PCR employs short (9–10 bp) primers with sequences chosen at random, removing the requirement for prior knowledge of the template DNA sequence. Purified DNA, a single colony, or a cell lysate can be used as the PCR template. Compared with other methods of molecular typing, RAPD-PCR is quick and relatively simple. Several studies have shown RAPD-PCR to be a highly discriminative method for rapid subtyping of L. monocytogenes (Boerlin et al. 1995). This technique has also been widely used in epidemiological studies and investigations of contamination events (Giovannacci et al. 1999). However, the relatively poor reproducibility of the method is often criticized by people.
The objective of this study was to investigate the genetic diversity L. monocytogenes from food samples. To accomplish this, we used 308 isolates taken from 1288 samples. Our samples were taken from six different food products and were typed using RAPD-PCR. From this we constructed RAPD-PCR fingerprint databases for L. monocytogenes strains.
Materials and methods
Sample collection
From January 2007 to April 2008, a total of 1288 food samples were purchased randomly from supermarkets in Baoding, Shijiazhuang and the surrounding areas of Hebei Province, China. These samples included: 500 pieces of raw meat, 338 frozen foods, 162 seafood products, 144 fruits or vegetables, 59 cooked meat products, and 25 dairy products. Samples were transported to the laboratory on ice and analyzed in a timely manner.
Isolation and identification of L. monocytogenes
Isolation and identification of L. monocytogenes was carried out according to a standard procedure (National Food Safety Standard, Food Microbiological Examination: Listeria monocytogenes, GB 4789.30-2010, China). Briefly, 25-g samples were aseptically added to 225 mL of enrichment broth (EB) and homogenized in a flask. The homogenized samples were incubated at 30 °C for 4 h, and selective agents (acridine flavin and nalidixic acid) were added following incubation. After incubating for an additional 48 h, aliquots from these EB-innoculated samples were streaked onto Oxford and PALCAM media. Typical Listeria-like colonies grown on Oxford and PALCAM media were purified on TSA-YE and verified using the biochemical and β-hemolysis assays. These isolates could be confirmed as L. monocytogenes based on the above results.
DNA extraction
DNA was prepared using the following methods. A colony was picked from the TSA culture and inoculated into 5 mL of TSB-YE. The culture was grown for 12 h with shaking at 120 rpm at 37 °C. One milliliter was removed from the TSB-YE culture and spun at 10,000 rpm for 5 min. The supernatant was discarded, and the pellet was washed in 1 mL of sterile distilled water two times. The suspension was centrifuged at 10,000 rpm for 5 min in a microcentrifuge to obtain the cell pellet. The pellet was resuspended in 250 μL lysozyme (1 mg/mL, Sigma, USA) and incubated in a water bath at 37 °C for 2 h. Two hundred microliters TE buffer (1 mM EDTA, 10 mM Tris–HCl, pH 8.0), 100 μL 10 % SDS (sodium dodecyl sulfate), 3 μL Proteinase K (20 mg/mL, Sigma, USA), and 60 μL NaCl (5 M) were then added to the tube with gentle agitation. To lyse the cells, the tube was then incubated in a 60 °C water bath for 20 min, or until the solution mixture became clear. Following lysis, 500 μL PCI mix (phenol-chloroform-isoamylalcohol, at a volume ratio of 25:24:1) was added to the lysate and mixed gently. The mixture was centrifuged at 12,000 rpm for 10 min. The upper aqueous layer (clear layer) was carefully transferred into a new sterile tube. Two hundred microliters CI mix (chloroform-isoamylalcohol, at a volume ratio of 24:1) was then added to the aqueous layer and mixed gently. This tube was centrifuged at 12,000 rpm for 10 min. The upper aqueous layer was transferred into a new sterile tube, and 20 μL potassium acetate and 400 μL cold ethanol (100 %) were added. The tube was incubated at −20 °C for 30 min and then centrifuged at 12,000 rpm for 10 min. The supernatant was discarded, and the pellet was washed twice with 500 μL of cold 70 % ethanol. After centrifugation at 12,000 rpm for 10 min, the ethanol solution was discarded and the pellet was air-dried and dissolved in 30 μL sterile distilled water. The DNA (1 μL) was used for the RAPD-PCR analysis.
