
Abstract
Human interleukin-3 (hIL-3) is a clinically important cytokine used to treat hematological malignancies, bone marrow transplantation, cytopenias, and immunological disorders. The cloning of hIL-3 gene was previously reported by our group, where its expression was optimized under methanol-inducible AOX1 promoter having N-terminal α mating factor signal sequence from Saccharomyces cerevisiae. This study investigated the role of glycosylation pattern on its molecular stability, secretion efficiency, and biological activity using the mutagenesis approach. The two N-linked glycosylation positions at N15th (Asn15) and N70th (Asn70) were sequentially mutated to generate three recombinant hIL-3 variants, i.e., N15A, N70A, and N15/70A. Asparagine at these positions was replaced with non-polar alanine amino acid (Ala, A). The alteration of N-linked glycosylation sites was disadvantageous to its efficient secretion in Pichia pastoris, where a 52.32%, 36.48%, 71.41% lower production was observed in N15A, N70A, and N15/70A mutants, respectively, as compared to native control. The fully glycosylated native hIL-3 protein showed higher thermal stability over its deglycosylated counterparts. The biological activity of native, N15A, N70A, and N15/70A hIL-3 protein was evaluated, where N15/70A mutant showed slightly higher proliferation efficacy than other combinations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The pleiotropic cytokine, human interleukin-3 (hIL-3), is released by antigen-activated monocytes, T-lymphocytes, endothelial, keratinocytes, and NK cells. It enables the maturation of megakaryocytes, mast cells, erythrocytes, and granulocytes from hematopoietic progenitor cells via differentiation and proliferation (Ding et al. 2003; Westers et al. 2006; Dagar et al. 2016b). Thus, it has enormous potential as a therapeutic agent to treat hematological malignancies, bone marrow transplantation, cytopenias, and immunological disorders (Su et al. 2010; Dagar et al. 2016b). The hIL-3 gene encodes for 152 amino acids which comprise a nineteen amino-acid hydrophobic signal sequence with N-linked glycosylation at the 15th and 70th amino-acid positions. The differentially glycosylated hIL-3 mature protein in a heterologous system produced a relatively higher molecular weight ranging from ~ 18 kDa to 35 kDa (Dagar et al. 2016b; Dagar and Khasa 2018).
The methylotrophic yeast P. pastoris is an excellent heterologous expression host exploited for laboratory and industrial applications over 20 years (Macauley-Patrick et al. 2005; Ahmad et al. 2014; Adivitiya et al. 2018; Liu et al. 2019). Pichia is favorably used as a heterologous expression platform because of its unique features, such as ease of genetic manipulations, the competence of performing post-translational modifications (PTMs), and endotoxins free recombinant protein production using high cell density fermentation processes on cost-effective media. The high-level product yields and selective secretion of recombinant proteins in the culture medium contribute to the designing of economical downstream processing strategies (Potvin et al. 2012; Ahmad et al. 2014; Looser et al. 2015; Liu et al. 2019).
The glycosylation process is a critical post-translational protein modifications event that immensely contributes to the stability, conformation, and biological activity of expressed proteins (Skropeta 2009; Wang et al. 2014; Radoman et al. 2021). The glycosylation frequency also affects the protein quality, cellular growth, and host’s production capabilities. Moreover, glycosylation is desirable for bio-therapeutics as it affects their solubility, bioactivity, in-vivo half-life, and immunogenicity (Skropeta 2009; Radoman et al. 2021).
Glycosylation is evolutionarily conserved in eukaryotes and is primarily of two types, i.e., N- and O-linked (Radoman et al. 2021). In the eukaryotic system, the amide group of the asparagine residues (~ 70–90%) has the N-linked glycans within consensus Asn-Xxx-Ser/Thr sequence where Xxx cannot be Proline. Moreover, N-linked glycosylation significantly contributes in the stability, secretion efficiency, and biological activity of heterologously expressed proteins (Lee et al. 2003; Skropeta 2009; Wang et al. 2014; Dotsenko et al. 2016). In many reports, the overall effects of individual glycosylation sites are also critical, where mutation of these sites can lead to undesirable changes in protein properties (Haraguchi et al. 1995; Newrzella and Stoffel 1996; Radoman et al. 2021). The removal of N-glycosylation site (N64Q) from the bovine enterokinase light chain (bEKL) significantly increased the catalytic efficiency, whereas the glycosylated bEKL is more thermostable over its non-glycosylated counterpart (Wang et al. 2018). In another report, the secretory production of recombinant streptokinase was significantly compromised (~ 46%) after the de-N-glycosylation of N14, N265, and N377 N-glycosylation residues sites, whereas a marginal decrease in its specific activity was noticed (Adivitiya et al. 2018). The gene of tetanus toxin fragment C (TetC) contains five potential N-linked glycosylation sites. The alteration of glycosylation sites of TetC was deleterious to its secretory production in P. pastoris (Wang et al. 2020).
The amino-acid sequence of hIL-3 consists of two potential N-glycosylation positions at the Asn-15 and Asn-70 amino-acid positions (N15 and N70). Previously, we had reported the expression of a differentially glycosylated hIL-3 protein in the Pichia expression system (Dagar et al. 2016b). Therefore, in the present work, a site-directed mutagenesis approach was employed by altering Asn to Ala amino acid at the glycosylation site to elucidate their role in molecular stability, secretion efficiency, and its biological activity. The recombinant hIL-3 protein variants were expressed in P. pastoris and compared.
