
Abstract
Citrus greening disease or huanglongbing (HLB) caused by Candidatus Liberibacter asiaticus (CLas) limits citrus production worldwide. CLas is transmitted by the Asian citrus psyllid (ACP), Diaphorina citri (Hemiptera: Psyllidae) in a persistent-propagative manner. Understanding the molecular interaction between CLas and ACP and interrupting the interrelationship can provide an alternative to insecticides for managing citrus greening disease. Transcriptome analysis of ACP in response to CLas showed differential expression of 3911 genes (2196 upregulated, and 1715 downregulated) including the key genes of ACP involved in cytoskeleton synthesis and nutrition-related proteins, such as vitellogenins, extensin, laminin, tropomyosin, troponin C, and flightin. Majority of the differentially expressed genes were categorized under molecular functions followed by cellular components and biological processes. KEGG pathway analysis showed differential regulation of carbohydrate, nucleotide, and energy metabolic pathways, the endocytotic pathway, and the defense-related pathways. Differential regulation of genes associated with the key pathways might favour CLas to become systemic and propagate in its insect vector. The study provides an understanding of genes involved in circulation of CLas in ACP. The candidate genes involved in key physiological processes and CLas transmission by ACP would be potential targets for sustainable management of ACP and CLas.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Citrus greening disease, also known as huanglongbing (HLB) is a major limiting factor in citrus production worldwide. The disease is incited by a fastidious, phloem-limited, α-proteobacterium Candidatus Liberibacter spp. Three species viz. Candidatus Liberibacter asiaticus (CLas), C. L. africanus (CLaf), and C. L. americanus (CLam) are reported to be associated with the disease (Bové 2006; Gottwald et al. 2007; Vyas et al. 2015). Among them, CLas is the most widespread destroying about 100 million citrus trees in Asia (Gottwald et al. 2007). Infection of HLB induces yellow shoots, leaves with blotchy mottles, and small lopsided fruits. HLB is transported both upward and downward through the tree but their distribution is patchy. The infected branches start to die back and the infected tree dries up gradually. CLas and CLam are transmitted by Asian citrus psyllid (ACP, Diaphorina citri Kuwayama, Hemiptera: Psyllidae), whereas African citrus psyllid, Trioza erytreae (Del Guercio, Hemiptera: Psyllidae) is the primary vector of CLaf in Africa (Ghosh et al. 2018). These two psyllids are the only known insect vectors of HLB and economic psyllid species on citrus in the world (Halbert and Manjunath 2004). ACP is thought to be originated in Asia, but it is also prevalent in parts of the Middle East, South, and Central America, Mexico, and the Caribbean. The relationship of CLas with ACP is a persistent-propagative type and there is evidence of transovarial transmission by ACP (Halbert and Manjunath 2004). CLas is acquired by ACP from infected citrus plants during nymphal or adult stages and carried in the hemolymph and salivary glands of psyllid. The infected ACP transfers the CLas to phloem of a healthy citrus plant during feeding sap. Similar to other insect-borne phytopathogens, CLas manipulates ACP to facilitate their spread and transmission either directly or through indirect mediation through plants (Galdeano et al. 2020). CLas infection induces karyorrhexis and apoptosis in the midgut of ACP (Mann et al. 2018). Largely, CLas exerts a positive effect on the fitness and behavior of ACP by enhancing the fecundity, fertility, and population growth of the latter (Pelz-Stelinski and Killiny 2016; Ren et al. 2016). Besides, immune responsive genes, such as catalase, cathepsin, transferrin, and metabolism-related genes like tropomyosin, ATPases are differentially regulated in CLas-infected ACP (Ramsey et al. 2017; Yu et al. 2019).
The primary strategy for management of HLB is to restrict the psyllid vectors. Application of insecticides is widely adopted to manage psyllid that exerts detrimental effects on the environment and nontarget organisms. Development of insecticide resistance in psyllids brings more complexity to the management of HLB. Understanding the intricate relationship of ACP with CLas may open up novel targets to manage the disease by interrupting the interrelationship. In the recent years, transcriptomics has emerged as a promising approach to study the global differential gene expression and also serves as a platform for gene discovery by facilitating comparative genomics. Antennal and abdominal transcriptome of ACP has helped to identify the potential chemosensory proteins (Wu et al. 2016). The differentially expressed transcripts of ACP in response to CLas may facilitate to identify putative genes involved in the transmission mechanism. In the present experiment, a transcriptome-wide response of ACP to CLas infection has been reported and expressions of few highly regulated genes have been validated. This knowledge may be useful in future studies to identify potential molecular targets to interfere with CLas transmission by ACP and management of the disease.