RAPD and gel electrophoresis
PCR amplification was performed with a T-Gradient Thermocycler (Biometra, Germany) in a 25 μL reaction mixture containing: 2.5 μL 10 × PCR buffer, 2 μL MgCl2, 1 μL dNTP mix, 1 μL Taq polymerase, 2.5 μL of a random primer RP1 (5’GGTGTGCTGT 3’), 0.5 μL of DNA template, and 15.5 μL ddH2O. The DNA amplification procedure was as follows: one cycle, 4 min at 94 °C for initial denaturation , 45 cycles of 1 min at 94 °C (denaturation), 1 min at 35 °C (annealing), and 1 min at 72 °C (elongation), followed by a final extension of 10 min at 72 °C. Following PCR, 7 μL of each PCR product was loaded into the wells of a 1.0 % agarose gel, electrophoresed at 100 V for 1 h, and stained with ethidium bromide. A 1 kb ladder and the DL2000 (TaKaRa, Japan) marker were used for reference and the gel was viewed with a BINTA 2020D (UVIT EC, UK). The experiments were repeated independently three times.
Analysis of RAPD-PCR fingerprinting
The bands on the RAPD-PCR fingerprint were measured in comparison to DNA standards (1 kb ladder and DL2000). The banding pattern obtained from the gel was scored in a binary data format. The scoring was based on the presence or absence of the band. A score of 0 indicated the absence of the band, whereas 1 indicated the presence of a particular band. The distance calculation for the different band profiles was carried out by an unweighted pair-group method analysis (UPGMA) and cluster analysis using statistical data software (NTSYS-PC version 2.1).
Results
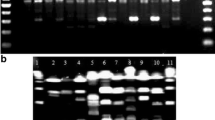
In this study, 308 isolates were analyzed using a RAPD-PCR assay with a single primer (RP1). The RAPD-PCR patterns from these isolates were compared, characterizing a total of 228 different patterns. This indicates that the L. monocytogenes isolates were genetically diverse. Analysis of the RAPD-PCR patterns (Fig. 1) revealed a high degree of polymorphism between L. monocytogenes isolates with gel band sizes from 0.1 to 3 kb. The gel bands were mostly distributed from 0.5 to 2 kb. By comparing the different gel bands, we found that the band sizes of 650 bp and 1.5 kb appeared with high frequency and were found within 77.27 and 66.88 % of total isolates, respectively. In addition, the isolates containing both 650 bp and 1.5 kb bands accounted for 57.79 % of total isolates (Table 1). Our analysis found that the 650 bp and 1.5 kb bands were widely distributed in most of the foodborne L. monocytogenes isolates. Importantly, isolates taken from dairy products were exceptions to this pattern and neither the 650 bp nor 1.5 kb bands were observed for these samples.

Agarose (1.0 %) gel electrophoresis of RAPD-PCR products from some L. monocytogenes isolates. Lanes: 1 kb, 1 kb ladder; DL2000, 2000 bp DNA marker; CK, negative control containing ddH2O in place of the DNA template; numbered lanes indicate different isolates of L. monocytogenes
Since the isolates with identical gel patterns were classified into the same category, a total of 228 different patterns were observed. Specifically, among the 308 isolates, 171 had individual patterns and the remaining 137 isolates shared the other 57 patterns. This indicates that there were two or more isolates with the same RAPD-PCR pattern among these 137 isolates. The main classification of the 137 isolates with shared patterns is summarized in Table 2. The results show that isolates derived from the same food product, sampling time, and sampling area (Group i) accounted for 47.45 % of the isolates. Those from different food sources, but the same sampling time and the same sampling area (Group ii) accounted for 34.31 % of the isolates. Isolates that originated from the same food type, but had different sampling times and sampling areas (Group iii) accounted for 11.68 % of the isolates. The samples taken from different food types, with different sampling times and areas (Group iv) accounted for 6.57 % of the isolates. These results indicate that the isolates derived from the same food source, the same sampling time or the same sampling area, were more closely related genetically.