Materials and methods
Expression vectors, host strains, and chemical reagents
The cloning and vector propagation was done in DH5α (Escherichia coli) strain. Yeast expression host (P. pastoris GS115) and pPICZαA vector were available in EasySelect™ Pichia Expression Kit from Invitrogen (Carlsbad, CA, USA). Zeocin was used as a selection antibiotic and also obtained from Invitrogen. The reagents used for PCR, cloning (restriction enzymes, T4 DNA ligase, Taq DNA polymerase, and dNTPs mix), de-N-glycosylation (PNGase F) were obtained from NEB (USA). The primers for PCR reaction and TF-1 cell line for biological activity assay were procured from Sigma-Aldrich (USA). The Ni–NTA Superflow resin was obtained from Qiagen (Hilden, Germany). The constituents for bacterial and yeast culture media were obtained from HiMedia (India).
Generation of recombinant constructs and site-directed mutagenesis
The generation of recombinant construct pPICZαA-hIL-3-cHis (native) has been described earlier (Dagar et al. 2016b) and used as a template to develop new constructs via site-directed mutagenesis (SDM). Briefly, the codon-optimized hIL-3 gene was cloned in Xho1 and Not1 restriction sites, containing a C-terminus 6 × His tag for downstream processing of hIL-3 protein. The desired amino acids were changed at the 15th and 70th positions by site-directed mutagenesis approach using manufacturer instructions (QuickChange II Site-Directed Mutagenesis Kit, Stratagene, USA). Primer FM15 (5’-GAAAACTTCTTGGGTGGCCTGCAGCAACATGATC-3’) and RM15 (5’-CGTCGATCATGTTGCTGCAGGCCACCCAAGAAG-3’) were used to mutate the 15th position, whereas FM70 (5’-GTTAAGTCTCTGCAGGCCGCGTCCGCTATTG-3’) and RM70 (5’-GCTTTCAATAGCGGACGCGGCCTGCAGAGAC-3’) were used to mutate the 70th position of N-glycosylation site. The double mutant (N15/70A) was developed using the FM70/RM70 mutagenic primer, where a confirmed single mutant construct (N15A) was used as a template. The PCR amplification was accomplished as per the conditions described earlier (Adivitiya et al. 2018). The impurity of methylated DNA used as a template was digested with the Dpn1 restriction enzyme from the PCR product. The treated PCR product was transformed into DH5α E. coli competent cells to amplify plasmids carrying desired mutations. Three mutant constructs viz. N15A (N15 mutated), N70A (N70 mutated), and N15/70A (both sites mutated) were developed in this work. All constructs were sequenced to check the incorporation of desired changes in the hIL-3 gene sequence (CIF, Delhi University-South Campus).
Transformation, screening, and copy number estimation
The Sac1 restriction enzyme was used for the linearization of all mutant constructs (N15A, N70A, and N15/70A). The linearized mutant recombinant constructs were transformed into Pichia GS115 expression host by electroporation using Bio-Rad Micropulser Electroporator (Bio-Rad, USA) as per the instruction manual of EasySelect™ Pichia Expression Kit (Invitrogen, USA). The transformed Pichia GS115 cells were plated on the YPDS agar plate (20 g/L D-glucose, 1 M Sorbitol, 20 g/L peptone, and 10 g/L yeast extract) containing 100 mg/L of Zeocin for 72 h in the incubator at 30 °C. The transformed Pichia cells were further patched on a YPD agar plate with 500 mg/L of Zeocin to select the over-expressing clones. The colonies were further screened with the PCR amplification method where alcohol oxidase 1 (AOX1) and hIL-3 primers were employed to validate hIL-3 expression cassette insertion into the Pichia genome. The isolation of genomic DNA and quantification of gene copy numbers in positive transformants were done via a qPCR strategy as per the protocol described earlier (Dagar et al. 2016b).
Small scale expression studies and protein quantification
A loopful culture of different recombinant Pichia clones containing native and mutant combinations from glycerol stock was inoculated in a 5 mL YPD seed medium. The primary culture was grown under the aerobic condition in the YPD medium, where seed culture was transferred into the 20 mL of BMGY medium for cell growth (Dagar et al. 2016). The culture broth was harvested after getting the desired OD600 of 8.0–10.0 (2000 rpm for 5 min). The cell’s pellet was subsequently resuspended into 20 mL of BMMY production medium, where glycerol was replaced by 0.5% methanol to induce protein expression. At every 12 h intervals, a 0.5% final concentration of methanol was added to maintain induction pressure as well as cellular growth. Culture broth samples were collected at regular intervals of 12 h till 0–96 h to quantify OD600, cell biomass, protein concentration, and product quality on 15% SDS-PAGE. The secreted recombinant protein into the culture supernatant was estimated via Bio-Rad protein Assay Kit (Bio-Rad, Hercules, USA), where Bovine Serum Albumin (BSA), supplied with the kit, was utilized as control (Bradford 1976).