Materials and methods
Establishing a homogeneous population of ACP, sample preparation, and nucleic acid isolation
The initial population of ACP was collected from the experimental field of Indian Agricultural Research Institute (IARI), New Delhi. A homogeneous CLas-free population of ACP was established on Murraya koenigii plant from a single female (supplementary Fig. 1). The population was maintained in controlled conditions at 25 ± 2 °C, 60 ± 10% RH, and 8 h dark. The next three generations were tested in PCR to ascertain the absence of CLas. Eggs were collected from the homogeneous population with a fine Camel hairbrush. Eggs were placed on healthy and CLas-infected citrus plants separately and reared up to the adult stage at the same environmental conditions. A few adult ACP was randomly collected from healthy and CLas-infected citrus plants for testing. The infection status of plants and psyllids were tested in PCR by amplifying the 809 bp region of outer membrane protein (omp) gene using Omp1218f and Omp2026r primers (Table 1) (Deng et al. 2008). Total DNA was isolated from citrus plants and ACP using GeneJet Plant Genomic DNA Purification Mini Kit (Thermo Fisher Scientific, MA, USA), and DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany), respectively following manufacturers' protocol. PCR was performed in 25 µl final reaction volume containing 1 × DyNAzyme buffer with 1.5 mM MgCl2, 0.5 µM of each forward and reverse primers, 0.5 mM of dNTPs, 50 ng of template DNA, and 1 unit of DyNAzyme II DNA polymerase (Thermo Fisher Scientific). The PCR conditions of 3 min of initial denaturation at 94 °C, followed by 35 cycles of 30 s of denaturation at 94 °C, 30 s of annealing at 52 °C, 45 s of extension at 72 °C, and a single final extension of 10 min at 72 °C were used in a thermal cycler (T-100, Bio-Rad, California, USA). PCR products were resolved on 1.6% agarose gel containing ethidium bromide and visualized in a UV transilluminator.
Two sets of CLas-infected adult ACP (designated as P + CLas1 and P + CLas2) and one set of healthy adult ACP (designated as P-CLas) were subjected to total RNA isolation using TRIzol (Thermo Fischer Scientific) following manufacturer's protocol. Each of the batches consisted of 10 adults of ACP. RNA concentration and quality were assessed using standard procedures as recommended for Illumina (Illumina, San Diego, CA, USA) sequencing. Another set of samples (both P + CLas, P-CLas) obtained from the same batch were preserved in -80 ºC for qRT-PCR experiments.
Library preparation and sequencing
In brief, 500 ng of total RNA was used to enrich mRNA using Magnetic mRNA Isolation Kit (NEB, MA, USA). The transcriptome library was prepared using NEB ultraII RNA library prep kit and sequenced using Illumina Next Seq 500 paired-end technology. The enriched mRNA was fragmented (approximately 200 bp) using a fragmentation buffer. Random hexamer primers were then added and hybridized to complementary RNA sequences. These short fragments were used as templates to synthesize the first-strand cDNA using reverse transcriptase and dNTPs. RNA in the synthesized DNA–RNA hybrids after first-strand cDNA synthesis was degraded using RNase H followed by conversion of single-stranded cDNA into double-stranded cDNA using E. coli DNA polymerase I. The double-stranded cDNA fragments were purified using 1.8X Ampure beads. The purified double-stranded cDNA was end-repaired to ensure that each molecule was free of overhangs and had 5′ phosphates and 3′ hydroxyls before the adaptor ligation. The adaptor-ligated DNA was purified using Ampure beads and was enriched using specific primers, compatible with sequencing on to the Illumina platforms. The final enriched library was purified and quantified by Qubit and the size was analyzed by Bioanalyzer.