The gel patterns were analyzed using statistical software (NTSYS-PC version 2.1) to generate a clustering dendrogram. The isolates were clustered into six genetic clusters (Fig. 2). Table 3 shows the statistical distribution of the 308 isolates in each genetic cluster. Table 3 shows that Cluster I contained 96 isolates, accounting for 31.17 % of the 308 isolates. Cluster II contained 153 isolates, accounting for 49.68 % of the total isolates. Other clusters were much smaller, suggesting that Clusters I and II were the dominant clusters in the regions sampled.

The clustering dendrogram of 228 L. monocytogenes isolates based on RAPD-PCR analysis
To investigate the changes in foodborne L. monocytogenes distribution among different food types, we analyzed 78 L. monocytogenes isolates obtained in July 2007 from Baoding. Table 4 shows that these isolates, taken from different food types, were distributed within the clusters named above, including isolates from: raw meat (I), quick-frozen food (III), seafood products (II), cooked meat (II), and fruits and vegetables (VI). These results suggest that genotype of L. monocytogenes differed between these different food types and foodborne L. monocytogenes prefers a particular host, although all samples were taken at the same sampling time and sampling area.
To investigate the change of foodborne L. monocytogenes distribution with time, we analyzed two batches of L. monocytogenes samples obtained from raw meat from Baoding. Table 5 shows that the samples taken in July 2007 were mainly distributed in Cluster I (56.10 %), and the samples taken in April 2008 were mainly distributed in Cluster II (65.96 %). This suggests that the L. monocytogenes genotype changed between the two sampling times, despite the fact that all samples were derived from the same food type and sampling area.
To investigate changes in the distribution of foodborne L. monocytogenes with respect to geographic location, we analyzed two batches of L. monocytogenes samples obtained in April 2008 from raw meat. Table 6 shows that most of the isolates from Shijiazhuang were found in Cluster I (47.06 %). In contrast, most of the isolates from Baoding were found in Cluster II (65.96 %). These results suggest that the genotype of L. monocytogenes differed between these different sampling locations.
In summary, this study indicates that foodborne L. monocytogenes prefers a particular food reservoir. Also, we found that the spread of foodborne L. monocytogenes can be characterized as regional and temporary.
To test the reproducibility of our experimental results, we repeated the experiments in our study three times. Representative RAPD-PCR patterns of replicate experiments are shown in Fig. 3. Figure 3 shows similar gel patterns both in Fig. 3a and b, results obtained from two completely independent, repeated trials. These results show strong reproducibility within our experiments.

Agarose (1.0 %) gel electrophoresis of representative RAPD-PCR patterns from replicate experiments. a and b: two completely independent, repeated trials. Lanes: M, 2000 bp DNA marker; CK, negative control, containing ddH2O in place of the DNA template; numbered lanes indicate different isolates of L. monocytogenes
Discussion
Traditional serotyping has been widely used in epidemiological and contamination investigations. However, compared with molecular typing, its discriminatory power is relatively low, and this limits the usefulness of this technique (Schraff 2012). Many molecular typing methods have been applied to L. monocytogenes, such as ribotyping (RT) (Gendel and Ulaszek 2000; Nadon et al. 2001; Suihko et al. 2002), restriction enzyme analysis (REA) (Giovannacci et al. 1999; Unnerstad et al. 1999; Jeffers et al. 2001), pulsed-field gel electrophoresis (PFGE) (Dauphin et al. 2001; Graves and Swaminathan 2001; Lundén et al. 2002), amplified fragment length polymorphism (AFLP) (Aarts et al. 1999; Guerra et al. 2002; Keto-Timonen et al. 2003), multi-locus sequence typing (MLST) (Chan et al. 2001; Cai et al. 2002), and RAPD-PCR. Each method has its advantages and disadvantages. The choice of method mainly depends on the method’s performance characteristics, such as discriminatory power, reproducibility, rapidity, ease of use, and the purpose of the subtyping experiment (Struelens 1996). The advantage of RAPD-PCR typing over other methods lies in its relative simplicity, speed, cost effectiveness, and high degree of discrimination between subtypes.