Purification of recombinant hIL-3 protein variants
Amicon Stirred Cell 3000 MWCO was used to concentrate and buffer exchange for different hIL-3 protein combinations, i.e., native and mutant from culture supernatant (Millipore, Billerica, MA, USA). The Ni–NTA Superflow resin was equilibrated using buffer-1 (20 mM imidazole, 50 mM potassium phosphate buffer, and 300 mM NaCl). The culture supernatant was loaded on the equilibrated column for the binding of the recombinant protein. The equilibration buffer-1 was also used to wash the column to remove the residual (unbound) protein and collected as a wash fraction. The column-bound recombinant protein was eluted using 250 mM imidazole in buffer-1 (10 mL), where 1 mL elution fractions were collected. Bradford assay was used to quantify the purified hIL-3 protein from the different elution fractions. The purity and glycosylation patterns were analyzed on 15% SDS-PAGE. The eluted fractions of recombinant hIL-3 protein variants were dialyzed and concentrated against pH 8.0, 10 mM Tris–HCl buffer solution for further experiments.
De-N-glycosylation and biological activity of purified recombinant hIL-3 protein variants
All the purified combinations were subjected to the de-N-glycosylation analysis to confirm the degree of glycosylation using PNGase F (Peptide-N-Glycosidase F), which cleaved the innermost N-acetyl-glucosamine (GlcNAc) and asparagine residues (Asn, N). Briefly, 10 μg of purified recombinant hIL-3 protein variants were denatured in 2.5 µL of glycoprotein denaturing buffer (1X) at 100 °C by boiling the mixture for 10 min and subsequently cooled to 25 °C. The reaction mixture was prepared by adding 5.0 µL of G7 buffer (10X), 5.0 µL of NP-40, and 1.0 µL of PNGase F enzyme followed by incubation at 37 °C for 4 h. The reaction mixture was studied on 15% SDS-PAGE to check the degree of glycosylation of recombinant hIL-3 protein variants.
The hIL-3 protein has the potential to proliferate human TF-1 cells, where its biological activity can be estimated by MTT assay. The different combinations of hIL-3 protein (purified recombinant hIL-3 protein variants) were tested on the TF-1 cell line for their proliferation efficacy. The detailed methodology and experimental arrangement for MTT assay had been reported earlier (Dagar et al. 2016b). The proliferation assay was done in triplicate and averaged for reporting.
Biophysical characterization of de-N-glycosylated recombinant hIL-3 variants
The fluorescence spectroscopy was done to evaluate the tertiary structure of the recombinant hIL-3 protein variants using a fluorescence spectrophotometer (Varian, Australia). A dedicated temperature controller was attached to the instrument to perform the reactions. The cuvette with a 1 cm path length having the excitation and emission slit width of 1 nm was used to record the spectra. The recombinant hIL-3 protein variants reconstituted in pH 8.0 buffer (10 mM Tris–HCl) was employed to get the fluorescence emission spectra. The Tryptophan (Trp) residue of the different proteins produced during this study was excited at 295 nm. The emission spectra were recorded from 300 to 500 nm wavelength.
The thermal stability of different protein combinations (recombinant hIL-3 protein variants) was determined by measuring its tryptophan fluorescence (344 nm) in the range of 20–95 °C at 5 °C intervals using fluorescence spectroscopy. At each temperature, the samples were equilibrated for 5 min, and scans were recorded. The fluorescence maxima were plotted against the temperature to calculate the melting temperature (Tm). The spectra of recombinant hIL-3 protein variants, were collected and averaged. The baseline of the spectra was corrected after the subtraction of blank buffer values.
Results
Development of de-N-glycosylated hIL-3 variants using site-directed mutagenesis and generation of recombinant Pichia strains
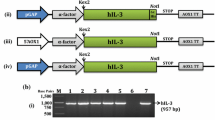
The generation of recombinant construct pPICZαA-hIL-3-cHis (native) has been described earlier (Dagar et al. 2016b). The codon-optimized and artificially synthesized hIL-3 gene was cloned under the methanol-inducible alcohol oxidase 1 (AOX1) promoter. The hIL-3 protein was targeted to the extracellular medium via α-mating factor signal sequence. The protein contained a C-terminal 6x-His tag for easy and cost-effective downstream processing. The hIL-3 protein sequence analysis using NetNGly 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/) showed two potent N-glycosylation sites at 15th and 70th (N15 & N70) positions (Ding et al. 2003). To evaluate the effect of each N-glycosylation position on hIL-3 properties during heterologous expression in the Pichia system, the amino acids at glycosylation positions were altered via site-directed mutagenesis strategy. The asparagine (Asn-15 and Asn-70) residues were replaced by small alanine (Ala-15 and Ala-70). It is a chemically inert, non-polar amino acid with a less-bulky methyl functional group. A total of three recombinant constructs were developed viz., N15A, N70A, and N15/70A and are schematically represented along with native construct designed in earlier work (Dagar et al. 2016b) (Fig. 1). The sequence accuracy of desired mutants was confirmed by DNA sequencing.

Schematic illustration showing the construction of different combinations of variants developed for hIL-3 production, i.e. A Native hIL-3 (developed and described earlier, Dagar et al. 2016b), B N15A, C N70A, D N15/70A construct
All construct combinations were linearized with Sac1 restriction enzyme and transformed into Pichia GS115 expression host to develop recombinant clones for the hIL-3 protein variants over-expression. The cells were screened on the different antibiotic concentrations from 100–500 mg/L of Zeocin, followed by colony PCR to select the positive transformants. The copy number of the different recombinant P. pastoris clones was determined by qPCR strategy (data not shown). For this work, the clones having one copy of the expression cassette were selected for shake flask expression studies.