Preprocessing of raw reads and differential gene expression analysis
The quality of raw reads was visualized using FASTQC v0.11.2. Raw reads were preprocessed by removing the adaptor sequences, duplicated sequences, ambiguous and low-quality reads (Phred score < 33) to obtain high-quality reads. After preprocessing, the quality of reads was ensured again using FASTQC. Preprocessed reads were mapped to the reference genome of D. citri (GCF_000475195.1) using TopHat v2.1.0 software with default parameters except for the mismatch parameter that was set to two. Gene expression was quantified using Subread v1.5.0 software with default parameters based on the Fragments Per Kilobase of transcript sequence per millions base pairs-sequenced (FPKM values). Average FPKM value of expressed genes from two P + CLas samples (designated as combined P + CLas) was compared with FPKM value of expressed genes of P-CLas sample for identification of differentially expresses genes (DEGs) using edgeR v1.24.0 software (without biological replicates). Genes having a normalized P value < 0.05 were only considered for differential expression analysis. Genes with FPKM values > 1 and < − 1 were regarded as up- and downregulated, respectively. Gene Ontology (GO) enrichment analysis of DEGs was performed (corrected p value < 0.05) using a web-based tool AgriGO (Du et al. 2010). KEGG pathway enrichment analysis of DEGs was performed to identify the pathways that were differentially regulated between combined P + CLas and P-CLas samples using the software KOBAS v3.0 with corrected p value < 0.05.
Quantitative real-time-PCR (qRT-PCR) analysis
To validate the most differentially regulated genes (vitellogenin-1, vitellogenin-2, troponin C and tropomyosin-1) of ACP, the top two genes each from up- and downregulated were selected. Primers were designed for the target genes using the web-based tool Primer3 v.0.4.0 (https://bioinfo.ut.ee/primer3-0.4.0/). The primer sequences used in this study are listed in Table 1. The annealing temperature of the primer sets was optimized in a gradient PCR. qRT-PCR was performed using the designed primers and cDNA from P + CLas and P-CLas as templates to ensure the specific amplification of the primers. qRT-PCR analysis was performed in Insta Q 48 m (Himedia) using the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific) and melting curves were analyzed for specificity of the reaction. α-tubulin was used as a reference for normalization of gene expression (Bin et al. 2019). Firstly, total RNA was isolated from P-CLas and P + CLas psyllid samples stored in – 80 °C using ZR Tissue and Insect RNA MicroPrep (ZymoResearch, California, USA). DNase treatment was performed using DNase I (Thermo Fisher Scientific) following the manufacturer's protocol. First-strand of cDNAs were synthesized with Oligo(dT)12–18 primer using First-strand cDNA synthesis kit (Thermo Fisher Scientific) following manufacturer's instructions. The 30 µl final qRT-PCR reaction mixture consisted of 1 × Maxima SYBR Green/ROX qPCR master mix (Thermo Fischer Scientific), 0.5 µM of each forward and reverse primers, and 300 ng of cDNA. Following thermal cycling was followed, initial denaturation at 94 °C for 3 min, then 30 cycles of 94 °C for 30 s, 62 °C for 30 s, and 72 °C for 30 s followed by a melting stage. A total of three biological and two technical replicates were used for qRT-PCR analysis. The relative gene expression was calculated using the \({2}^{{ - \Delta \Delta C_{{\text{T}}} }}\) method (Livak and Schmittgen 2001). Statistical significance of the qRT-PCR data was analyzed using Tukey's multiple comparison tests. Values with p < 0.05 were considered to be statistically significant.
Results
CLas infection in ACP and citrus plant
PCR with CLas-infected citrus plant and ACP yielded corresponding amplification of 809 bp in agarose gel electrophoresis (Supplementary Fig. 2). No amplification was observed in the case of CLas-free citrus plant and ACP exposed to healthy citrus plants.
Pre-processing of raw data and mapping of clean reads to reference genome
Raw reads obtained from three libraries (P + CLas1, P+CLas2, and P-CLas) ranged from 63.96 to 77.18 million with an average of 68.82 million. 98.93–99.21% of the total raw reads in three libraries qualified as clean reads after preprocessing steps. The proportion of clean reads that mapped to ACP genome ranged from 62.8 to 48.4% across libraries with an average of 58.7%. Among the mapped reads, 39.2–47.5% of the total reads were uniquely mapped while 9.2–15.3% of the total reads were mapped to multiple loci in the genome (Table 2).
Global gene expression analysis
Genes having FPKM values greater than or equal to 1 were considered to be expressed. A total of 17,437 and 17,059 expressed genes were found in P + CLas1 and P + CLas2 samples, respectively while 15,444 genes were expressed in P-CLas. In DEG analysis using edgeR with BH correction, a heatmap of significantly expressed genes was generated. It was observed that the P + CLas samples clustered together as their gene expression patterns were nearly similar. However, both the samples differed from P-CLas indicating their differential gene expression pattern from the latter (Fig. 1).