With a lack of prior knowledge of template DNA sequences, it is tedious and difficult to select an appropriate primer and optimize PCR conditions. A suitable primer is essential to RAPD-PCR analysis (Blixt et al. 2003) and can generate RAPD-PCR fingerprinting patterns with rich bands and good discrimination. To enhance the discrimination, a larger number of primers have been used in some studies (Yoshida et al. 1999; Samuel et al. 2011; Lee et al. 2011). In the present study, only one oligomer (RP1) was used as a primer in typing 308 strains of foodborne L. monocytogenes and 228 different RAPD-PCR fingerprinting patterns were observed. Obtaining such a great diversity of RAPD-PCR patterns shows that the RP1 primer discriminates well, and was able to distinguish isolates of L. monocytogenes.
The main limitation of RAPD-PCR is its poor reproducibility (Allerberger et al. 1997; Wagner et al. 1999; Malak et al. 2001). However, in this study, the experimental results were reproducible through the three replicate experiments. This suggests that as long as the reagents, laboratory equipment and test procedure are consistent, good reproducibility can be achieved. We also found that the quality of template was directly related to experimental consistency and that, purified DNA is more suitable for the RAPD-PCR template than a cell lysate or a single colony. Our results show that RAPD-PCR is a simple, rapid, and reliable method for use with L. monocytogenes in both epidemiological studies and investigations of bacterial contamination.
Conclusion
In this study, a RAPD-PCR fingerprint database of Listeria monocytogenes isolates was constructed using only one oligomer (RP1) as the primer. A dendrogram showing the genetic relationship of the 308 foodborne L. monocytogenes isolates was created. The results of our clustering analysis show that the 308 isolates of L. monocytogenes could be grouped into six genetic clusters. Among them, Clusters I and II were the dominant groups in the Baoding and Shijiazhuang areas of Hebei Province, China. It was also found that there were many differences between each cluster that were related to the food type, sampling time and region. The great diversity found within the RAPD-PCR fingerprinting patterns obtained from the six food types sampled indicates that the L. monocytogenes isolates are highly diverse.
References
Aarts HJ, Hakemulder LE, Van Hoef AM (1999) Genomic typing of Listeria monocytogenes strains by automated laser fluorescence analysis of amplified fragment length polymorphism fingerprint patterns. Int J Food Microbiol 49:95–102. doi:10.1016/S0168-1605(99)00057-4
Allerberger F, Dierich MP, Grundmann H, Hartung D, Bannerman E, Bille J (1997) Typing of Austrian Listeria monocytogenes isolates by automated laser fluorescence analysis of randomly amplified polymorphic DNA. Zentralbl Bakteriol 286:33–40. doi:10.1016/S0934-8840(97)80072-2
Aurora R, Prakash A, Prakash S (2009) Genotypic characterization of Listeria monocytogenes isolated from milk and ready-to-eat indegenous milk products. Food Control 20:835–839. doi:10.1016/j.foodcont.2008.10.017
Blixt Y, Knutsson R, Borch E, Rådström P (2003) Interlaboratory random amplified polymorphic DNA typing of Yersinia enterocolitica and Y. enterocoliticalike bacteria. Int J Food Microbiol 83:15–26. doi:10.1016/S0168-1605(02)00319-7