Effect of de-N-glycosylation on hIL-3 secretion and expression yield
The small-scale expression studies were conducted at the shake flask level using single copy recombinant Pichia clones of hIL-3 varinats to elucidate the effect of N-linked glycosylation on the hIL-3 protein secretion. The cell growth was unhindered in hIL-3 protein variants clones after recombinant protein expression (Fig. 2A). Bradford assay was employed to determine the product concentration in the extracellular medium. Recombinant hIL-3 expression level decreased substantially in all mutants compared to the native construct. The maximum hIL-3 production level reached up to 135 mg/L in native, 64.36 mg/L in N15A, 85.74 mg/L in N70A, and 38.59 mg/L in N15/70A after 72 h of methanol induction (Fig. 2B). These results showed that an alteration in N-linked glycosylation sites deleteriously affected the hIL-3 production in P. pastoris. A decrease in secretion efficiency of 52.32% in the N15A, 36.48% in N70A, and 71.41% in N15/70A mutants was observed as compared to native control, confirming the essentiality of glycosylation to achieve optimal extracellular expression.

Cell growth and production profile of Pichia host expressing the recombinant hIL-3 protein variants. A Cell growth profile, B hIL-3 protein concentrationon (mg/L)
Purification and de-N-glycosylation analysis of recombinant hIL-3 variants
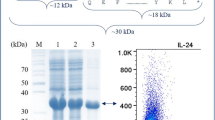
The native and mutant hIL-3 proteins having C-terminal 6x-His tag from P. pastoris GS115 were purified via affinity chromatography using Ni–NTA agarose resin from the culture supernatant. The SDS-PAGE results demonstrated that the native hIL-3 protein revealed a hyper-glycosylated band migrating at an MW of ~ 22–35 kDa, whereas it was ~ 18–35 kDa in N15A, ~ 22–35 kDa in N70A, and ~ 18 kDa in N15/70A mutant hIL-3 proteins (Fig. 3A). These results suggested a maximal contribution by N15 residue towards the hIL-3 hyper-glycosylation compared to position N70. These results are well in agreement with the prediction of the NetNGly 1.0 server where the N-glycosylation potential of N15 (0.70) was higher as compared to N70 (0.59) position (Gupta and Brunak 2002).

Purification and de-N-glycosylation analysis of recombinant hIL-3 protein variants (A). Purification of recombinant hIL-3 protein variants, where Lane M: MW marker, Lane 1: Native hIL-3 protein, Lane 2: N15A, Lane 3: N70A, Lane 4: N15/70A, and Lane 5: Fully non-glycosylated hIL-3 protein expressed in E. coli. B De-N-glycosylation analysis of recombinant hIL-3 protein variants, where Lane M: MW marker, Lanes 1–2: Analysis of native rhIL-3 protein, Lanes 3–4: N15A protein, Lanes 5–6: N70A protein, and Lanes 7–8: N15/70A protein before and after PNGase F digestion (− No PNGase, + With PNGase)
The purified recombinant hIL-3 protein variants were subjected to de-N-glycosylation analysis using the PNGase F enzyme. A sharp hIL-3 protein band was detected at the MW of ~ 18 kDa after PNGase F treatment in all the proteins (Fig. 3B). The de-N-glycosylated protein samples showed a comparable molecular weight on SDS-PAGE with the non-glycosylated hIL-3 protein obtained from E. coli (Dagar et al. 2016b).
Biophysical characterization of de-N-glycosylated recombinant hIL-3 variants
The fluorescence spectroscopy was used to study the intrinsic fluorescence property of recombinant hIL-3 protein variants expressed in the Pichia system. The hIL-3 protein contained two tryptophan and one tyrosine residue, which were responsible for its fluorescent properties. After excitation of recombinant hIL-3 protein variants at 295 nm, the maximum fluorescence emission was detected at 344 nm (Fig. 4).

Fluorescence emission spectra of recombinant hIL-3 protein variants expressed in P. pastoris
The thermal stability of proteins was assessed by studying the effect of temperature on their tertiary structure. The purified hIL-3 proteins were treated in a temperature range of 20–95 °C, and simultaneously, the fluorescence spectra were recorded at 344 nm. The graphs of fluorescence maxima were plotted against temperature (Fig. 5). The curve fitting showed that the apparent Tm was 52 °C for native hIL-3, 50 °C for N15A and N70A, whereas it was 47 °C for N15/70A mutant proteins (Fig. 5A-D). These results demonstrated that the fully glycosylated native hIL-3 protein showed a moderately high thermostability than its fully non-glycosylated (N15/70A) counterpart expressed in the Pichia expression system.