The overall results of FPKM cluster analysis, clustered using the log2 (FPKM + 1) value. Red denotes genes with high expression levels, and blue denotes genes with low expression levels. The color range from blue to red represents the log2 (FPKM + 1) value from large to small. P-CLs denotes the ACP population without an exposure to CLas, while P + CLas is CLas-infected ACP population
Differential gene expression analysis
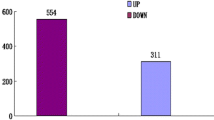
In total, 3911 genes (27%) were found to be differentially expressed between combined P + CLas and P-CLas samples while the expression of 73% of the genes remained unaltered (Fig. 2a). Among the DEGs, 2196 and 1715 genes were upregulated and downregulated, respectively in combined P + CLas as compared to P-CLas (Fig. 2b). The top 20 differentially expressed (up and downregulated) genes in CLas-infected ACP have been listed in Table 3. Nutrition-related genes of ACP like vitellogenin-1, -2, and -3, and extensin were found highly overexpressed post exposure to CLas. Significant downregulation of cytoskeleton-related genes of ACP, such as myosin heavy chain, troponin C, tropomyosin-1, E3 ubiquitin, and flightin was also recorded. Besides, few genes possibly involved in CLas-ACP interaction were differentially expressed, such as laminin, papilin, integrin, talin, phenoloxidase, serine proteases, hemocytin, cathepsin B, and ABC transporters.

Differential gene expression analysis. a Proportion of differentially expressed genes in the total transcriptome. b Volcano plot for differentially expressed genes. The x axis shows the fold change in gene expression between different samples, and the y axis shows the statistical significance of the differences. Significantly up- and downregulated genes are highlighted in red and green, respectively
Functional analysis of DEGs
Gene ontology (GO) terms were assigned to the 3911 DEGs. Based on the GO terms, DEGs were broadly grouped into three categories viz. genes associated with cellular components, biological processes, and molecular functions. The majority of the DEGs were categorized under molecular functions (50%) followed by cellular components (33%) and biological processes (17%) (Fig. 3a). In molecular function category, genes associated with catalytic activity, binding, and nucleotide-binding were mostly enriched. Genes associated with cell, cell parts, membrane subcategories were differentially enriched in cellular component category. In the biological processes, genes associated with metabolic, cellular, and primary metabolic processes were the most enriched (Fig. 3b). Figure 4 shows the highly enriched molecular functions in CLas-infected ACP.

GO enrichment analysis of DEGs. a Proportion of differentially expressed genes that participate in biological processes, cellular components and molecular functions. b Major processes and functions that are affected in CLas-infected ACP. In molecular function category, genes associated with catalytic activity, binding, and nucleotide-binding were mostly enriched. Genes associated with cell, cell parts, membrane sub-categories were differentially enriched in cellular component category. In the biological processes, genes associated with metabolic, cellular, and primary metabolic processes were the most enriched

Illustration of top GO DAG under molecular functions category. Each node represents a GO term, the darker the color is, the higher is the enrichment level of the term. The name and p value of each term are present on the node
KEGG pathway analysis of DEGs
Based on the KEGG pathway analysis, the DEGs were involved in metabolism, environmental and nucleotide signal processing, organismal systems, and cellular processes. The major metabolic pathways affected include carbohydrate, nucleotide, and energy metabolism while transcription, translation, folding, sorting, and degradation were differentially regulated nucleotide signal processing pathways. Genes involved in signal transduction and aging that aid in environmental signal processing and organismal systems, respectively, were also differentially regulated. Transport and catabolism related pathways remained the most affected cellular processes (Fig. 5). The results of KEGG pathway analysis were consistent with the pathways identified in GO enrichment analysis.

KEGG pathway analysis of DEGs in D. citri upon CLas infection. Major pathways affected in CLas infected ACP are shown. DEGs were involved in metabolism, environmental, and nucleotide signal processing, organismal systems, and cellular processes
Validation of DEGs in qRT-PCR
The expression of vitellogenin-1 and vitellogenin-2 genes was 8.0 and 8.4-fold higher, respectively, in P + CLas sample as compared to P-CLas. However, troponin C and tropomyosin-1 genes were downregulated to 6.0 and 5.4-fold respectively in P + CLas sample in comparison with P-CLas samples. Similarly, RNA-seq analysis showed that expression of vitellogenin-1 and vitellogenin-2 genes were upregulated to 18.4 and 17.0-fold, respectively, while the troponin C and tropomyosin-1 genes were downregulated to 5.8 and 5.0-fold, respectively, in CLas-infected ACP samples (Fig. 6). Melt curve analysis in qRT-PCR confirmed the specificity of the reactions (Supplementary Fig. 3).

