
Abstract
The current study deals with the investigation of the antioxidant, anti-inflammatory and immunomodulatory properties of the essential oil from Datura stramonium leaves (D. oil). The GC-MS analysis showed that the dominant compounds present in the D. oil were neophytadiene (Phytol acetate) (10.76%), β-damascenone (9.67%), and β- eudesmol (7.2%). D. oil exhibited in vitro scavenging potential of free radicals by DPPH and ABTS assays (IC50 values 71.35 ±1.06 μg/ml and 61.01 ± 1.07 μg/ml, respectively). We found that D. oil decreased the nitric oxide production in LPS-stimulated J774A.1 cells by 52.43% without affecting their cell viability. D. oil was found to stimulate the proliferation of human peripheral blood mononuclear cells (PBMC) and, also enhanced the secretion of IL-2, IFN-γ and TNF-α. Furthermore, D. oil treatment of PBMC induced the expression of CD3, CD8, and CD56 and intracellular granulysin levels in the immune cells. The treatment of human lymphocytes by D. oil enhanced their ability to kill colon cancer cells HCT-116 (51.09 ± 7.5%) and SW620 (48.57 ± 8.08%) at 20:1 (effector: target ratio). Moreover, these activated lymphocytes cause target cell death by reactive oxygen species and by damaging mitochondrial membrane potential of these cells. Taken together, the current findings showed D. oil as immunotherapeutic agent which can be used for colon cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Essential oils (EOs) possess a wide variety of therapeutic properties with promising outcomes that could be implemented as alternative treatments to diverse disorders. EOs are composites of hydrocarbons and their oxygenated byproducts derived from two different isoprenoid pathways. EOs are largely produced and secreted by specialized secretory tissues, glandular trichomes which are diffused in tissues of flowers and leaves (Iriti et al. 2006). Datura stramonium L. (Solanaceae) is a widespread plant with hairy leaves, round thorny fruit and white flowers. Datura genus is widely known for tropane alkaloid content and it is extensively studied for alkaloid identification and quantification (Berkov et al. 2006). In addition to its alkaloids, various flavonoids and withanolides/lactones have been isolated and analyzed for their diverse biological applications (Guo et al. 2019). Earlier, D. stramonium essential oil (from seeds and leaves) had been reported to exhibit anti-inflammatory activity in albumin induced rats (Aboluwodi et al. 2017). Conventional methods or modern technologies are used by the pharmaceutical industry to extract essential oils having multi-faceted medicinal activities. Datura essential oil is one of the most effective commercial oil isolated at industrial level due to its high medicinal value.
Colorectal cancer (CRC) is the fourth leading cause of cancer morbidity worldwide in both men and women of various age groups, and these cases are steadily increasing in the developing nations (Bray et al. 2018). Treatments for primary and metastatic colorectal cancer include laparoscopic surgery, radiotherapy, neoadjuvant, bio-marker guided therapy and chemotherapy (Kuipers et al. 2015; Sveen et al. 2020). Chronic inflammation causes damage to nucleic acids, proteins, and lipids via reactive oxygen/nitrogen species (ROS/RNS) generation, resulting in tissue damage and thereby leading to serious disorders like cancer (Ohshima et al. 2003). Plant-based therapeutics have gathered attention of researchers due to their promising potential as immunomodulatory agents. Many experimental studies on EOs or their constituents in cancer models confirmed their efficiency as anticancer agents (Zuzarte et al. 2015; Hata et al. 2003). EOs boost immune system and thus could serve as alternative to conventional treatment (Orhan 2016; Lin et al. 2011). Recently, immunotherapy has become a key therapeutic alternative which include components of the immune system such as antibodies, cytokines, and dendritic cells to treat cancer, allergies and inflammatory diseases (Ganesh et al. 2019; Kumar et al. 2017).
Herein, we have investigated antioxidant, anti-inflammatory and immunomodulatory activities of essential oil extracted from D. stramonium (D. oil) leaves. The chemical composition of D. oil was analyzed by GC-MS. This is the first report which shows the immunostimulatory potential of D. oil on human peripheral blood lymphocytes. The immunoenhancing and anticancer properties of human lymphocytes was evaluated using colon carcinomas via co-culture assay which revealed the mitochondrial depolarization and ROS generation mediated killing of cancer cells. The immune-enhancing potential of D. oil was also evaluated by analyzing the cytokines production and studying the expression of immunomarkers in activated lymphocytes.
Materials and methodology
Materials
Methanol (34860), hexane (270504) and acetonitrile (271004) were procured from Merck chemicals, India. Sulphanilamide-S9251, N-1-napthylethylenediamine dihydrochloride (222488), diclofenac sodium (D6899), Bovine Serum Albumin (BSA, A8806), 1,1-diphenyl-2- picrylhydrazyl (DPPH, D9132), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS, A1888), and lipopolysaccharide (LPS, L2630) were procured from Sigma, India.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), CT02, Dulbecco’s Modified Eagle’s Medium (DMEM), Roswell Park Memorial Institute-1640 medium (RPMI-1640), fetal bovine serum (FBS, RM10685), dimethyl sulphoxide (DMSO), lymphocyte separation media (LSM)-1066, phosphate buffer saline (PBS) and Concanavalin A (Con A, C5275) were purchased from Himedia (Mumbai, India), while rhodamine 123 (Rh 123, R8004) and 2,7-dichlorofluorescin diacetate (DCF-DA D6883) were procured from Sigma-Aldrich (USA). ELISA kits for detecting IL-2, IFN-γ and TNF-α (Elisa Max, Biolegend) were procured from BD Oncomark™ CD8 FITC/CD56 PE/CD3 PerCP-Cy™5, APC Mouse Anti-Human CD14 (BD Pharmigen). HCT-116 (human colorectal cancer) and SW620 (human Colon cancer) were purchased from the National Centre for Cell Sciences, Pune, India.