Thermal stability analysis of recombinant hIL-3 protein variants using fluorescence spectroscopy. A Native hIL-3 protein, B N15A, C N70A and D N15/70A. Melting temperature (Tm) values were indicated by a line
Biological activity of recombinant hIL-3 variants on TF-1 cell line
Different combinations of purified hIL-3 proteins, i.e., native, N15A, N70A, and N15/70A expressed during this study, were tested for their biological activity using MTT assay on TF-1 cells. The MTT assay was performed with appropriate negative and positive controls as described earlier (Kitamura et al. 1989; Dagar et al. 2016b). The maximum proliferation efficacy was observed in N15/70A, followed by N15A, N70A, and native hIL-3 protein. The overall proliferation efficiency of Pichia-produced hIL-3 protein variants was moderately better than the positive control (ProSpac, Israel). These observations established that the glycosylation of hIL-3 protein did not significantly favor its higher biological activity when expressed in P. pastoris (Fig. 6).

Biological activity measurement of recombinant hIL-3 protein variants expressed in P. pastoris. Various concentrations (0–32 ng/mL) of recombinanant hIL-3 protein variants were taken in 96 wells plate, and cell proliferation was measured using MTT assay
Discussion
The hIL-3 is a glycoprotein that participates in the differentiation and proliferation of hematopoietic progenitor cells (Ding et al. 2003; Westers et al. 2006; Dagar and Khasa 2018). It has therapeutic potential in bone marrow transplantation, immunodeficiency disorders, cytopenias, and hematological malignancies (Su et al. 2010; Dagar and Khasa 2018). Two potential N-linked glycosylation sites are present on the hIL-3 protein at the 15th and 70th amino-acid positions (Ding et al. 2003; Li et al. 2011). Protein glycosylation is an essential post-translational protein modification required for protein stability, conformation, and biological activity. The glycosylation also affects protein quality, in-vivo half-life, immunogenicity, as well as cellular growth and secretion efficiency. The N- and O-linked glycosylation are evolutionarily conserved in fungi and animals (Trombetta 2003; Macauley-Patrick et al. 2005; Skropeta 2009; Radoman et al. 2021). The N-glycosylation is common PTM of eukaryotic proteins expressed in the Pichia expression system that plays an essential role in the stability, secretion, and biological activity of heterologously expressed proteins (Lee et al. 2003; Dotsenko et al. 2016; Radoman et al. 2021). In the eukaryotic system, the amide group of the asparagine residues (~ 70–90%) has the N-linked glycans within the Asn-Xxx-Ser/Thr sequence (Trombetta 2003; Macauley-Patrick et al. 2005; Skropeta 2009; Radoman et al. 2021). It is reported in the literature that the alteration of the glycosylation site will change the properties of the expressed proteins (Haraguchi et al. 1995; Newrzella and Stoffel 1996; Radoman et al. 2021). The enzymatic activity and thermostability of glycosylated laccase Lcc9 (rLcc9) from Coprinopsis cinerea was slightly decreased as compared to deglycosylated rLec9 when produced in P. pastoris (Xu et al. 2019). Yoshimasu and co-worker reported that the Pichia-produced glycosylated porcine pepsin showed better structural stability and catalytic efficiency compared to its deglycosylated form (Yoshimasu et al. 2004). The AXE1 (acetyl xylan esterase) has two potential N-linked glycosylation sites. The secretion of deglycosylated AXE1 in P. pastoris was drastically reduced, whereas catalytic activity and stability were slightly affected compared to glycosylated control (Tian et al. 2012). The amino-acids sequence of hIL-3 consists of two sites, i.e., Asn-15 and Asn-70 (N15 and N70) for N-linked glycosylation. In an earlier report, we developed an efficient Pichia expression platform to produce a glycosylated hIL-3 protein (Dagar et al. 2016b). Herein, we report the effects of N-glycosylation on the secretion, stability, and biological activity of recombinant hIL-3 proteins produced in P. pastoris. Mutations were incorporated at the desired position at N15A, N70A, and N15/70A to investigate the effects of these glycosylation sites on protein secretion, stability, and biological activity.
Quantitative real-time PCR (qPCR) is one of the most commonly used techniques for quantifying the number of expression cassettes in the Pichia genome (Nordén et al. 2011; Dagar and Khasa 2018). Therefore, recombinant Pichia clones containing one copy of the expression cassette in the genome were selected for the small-scale expression studies at the shake flask level. The fully glycosylated native hIL-3 protein migrated at the molecular weight range of ~ 22–35 kDa on SDS-PAGE. Similarly, the mutated proteins migrated at the MW of ~ 18–35 kDa (N15A), ~ 22–35 kDa (N70A), and ~ 18 kDa (N15/70A), demonstrating the role of glycosylation on the differential molecular weight for recombinant proteins. These findings were well in agreement with the earlier results, where preferential glycosylation at the N15 position was reported (Ding et al. 2003; Dagar and Khasa 2018).