Validation of the differential expression of selected genes related to insect cytoskeleton and nutrition using qRT-PCR. The expression of DEGs in qPCR (blue) and RNA-Seq (green) is shown. A total of three biological and two technical replicates were used for qRT-PCR analysis. The data are mean ± standard error. Error bars are standard error of mean (SEM)
Discussion
A host–pathogen–vector relationship involves complex direct and indirect interactions where pathogen and vector compete with each other for a shared host. They can affect each other by altering the biochemical traits of the host. A hypothesis of the manipulation of vectors by the pathogens has been proposed by Ingwell (2012). This could occur by modifying the physiological processes of the vector directly (Fiebig et al. 2004; Belliure et al. 2008). Both positive and negative effects of plant pathogens on their vectors have been recorded (Belliure et al. 2008; Ghosh et al. 2016, 2019). CLas is transmitted by ACP in a persistent-propagative manner (Halbert and Manjunath 2004). Multiplication of CLas in ACP suggests a potential alteration in the physiological process of ACP. ACP nymphs acquire CLas from infected hosts efficiently than the adults and become viruliferous in late instars or after emergence as adults. ACP adults can also acquire the virus in 15–30 min feeding on infected host plants, but an increase in titer of CLas was found when acquired by nymphs (Tufail et al. 2014). Once acquired, CLas crosses the mid-gut barrier and reaches hemolymph of ACP. It is thought to be transported to the salivary glands and accumulates there to infect healthy hosts during feeding and salivation. CLas also invades reproductive organ of ACP and carried to the next generation through eggs (Halbert and Manjunath 2004). The molecular mechanism of interaction between ACP and CLas is still unknown. Transcriptomic studies have been used to understand the interaction between pathogen and vector at the molecular level (Tufail et al. 2014). The present study was performed to understand the differential gene expression pattern in ACP in response to CLas infection. Cluster analysis of expressed genes grouped the two sets of ACP samples infected with CLas and both these samples were different from the healthy ACP sample indicating the differential gene expression pattern between CLas-infected and CLas-free psyllids.
A group of vitellogenin genes of ACP was found upregulated post CLas exposure in the current study. Vitellogenin proteins are the major egg yolk protein precursors in insects (Tufail et al. 2014). Exposure to CLas increases the fecundity of ACP females (Pelz-Stelinski and Killiny 2016; Ren et al. 2016). CLas-infected ACP produces more female adults per day than CLas-free ACP (Galdeano et al. 2020). Upregulation of vitellogenin related genes in CLas-exposed ACP in the current study supports the positive effects of CLas on the fecundity of ACP. However, the expression of vitellogenin gene in Apis mellifera males suggests their possible role other than yolk formation (Piulachs et al. 2003). Role of vitellogenins as antioxidants in A. mellifera and Caenorhabditis elegans (Nakamura et al. 1999; Seehuus et al. 2006) and as antibacterial agents in Bombyx mori (Singh et al. 2013) has been elucidated. Huo et al. (2018) uncovered a positive correlation between the presence of vitellogenin and in-hemolymph survival of rice stripe virus (RSV) in Laodelphax striatellus. The study showed that vitellogenin interacted with RSV to protect the latter in the unhostile hemolymph environment of L. striatellus. The upregulation of vitellogenin-like genes in the current study, thus, suggests the putative role of these proteins in protecting CLas in the hemolymph of ACP to systemically colonize various tissues of the insect vector. Besides, extensin-like proteins that are involved in pathogenic stress response in insects (Sinha et al. 2016) were upregulated in CLas infected ACP. Laminin, one of the major constituents of the midgut and salivary gland basal lamina, functions as receptors for parasites in invertebrate systems (de Almeida et al. 2012). Upregulation of laminin in CLas-exposed ACP in the current study suggests its role to bind with basal lamina to cross the midgut and salivary gland barriers and is consistent with the observation by Vyas et al. (2015).