Boerlin P, Bannerman E, Ischer F, Rocourt J, Bille J (1995) Typing Listeria monocytogenes: a comparison of random amplification of polymorphic DNA with 5 other methods. Res Microbiol 146:35–49. doi:10.1016/0923-2508(96)80269-5
Cai S, Kabuki DY, Kuaye AY, Cargioli TG, Chung MS, Nielsen R, Wiedmann M (2002) Rational design of DNA sequence-based strategies for subtyping Listeria monocytogenes. J Clin Microbiol 40:3319–3325. doi:10.1128/JCM.40.9.3319-3325.2002
Chan MS, Maiden MC, Spratt BG (2001) Database-driven multi locus sequence typing (MLST) of bacterial pathogens. Bioinformatics 17:1077–1083. doi:10.1093/bioinformatics/17.11.1077
Dauphin G, Ragimbeau C, Malle P (2001) Use of PFGE typing for tracing contamination with Listeria monocytogenes in three cold-smoked salmon processing plants. Int J Food Microbiol 64:51–61. doi:10.1016/S0168-1605(00)00442-6
EFSA (2010) The community summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in the European Union in 2008. EFSA J 8(1496):138–141. doi:10.2903/j.efsa.2010.1496
Gandhi M, Chikindas ML (2007) Listeria: a foodborne pathogen that knows how to survive. Int J Food Microbiol 113:1–15. doi:10.1016/j.ijfoodmicro.2006.07.008
Gendel SM, Ulaszek J (2000) Ribotype analysis of strain distribution in Listeria monocytogenes. J Food Prot 63:179–185
Giovannacci I, Ragimbeau C, Oueguiner S, Salvat G, Vendeuvre JL, Carlier V, Ermel G (1999) Listeria monocytogenes in pork slaughtering and cutting plants: use of RAPD, PFGE and PCR-REA for tracing and molecular epidemiology. Int J Food Microbiol 53:127–140. doi:10.1016/S0168-1605(99)00141-5
Graves LM, Swaminathan B (2001) PulseNet standardized protocol for subtyping Listeria monocytogenes by macrorestriction and pulsed-field gel electrophoresis. Int J Food Microbiol 65:55–62. doi:10.1016/S0168-1605(00)00501-8
Guerra MM, Bernardo F, McLauchlin J (2002) Amplified fragment length polymorphism (AFLP) analysis of Listeria monocytogenes. Syst Appl Microbiol 25:456–461. doi:10.1078/0723-2020-00133
Jeffers GT, Bruce JL, McDonough PL, Scarlett J, Boor KJ, Wiedmann M (2001) Comparative genetic characterization of Listeria monocytogenes isolates from human and animal listeriosis cases. Microbiology 147:1095–1104
Keto-Timonen RO, Autio TJ, Korkeala HJ (2003) An improved amplified fragment length polymorphism (AFLP) protocol for discrimination of Listeria isolates. Syst Appl Microbiol 26:236–244. doi:10.1078/072320203322346083
Lee HY, Chai LC, Pui CF, Tunung R, Wong WC, Shuhaimi M, Cheah YK, Farinazleen MG, Nishibuchi M, Son R (2011) Using RAPD-PCR as molecular assessment on the performance of CHROMAgar™ Listeria and PALCAM agar on isolation of Listeria spp. and L. monocytogenes from foods. Int Food Res J 18:501–506. www.ifrj.upm.edu.my/18%20(02)%202011/(5)%20IFRJ-2010-250.pdf
Lundén JM, Autio TJ, Korkeala HJ (2002) Transfer of persistent Listeria monocytogenes contamination between food-processing plants associated with a dicing machine. J Food Prot 65:1129–1133
Malak M, Vivier A, André P, Decallonne J, Gilot P (2001) RAPD analysis, serotyping, and esterase typing indicate that the population of Listeria monocytogenes strains recovered from cheese and from patients with listeriosis in Belgium are different. Can J Microbiol 47:883–887