Further, we evaluated the effect of N-linked glycosylation on hIL-3 extracellular production using three mutants, i.e., N15A, N70A, and N15/70A and compared it with native control. The mutation at N15A, N70A, and N15/70A significantly decreased the protein secretion by 52.32%, 36.48%, and 71.41%, respectively, as compared to native control. The secretion level of N15A and N70A was moderately varied when compared to each other. The comparison of fully glycosylated (Native hIL-3) and fully non-glycosylated (N15/70A) showed a considerable difference in the production level. The drastic decrease in the expression level of hIL-3 protein variants might be due to the N-glycans, which influence the correct folding or proper translation of the protein. These findings established that the alterations of N-linked glycosylation sites in hIL-3 protein were deleterious to its production in P. pastoris. Several other reports also indicated similar behavior in the Pichia system (Skropeta 2009). The alteration of glycosylation site in elastase significantly affected its secretory production in P. pastoris, where it decreased to 23.9%, 63.6%, 63.7% in N43Q, N212Q, N280Q mutant, respectively, in comparison to wild-type control (Han et al. 2014). The expression of deglycosylated hydroxynitrile lyase (HNL) enzymes from Chamberlinius hualienensis in P. pastoris was drastically decreased in contrast to its wild-type control (Zhai et al. 2019). The secretion of other molecules such as ovalbumin and lipase also reduced significantly when expressed in P. pastoris after removing their N-linked glycosylation sites (Ito et al. 2007; Yang et al. 2015). Human epithelial lipase (EL) contains five potential N-linked glycosylation sites, where removal of Asn-373 N-glycosylation site resulted in a three fold decrease in production titer compared to wild-type EL (Gwen et al. 2004).
The Ni–NTA affinity chromatography was used to purify recombinant hIL-3 protein variants. The band arrangement of native hIL-3 protein (fully glycosylated) was similar to the earlier report (Li et al. 2011; Dagar et al. 2016b). These outcomes were further corroborated by the de-N-glycosylation analysis of recombinant proteins using PNGase F, where a protein band at a molecular weight position of ~ 18 kDa established the presence of N-linked glycosylation on hIL-3 protein (Bretthauer and Castellino 1999; Dagar et al. 2016b). The de-N-glycosylation data also depicts that the N15 glycosylation site has an added contribution compared to the N70 site. The band of the hIL-3 protein smeared due to differential glycosylation and was higher than its calculated MW. The varying conformations of the carbohydrate moieties can alter the interactions with gel or SDS binding, which does not reflect the correct size of the corresponding protein (Durchschlag et al. 1991).
The structural properties of native and mutant hIL-3 protein were confirmed by fluorescence spectroscopy. The fluorescence emission spectra maxima of all combinations was observed at 344 nm. These findings are also well supported by the earlier reports (Freeman et al. 1991; Curtis et al. 1991; Klein et al. 1999; Dagar et al. 2016a). The mutations in the hIL-3 protein did not affect its structural property. Wang and co-workers also reported that the confirmation of the polypeptide backbone was not affected by glycan modifications (Wang et al. 1996).
The presence of N-linked glycans enhances protein stability, plasma residence time, non-specific interactions and provides steric protection from proteases (Rudd et al. 2001; Hoffmeister et al. 2003; Sinclair and Elliott 2005; Skropeta 2009). In our study, the hIL-3 protein variants were evaluated for their thermal stability via fluorescence spectroscopy, where fully glycosylated hIL-3 (native hIL-3) protein showed higher thermal stability than its non-glycosylated counterparts (N15/70A). The partially glycosylated hIL-3 protein variants (N15A and N70A) showed almost similar thermostability, as both variants had single glycosylation site. The differential glycosylation pattern was reported in the literature because of the N-linked glycosylation potential of N15 (0.70) was higher as compared to N70 (0.59) position. Ding et al 2003 reported the differential glycosylation pattern of hIL-3 protein when expressed in the Baculovirus expression system. The purified hIL-3 protein was evaluated through HPLC-ESI-mass spectrometry, where the tryptic peptide containing N15 was found in its fully glycosylated form. In contrast, about 65% of the peptide bearing the N70 glycosylation site was present in its non-glycosylated form (Ding et al. 2003). Li et al (2011) reported the expression of native hIL-3 protein using P. pastoris expression system, where the protein was expressed in two forms with different degrees of N-glycosylation. The same was confirmed by MALDI-TOF-TOF mass spectrometry (Li et al. 2011). These results agreed well with an earlier report where Rhizopus chinensis lipase (RCL) showed higher thermal stability with the more N-linked glycosylation sites (Yang et al. 2015). The biological activity analysis of recombinant hIL-3 protein variants was evaluated using MTT assay on TF-1 cells (Kitamura et al. 1989). The mutant proteins showed a slightly better proliferation efficacy as compared to fully glycosylated hIL-3 protein. Similar results were observed with rhIL-4 protein, where deglycosylated rhIL-4 showed higher biological activity compared to its glycosylated form (Li et al. 2014). In conclusion, we successfully demonstrated the role of glycosylation on recombinant hIL-3 molecular integrity and enhanced secretion in heterologous expression hosts like Pichia pastoris. The N-glycosylation of recombinant protein enhanced its thermal stability. The proliferation efficacy of the non-glycosylated hIL-3 protein was slightly higher than its glycosylated counterpart. The present investigation will help the researchers to develop the technology for producing hIL-3 protein for its commercialization.