Genes like tropomyosin and troponin C of ACP have been recorded to be downregulated post exposure to CLas. Tropomyosins are evolutionally conserved proteins that bind to the actin filaments and participate in a range of cellular processes. In insects, tropomyosin in collaboration with the troponin complex regulates skeletal muscle contraction and relaxation; thereby, playing an important role in the regulation of insect survival, feeding, and breeding (Goins and Mullins 2015). In a recent study, Lan et al. (2018) showed that rice dwarf virus (RDV) reduced the survival and fecundity of its vector, Nephotettix cincticeps by downregulating the expression of troponin C. Similarly, CLas infection of ACP was shown to downregulate the cytoskeletal proteins to induce changes in cytoskeletal configuration (Ramsey et al. 2015). Lu and Killiny (2017) and Lu et al. (2019) have demonstrated the downregulation of tropomyosin in CLas-infected ACP. Downregulation of troponin C and tropomyosin-1 genes in CLas-infected ACP in the current study also suggests the possible cytoskeletal modification in cells of ACP to mediate the circulation of CLas within ACP. Besides flightin, a myosin-binding protein that facilitates thick filament assembly and muscle integrity (Barton et al. 2005), also showed reduced expression in CLas-infected ACP. Taken together, all these might account for the morphological abnormalities of the CLas-infected ACP as demonstrated by Ghanim et al. (2016). Among the insect immune responses, ubiquitination plays a pivotal role in recognition and elimination of the invading microbes. Several of the ubiquitination-related genes were differentially expressed in the midgut of CLas-infected ACP. It has been observed that two of the three E3 ubiquitin ligase class of genes were downregulated in CLas-infected ACP though E3 ubiquitin ligase gene was upregulated (Yu et al. 2020). In our study, E3 ubiquitin ligase gene was downregulated in CLas-infected ACP and a similar observation was made by Ramsey et al. (2015). We speculate the possible manipulation of 26S proteasomal degradation pathway of ACP by CLas to prevent proteins of the later from degradation (Ramsey et al. 2015). Similarly, downregulation of defense-related genes like phenoloxidase enzyme involved in melanization, proteins involved in insect humoral responses-serine proteases, hemocytin, and cathepsin B, a proteolytic enzyme, associated with insect stress suggests the subversion of ACP's defense by CLas. ABC transporters involved in solute transportation across membranes via ATP hydrolysis were under-expressed in CLas-infected ACP to gain an advantage of the membrane dysfunction. Suppression of the defense-related genes of ACP by CLas is consistent with the findings of Vyas et al. (2015). Differential expression of a cell adhesion protein integrin, its binding protein talin, and basal membrane formation protein papilin in CLas-infected ACP may mediate cell shape change during adhesion and invasion process as speculated by Das et al. (2014) and Vyas et al. (2015).
Among the major metabolic pathways of ACP affected by CLas, carbohydrate, energy, and nucleotide metabolism were important. To meet out the energy and nucleotide requirements, CLas manipulates the respective pathways of the vector (Killiny et al. 2017). Considering the nonmetabolic pathways, endocytotic pathway plays an inevitable role in host invasion by CLas (Vyas et al. 2015). Enrichment of genes associated with cellular endocytosis in ACP suggests possible manipulation of endocytotic pathway upon CLas infection.
In conclusion, a total of 3911 genes were differentially expressed including 2196 upregulated and 1715 downregulated genes. Most DEGs included vitellogenins, extensin, cytoskeleton, and endocytotic pathway-related genes. Molecular functions were highly enriched in CLas-infected ACP. Carbohydrate, energy, and nucleotide metabolism were the major metabolic pathways which got affected. Endocytotic and defense-related pathways that facilitate the circulation and multiplication of CLas in ACP were also affected. The putative genes of ACP that are highly regulated in response to CLas can be targeted for the effective management of CLas.
Data availability
The datasets generated during the current study are available in the NCBI with BioProject ID PRJNA634436.