McLauchlin J, Mitchell RT, Smerdon WJ, Jewell K (2004) Listeria monocytogenes and listeriosis: a review of hazard characterisation for use in microbiological risk assessment of foods. Int J Food Microbiol 92:15–33. doi:10.1016/S0168-1605(03)00326-X
Mead PS, Dunne EF, Graves L, Wiedmann M, Patrick M, Hunter S, Salehi E, Mostashari F, Craig A, Mshar P, Bannerman T, Sauders BD, Hayes P, Dewitt W, Sparling P, Griffin P, Morse D, Slutsker L, Swaminathan B (2006) Nationwide outbreak of listeriosis due to contaminated meat. Epidemiol Infect 134:744–751. doi:10.1017/S0950268805005376
Nadon CA, Woodward DL, Young C, Rodgers FG, Wiedmann M (2001) Correlations between molecular subtyping and serotyping of Listeria monocytogenes. J Clin Microbiol 39:2704–2707. doi:10.1128/JCM.39.7.2704-2707.2001
Ryser T, Marth EH (2007) Listeria, listeriosis and food safety, 3rd edn. CRC Press, Boca Raton, pp 21–53
Samuel L, Marian MM, Apun K, Lesley MB, Son R (2011) Characterization of Escherichia coli isolated from cultured catfish by antibiotic resistance and RAPD analysis. Int Food Res J 18(3):971–976. www.ifrj.upm.edu.my/18%20(03)%202011/(16)IFRJ-2010-089.pdf
Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, Roy SL, Jones JL, Griffin PM (2011) Foodborne illness acquired in the United States--major pathogens. Emerg Infect Dis 17:7–15. doi:10.3201/eid1701.091101p1
Schraff RL (2012) Economic burden from health losses due to foodborne illness in the United States. J Food Prot 75(1):123–131. doi:10.4315/0362-028X.JFP-11-058
Struelens MJ (1996) Consensus guidelines for appropriate use and evaluation of microbial epidemiologic typing systems. Clin Microbiol Infect 2:2–11. doi:10.1111/j.1469-0691.1996.tb00193.x
Suihko ML, Salo S, Niclasen O, Gudbjörnsdóttir B, Torkelsson G, Bredholt S, Sjöberg AM, Gustavsson P (2002) Characterization of Listeria monocytogenes isolates from the meat, poultry and seafood industries by automated ribotyping. Int J Food Microbiol 72:137–146. doi:10.1016/S0168-1605(01)00631-6
Unnerstad H, Nilsson I, Ericsson H, Danielsson-Tham ML, Bille J, Bannerman E, Tham W (1999) Division of Listeria monocytogenes serovar 1/2a strains into two groups by PCR and restriction enzyme analysis. Appl Environ Microbiol 65:2054–2056
Wagner M, Maderner A, Brandl E (1999) Development of a multiple primer RAPD assay as a tool for phylogenetic analysis in Listeria spp. strains isolated from milk product associated epidemics, sporadic cases of listeriosis and dairy environments. Int J Food Microbiol 52:29–37. doi:10.1016/S0168-1605(99)00087-2
Welsh J, McClelland M (1990) Fingerprinting genomes using PCR with arbitraty primers. Nucleic Acids Res 18:7213–7218. doi:10.1093/nar/18.24.7213
Yoshida T, Takeuchi M, Sato M, Hirai K (1999) Typing Listeria monocytogenes by Random Amplified Polymorphic DNA (RAPD) fingerprinting. J Vet Med Sci 61(7):857–860. doi:10.1292/jvms.61.857
Acknowledgments
This study is supported by the Natural Science Foundation of Hebei Province (No.C2006000514).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Kang, Cy., Yu, Hw., Guo, Rf. et al. Genetic diversity of isolates of foodborne Listeria monocytogenes by RAPD-PCR. Ann Microbiol 66, 1057–1064 (2016). https://doi.org/10.1007/s13213-015-1186-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-015-1186-y