References
Adivitiya B, Mohanty S, Khasa YP (2018) Engineering of deglycosylated and plasmin resistant variants of recombinant streptokinase in Pichia pastoris. Appl Microbiol Biotechnol 102:10561–10577. https://doi.org/10.1007/s00253-018-9402-x
Ahmad M, Hirz M, Pichler H, Schwab H (2014) Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98:5301–5317. https://doi.org/10.1007/s00253-014-5732-5
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Bretthauer RK, Castellino FJ (1999) Glycosylation of Pichia pastoris-derived proteins. Biotechnol Appl Biochem 30(Pt 3):193–200
Curtis BM, Williams DE, Broxmeyer HE et al (1991) Enhanced hematopoietic activity of a human granulocyte/macrophage colony-stimulating factor-interleukin 3 fusion protein. Proc Natl Acad Sci U S A 88:5809–5813
Dagar VK, Adivitiya null, Khasa YP (2016) High-level expression and efficient refolding of therapeutically important recombinant human Interleukin-3 (hIL-3) in E. coli. Protein Expr Purif 131:51–59. https://doi.org/10.1016/j.pep.2016.11.005
Dagar VK, Khasa YP (2018) Combined effect of gene dosage and process optimization strategies on high-level production of recombinant human interleukin-3 (hIL-3) in Pichia pastoris fed-batch culture. Int J Biol Macromol 108:999–1009. https://doi.org/10.1016/j.ijbiomac.2017.11.008
Dagar VK, Adivitiya DN, Khasa YP (2016) Bioprocess development for extracellular production of recombinant human interleukin-3 (hIL-3) in Pichia pastoris. J Ind Microbiol Biotechnol 43:1373–1386. https://doi.org/10.1007/s10295-016-1816-9
Ding H, Griesel C, Nimtz M et al (2003) Molecular cloning, expression, purification, and characterization of soluble full-length, human interleukin-3 with a baculovirus–insect cell expression system. Protein Expr Purif 31:34–41. https://doi.org/10.1016/S1046-5928(03)00138-4
Dotsenko AS, Gusakov AV, Volkov PV et al (2016) N-linked glycosylation of recombinant cellobiohydrolase I (Cel7A) from Penicillium verruculosum and its effect on the enzyme activity. Biotechnol Bioeng 113:283–291. https://doi.org/10.1002/bit.25812
Durchschlag H, Christl R, Jaenicke R (1991) Comparative determination of the particle weight of glycoproteins by SDS-PAGE and analytical ultracentrifugation. In: Borchard W (ed) Progress in analytical ultracentrifugation. Steinkopff, Darmstadt, pp 41–56
Freeman JJ, Parr GR, Hecht RI et al (1991) Secondary structure of human interleukin-3. Int J Biochem 23:353–360
Gupta R, Brunak S (2002) Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput 7:310–322
Gwen M, Cj L, Ed B, et al (2004) Role of N-linked glycosylation in the secretion and activity of endothelial lipase. J. Lipid Res. https://pubmed.ncbi.nlm.nih.gov/15342690/.
Han M, Wang X, Ding H et al (2014) The role of N-glycosylation sites in the activity, stability, and expression of the recombinant elastase expressed by Pichia pastoris. Enzyme Microb Technol 54:32–37. https://doi.org/10.1016/j.enzmictec.2013.09.014
Haraguchi M, Yamashiro S, Furukawa K et al (1995) The effects of the site-directed removal of N-glycosylation sites from beta-1,4-N-acetylgalactosaminyltransferase on its function. Biochem J 312:273–280
Hoffmeister KM, Josefsson EC, Isaac NA et al (2003) Glycosylation restores survival of chilled blood platelets. Science 301:1531–1534. https://doi.org/10.1126/science.1085322
Ito K, Ishimaru T, Kimura F, Matsudomi N (2007) Importance of N-glycosylation positioning for secretion and folding of ovalbumin. Biochem Biophys Res Commun 361:725–731. https://doi.org/10.1016/j.bbrc.2007.07.066
Kitamura T, Tange T, Terasawa T et al (1989) Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol 140:323–334. https://doi.org/10.1002/jcp.1041400219
Klein BK, Olins PO, Bauer SC et al (1999) Use of combinatorial mutagenesis to select for multiply substituted human interleukin-3 variants with improved pharmacologic properties. Exp Hematol 27:1746–1756
Lee J, Park J-S, Moon J-Y et al (2003) The influence of glycosylation on secretion, stability, and immunogenicity of recombinant HBV pre-S antigen synthesized in Saccharomyces cerevisiae. Biochem Biophys Res Commun 303:427–432
Li H, Li N, Gao X et al (2011) High level expression of active recombinant human interleukin-3 in Pichia pastoris. Protein Expr Purif 80:185–193. https://doi.org/10.1016/j.pep.2011.08.027
Li R, Xie C, Zhang Y et al (2014) Expression of recombinant human IL-4 in Pichia pastoris and relationship between its glycosylation and biological activity. Protein Expr Purif 96:1–7. https://doi.org/10.1016/j.pep.2014.01.005
Liu W-C, Inwood S, Gong T et al (2019) Fed-batch high-cell-density fermentation strategies for Pichia pastoris growth and production. Crit Rev Biotechnol 39:258–271. https://doi.org/10.1080/07388551.2018.1554620
Looser V, Bruhlmann B, Bumbak F et al (2015) Cultivation strategies to enhance productivity of Pichia pastoris: a review. Biotechnol Adv 33:1177–1193. https://doi.org/10.1016/j.biotechadv.2015.05.008