References
Barton B, Ayer G, Heymann N, Maughan DW, Lehmann FO, Vigoreaux JO (2005) Flight muscle properties and aerodynamic performance of Drosophila expressing a flightin transgene. J Exp Biol 208:549–560
Belliure B, Janssen A, Sabelis MW (2008) Herbivore benefits from vectoring plant virus through reduction of period of vulnerability to predation. Oecologia 156:797–806
Bin S, Pu X, Shu B, KangC LuoS, Tang Y, Wu Z, Lin J (2019) Selection of reference genes for optimal normalization of quantitative real-time polymerase chain reaction results for Diaphorina citri adults. J Econ Entomol 112:355–363
Bové JM (2006) Huanglongbing: a destructive, newly-emerging, century-old disease of citrus. J Plant Pathol 88:7–37
Das M, Ithychanda SS, Qin J, Plow EF (2014) Mechanisms of talin-dependent integrin signaling and crosstalk. Biochim Biophys Acta Biomembr 1838:579–588
De Almeida DF, Dos Santos ALS, Lery LMS, Silva E, TLA, Oliveira MM, Bisch PM, Saraiva EM, Souto-Padron TC, Lopes AH, (2012) Evidence that a laminin-like insect protein mediates early events in the interaction of a Phytoparasite with its vector’s salivary gland. PLoS ONE 7:e48170
Deng X, Chen J, Feng Z, Shan Z, Guo H, Zhu J, Li H, Civerolo EÁ (2008) Identification and characterization of the Huanglongbing bacterium in pummelo from multiple locations in Guangdong, PR China. Plant Dis 92:513–518
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70
Fiebig M, Poehling HM, Borgemeister C (2004) Barley yellow dwarf virus, wheat, and Sitobion avenae: a case of trilateral interactions. Entomol Exp Appl 110:11–21
Galdeano DM, de Souza PI, Alves GR, Granato LM, Rashidi M, Turner D, Levy A, Machado MA (2020) Friend or foe? relationship between ‘Candidatus Liberibacter asiaticus’ and Diaphorina citri. Trop Plant Pathol 45:559–571
Ghanim M, Fattah-Hosseini S, Levy A, Cilia M (2016) Morphological abnormalities and cell death in the Asian citrus psyllid (Diaphorina citri) midgut associated with Candidatus Liberibacter asiaticus. Sci Rep 6:33418
Ghosh A, Das A, Vijayanandraj S, Mandal B (2016) Cardamom bushy dwarf virus infection in large cardamom alters plant selection preference, life stages, and fecundity of aphid vector, Micromyzus kalimpongensis (Hemiptera: Aphididae). Environ Entomol 45:178–184
Ghosh DK, Motghare M, Gowda S (2018) Citrus Greening: overview of the most severe disease of citrus. Adv Agric Res Technol J 2(1):83–100
Ghosh A, Basavaraj YB, Jangra S, Das A (2019) Exposure to watermelon bud necrosis virus and groundnut bud necrosis virus alters the life history traits of their vector, Thrips palmi (Thysanoptera: Thripidae). Arch Virol 164:2799–2804
Goins LM, Mullins RD (2015) A novel tropomyosin isoform functions at the mitotic spindle and Golgi in Drosophila. Mol Biol Cell 26:2491–2504
Gottwald TR, Graça JVD, Bassanezi RB (2007) Citrus huanglongbing: the pathogen and its impact. Plant Health Prog 8:31
Halbert SE, Manjunath KL (2004) Asian citrus psyllids (Sternorrhyncha: Psyllidae) and greening disease of citrus: a literature review and assessment of risk in Florida. Fla Entomol 87:330–353
Huo Y, Yu Y, Chen L, Li Q, Zhang M, Song Z, Chen X, Fang R, Zhang L (2018) Insect tissue-specific vitellogenin facilitates transmission of plant virus. Plos Pathog 14:e1006909
Ingwell LL, Eigenbrode SD, Bosque-Pérez NA (2012) Plant viruses alter insect behavior to enhance their spread. Sci Rep 2:1–6
Killiny N, Hijaz F, Ebert TA, Rogers ME (2017) A plant bacterial pathogen manipulates its insect vector’s energy metabolism. Appl Environ Microbiol 83:e03005-e3016
Lan H, Hong X, Huang R, Lin X, Li Q, Li K, Zhou T (2018) RNA interference-mediated knockdown and virus-induced suppression of Troponin C gene adversely affect the behavior or fitness of the green rice leafhopper, Nephotettix cincticeps. Arch Insect Biochem Physiol 97:21438
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Lu Z, Killiny N (2017) Huanglongbing pathogen Candidatus Liberibacter asiaticus exploits the energy metabolism and host defence responses of its vector Diaphorina citri. Physiol Entomol 42:319–335
Jun LZ, Hua ZC, Zhong YH et al (2019) Potential roles of insect Tropomyosin1-X1 isoform in the process of Candidatus Liberibacter asiaticus infection of Diaphorina citri. J Insect Physiol 114:125–135