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM (2005) Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270. https://doi.org/10.1002/yea.1208
Newrzella D, Stoffel W (1996) Functional analysis of the glycosylation of murine acid sphingomyelinase. J Biol Chem 271:32089–32095
Nordén K, Agemark M, Danielson JÅ et al (2011) Increasing gene dosage greatly enhances recombinant expression of aquaporins in Pichia pastoris. BMC Biotechnol 11:47. https://doi.org/10.1186/1472-6750-11-47
Potvin G, Ahmad A, Zhang Z (2012) Bioprocess engineering aspects of heterologous protein production in Pichia pastoris: a review. Biochem Eng J 64:91–105. https://doi.org/10.1016/j.bej.2010.07.017
Radoman B, Grünwald-Gruber C, Schmelzer B et al (2021) The degree and length of O-Glycosylation of recombinant proteins produced in Pichia pastoris depends on the nature of the protein and the process type. Biotechnol J 16:e2000266. https://doi.org/10.1002/biot.202000266
Rudd PM, Elliott T, Cresswell P et al (2001) Glycosylation and the immune system. Science 291:2370–2376
Sinclair AM, Elliott S (2005) Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci 94:1626–1635. https://doi.org/10.1002/jps.20319
Skropeta D (2009) The effect of individual N-glycans on enzyme activity. Bioorg Med Chem 17:2645–2653. https://doi.org/10.1016/j.bmc.2009.02.037
Su Y, Li S-Y, Ghosh S et al (2010) Characterization of variant diphtheria toxin-interleukin-3 fusion protein, DTIL3K116W, for phase I clinical trials. Biol J Int Assoc Biol Stand 38:144–149. https://doi.org/10.1016/j.biologicals.2009.08.016
Tian B, Chen Y, Ding S (2012) A combined approach for improving alkaline acetyl xylan esterase production in Pichia pastoris, and effects of glycosylation on enzyme secretion, activity and stability. Protein Expr Purif 85:44–50. https://doi.org/10.1016/j.pep.2012.06.008
Trombetta ES (2003) The contribution of N-glycans and their processing in the endoplasmic reticulum to glycoprotein biosynthesis. Glycobiology 13:77R-91R. https://doi.org/10.1093/glycob/cwg075
Wang C, Eufemi M, Turano C, Giartosio A (1996) Influence of the carbohydrate moiety on the stability of glycoproteins. Biochemistry 35:7299–7307. https://doi.org/10.1021/bi9517704
Wang XJ, Wang XM, Teng D et al (2014) Recombinant production of the antimicrobial peptide NZ17074 in Pichia pastoris using SUMO3 as a fusion partner. Lett Appl Microbiol 59:71–78. https://doi.org/10.1111/lam.12246
Wang Z, Guo C, Liu L, Huang H (2018) Effects of N-glycosylation on the biochemical properties of recombinant bEKL expressed in Pichia pastoris. Enzyme Microb Technol 114:40–47. https://doi.org/10.1016/j.enzmictec.2018.03.004
Wang N, Wang KY, Xu F et al (2020) The effect of N-glycosylation on the expression of the tetanus toxin fragment C in Pichia pastoris. Protein Expr Purif 166:105503. https://doi.org/10.1016/j.pep.2019.105503
Westers L, Dijkstra DS, Westers H et al (2006) Secretion of functional human interleukin-3 from Bacillus subtilis. J Biotechnol 123:211–224. https://doi.org/10.1016/j.jbiotec.2005.11.007
Xu G, Wu Y, Zhang Y et al (2019) Role of N-glycosylation on the specific activity of a Coprinopsis cinerea laccase Lcc9 expressed in Pichia pastoris. J Biosci Bioeng 128:518–524. https://doi.org/10.1016/j.jbiosc.2019.05.004
Yang M, Yu X-W, Zheng H et al (2015) Role of N-linked glycosylation in the secretion and enzymatic properties of Rhizopus chinensis lipase expressed in Pichia pastoris. Microb Cell Factories 14:40. https://doi.org/10.1186/s12934-015-0225-5
Yoshimasu MA, Tanaka T, Ahn J-K, Yada RY (2004) Effect of N-linked glycosylation on the aspartic proteinase porcine pepsin expressed from Pichia pastoris. Glycobiology 14:417–429. https://doi.org/10.1093/glycob/cwh024
Zhai Z, Nuylert A, Isobe K, Asano Y (2019) Effects of codon optimization and glycosylation on the high-level production of hydroxynitrile lyase from Chamberlinius hualienensis in Pichia pastoris. J Ind Microbiol Biotechnol 46:887–898. https://doi.org/10.1007/s10295-019-02162-w
Funding
The research work was supported by Department of Biotechnology (DBT), Government of India, New Delhi via Grant No. BT/PR5822/PID/6/684/2012 to Dr. Yogender Pal Khasa (Project Investigator). Vikas Kumar Dagar, acknowledges the financial support from the Indian Council of Medical Research (ICMR, Govt. of India) New Delhi as he received a senior research fellowship (Sanction No. 3/1/3-JRF-2009/MPD-89).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dagar, V.K., Babbal, Mohanty, S. et al. Effect of N-glycosylation on secretion, stability, and biological activity of recombinant human interleukin-3 (hIL-3) in Pichia pastoris. 3 Biotech 12, 221 (2022). https://doi.org/10.1007/s13205-022-03293-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-022-03293-1