Mann M, Fattah-Hosseini S, Ammar ED et al (2018) Diaphorina citri nymphs are resistant to morphological changes induced by Trop. plant pathol. (2020) 45:559–571 569 “Candidatus Liberibacter asiaticus” in Midgut Epithelial Cells. Infect Immun 86:1–19
Nakamura A, Yasuda K, Adachi H, Sakurai Y, Ishii N, Goto S (1999) Vitellogenin-6 is a major carbonylated protein in aged nematode, Caenorhabditis elegans. Biochem Biophys Res Commun 264:580–583
Pelz-Stelinski KS, Killiny N (2016) Better together: association with 'Candidatus Liberibacter asiaticus’ increases the reproductive fitness of its insect vector, Diaphorina citri (Hemiptera: Liviidae). Ann Entomol Soc Am 109:371–376
Piulachs MD, Guidugli KR, Barchuk AR, Cruz J, Simoes ZLP, Belles X (2003) The vitellogenin of the honey bee, Apis mellifera: structural analysis of the cDNA and expression studies. Insect Biochem Mol Biol 33:459–465
Ramsey JS, Johnson RS, Hoki JS, Kruse A, Mahoney J, Hilf ME, Hunter WB, Hall DG, Schroeder FC, MacCoss MJ, Cilia M (2015) Metabolic interplay between the Asian citrus psyllid and its Profftella symbiont: an Achilles’ heel of the citrus greening insect vector. PLoS ONE 10:e0140826
Ramsey JS, Chavez JD, Johnson R et al (2017) Protein interaction networks at the host–microbe interface in Diaphorina citri, the insect vector of the citrus greening pathogen. R Soci Open Sci 4:160545
Ren S-L, Li Y-H, Zhou Y-T et al (2016) Effects of Candidatus Liberibacter asiaticus on the fitness of the vector Diaphorina citri. J Appl Microbiol 121:1718–1726
Seehuus SC, Norberg K, Gimsa U, Krekling T, Amdam GV (2006) Reproductive protein protects functionally sterile honey bee workers from oxidative stress. Proc Natl Acad Sci 103:962–967
Singh NK, Pakkianathan BC, Kumar M, Prasad T, Kannan M, König S, Krishnan M (2013) Vitellogenin from the silkworm, Bombyx mori: an effective anti-bacterial agent. PLoS ONE 8:e73005
Sinha DK, Chandran P, Timm AE, Aguirre-Rojas L, Smith CM (2016) Virulent Diuraphis noxia aphids over-express calcium signaling proteins to overcome defenses of aphid-resistant wheat plants. PLoS ONE 11:e0146809
Tufail M, Nagaba Y, Elgendy AM, Takeda M (2014) Regulation of vitellogenin genes in insects. Entomol Sci 17:269–282
Vyas M, Fisher TW, He R, Nelson W, Yin G, Cicero JM, Willer M, Kim R, Kramer R, May GA, Crow JA (2015) Asian citrus psyllid expression profiles suggest Candidatus Liberibacter asiaticus-mediated alteration of adult nutrition and metabolism, and of nymphal development and immunity. PLoS ONE 10:e0130328
Wu Z, Zhang H, Bin S, Chen L, Han Q, Lin J (2016) Antennal and abdominal transcriptomes reveal chemosensory genes in the Asian citrus psyllid Diaphorina citri. PLoS ONE 11:e0159372
Yu H-Z, Huang Y-L, Li N-Y et al (2019) Potential roles of two Cathepsin genes, DcCath-L and DcCath-O in the innate immune response of Diaphorina citri. J Asia Pac Entomol 22:1060–1069
Yu H-Z, Li NY, Zeng XD, Song JC, Yu XD, Su HN, Chen CX, Yi L, Lu ZJ (2020) Transcriptome analyses of Diaphorina citri midgut responses to Candidatus Liberibacter asiaticus infection. Insects 11:171
Acknowledgements
The sequencing service provided by Nucleome Informatics Pvt. Ltd. Hyderabad, India is thankfully acknowledged. We also acknowledged the funding received from IARI and DBT. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Funding
Indian Agricultural Research Institute, New Delhi. Department of Biotechnology, Government of India.
Author information
Authors and Affiliations
Contributions
AG and VKB conceived and designed the research. AG prepared biological samples. DJ and RR carried out the wet lab experiments. AG, VKS analyzed data. SKS provided HLB isolate. NC reviewed the results. DJ and VKS wrote draft manuscript. VKB and AG edited final manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that there are no conflicts of interest in regard to the work reported in this paper.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jaiswal, D., Sidharthan, V.K., Sharma, S.K. et al. Candidatus Liberibacter asiaticus manipulates the expression of vitellogenin, cytoskeleton, and endocytotic pathway-related genes to become circulative in its vector, Diaphorina citri (Hemiptera: Psyllidae). 3 Biotech 11, 88 (2021). https://doi.org/10.1007/s13205-021-02641-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-021-02641-x