Collection of plant material
Fresh Leaves of D. stramonium were collected from Solan (30.904486 °N, 77.096733 °E) region of Himachal Pradesh, India and authenticated by Department of Forest Products, Dr. Y.S. Parmar University of Horticulture and Forestry, Nauni, Solan,H. P. India (Herbarium Field book no. 2916, Receipt No. 072). The dried plant material was stored in the laboratory at room temperature (25 °C), protected from light before extraction of EOs.
Isolation of the essential oil
D. oil was extracted from the leaves of D. stramonium by hydro-distillation method using the Clevenger Apparatus. The extraction was carried out for 4 h using 1 kg of leaves in 2000 ml of distilled water at 60 °C. The vapours of D. oil were condensed and dried with anhydrous sodium sulphate and, concentrated under reduced pressure by rotatory evaporator. The yield of D. oil was calculated by measuring the oil (in ml) per 1000 g of raw material (Pathania et al. 2013). The isolated pure D. oil was stored at 4 °C.
Analysis of the D. oil
Using the gas chromatography mass spectrometry (GC-MS), the chemical composition of D. oil was assessed and the identification of the main constituents was done at Laboratory of Natural products chemistry, Council of Scientific and Industrial Research (CSIR)-Indian Institute of Integrative Medicine (IIIM), Jammu, India. The D. oil (20 µl) was sonicated for 30 min and then sample was filtered through an ultra-membrane filter (pore size 0.45 µm) prior to injection in the sample loop. Filtrate was used for GC-MS analysis (Agilent GC 7890A) with ion trap gas-chromatograph equipped with HP5 capillary column (30 m × 0.25 mm; coating thickness 0.25 µm). The MS conditions included an EI ion source temperature of 230 °C, an ionization energy of 70 eV, and a mass scan range of 40–600 amu. Analytical conditions: Maximum temperature was 300 °C; carrier gas, helium at 1 ml/min; injection 0.2 µl of (10% hexane solution). Running conditions were as follows: injection volume HS 2.5 mL syringe, HS SPME injection technique. The identification of the components was performed by comparing their retention times with the pure authentic samples and, by mean of their liner retention indices and by matching against commercial and home-made library mass spectra built up from pure substances and components of known oils which were in-built in the software of GC-MS and compared with literature (Verma et al. 2010).
Antioxidant activity of D. oil
DPPH radical scavenging activity
The antioxidant activity of the D. oil was analyzed using DPPH free radicals (Brand-Williams et al. 1995). A volume of 50 μl of D. oil prepared at different concentrations (10–80 μg/ml) was mixed with 150 μl of DPPH free radical solution. The disappearance of the DPPH was measured after 30 min of incubation at 517 nm. Ascorbic acid (10-80 μg/ml) was used as a positive control. The inhibition percentage of the DPPH radical by the essential oil was estimated using the following equation: scavenging percentage (%) = [100 × (AC − AS/Ac)], where Ac is the absorbance of the control reaction (containing all reagents except the test sample) and AS the absorbance of the tested sample. The concentration of oil that could scavenge 50% of the DPPH radicals (IC50) was calculated by using Graphpad prism 5.
ABTS radical scavenging activity
ABTS radical scavenging activity of D. oil was measured by the ABTS cation decolorization assay (Chahal et al. 2019). The ABTS radicals (ABTS•+) were produced by reaction of 7 mM stock solution of ABTS with 2.45 mM potassium persulfate and allowing the mixture to stand in dark at room temperature (25 °C) for 12 h before use. 50 μl of essential oil at different concentrations (10–80 μg/ml) was allowed to react with 150 μl of the ABTS•+ solution and the absorbance was measured at 734 nm. Ascorbic acid was used as positive control. The percentage inhibition of ABTS∙+ was calculated using the following formula:
where, Ao is the absorbance of the radical cation of the sample and Af is the absorbance after addition of sample.
Anti-inflammatory activity of D. oil
Inhibition of heat induced hemolysis
Fresh human blood (4 ml) was collected as per Institutional ethical committee guidelines (IEC no. SUIEC/14/01) and kept in EDTA coated vials to avoid coagulation. The blood was washed 3-4 times by using 5 ml of normal saline. The blood suspension was maintained as 10% v/v in normal saline for the experiment. 1 ml of D. oil at various concentrations (10–80 μg/ml) and 1 ml of 10% (v/v) RBCs suspension were treated at 56 °C for 30 min for RBCs lysis. The tubes were then cooled down, centrifuged (3000 rpm for 5 min) and the supernatants were taken for absorbance at 560 nm using varioskan multiskan spectrophotometer (Sharma et al. 2016). Diclofenac sodium (100 μg/ml) was used as a positive control and normal saline was taken as negative control. The experiment was performed in triplicate and the percentage inhibition of haemolysis was calculated as follows:
Inhibition of albumin denaturation
Bovine Serum Albumin (BSA) was used to study the anti-denaturation activity of D. oil. Briefly, 2 ml of reaction mixture comprising of 0.2 ml of BSA and varying concentration of D. oil (10-80 μg/ml) was used in PBS. Distilled water was taken as a negative control and diclofenac sodium was used as a standard drug. The reaction mixture was incubated at 37 °C for 15 min and further treated for BSA denaturation at 70 °C for 5 min. After cooling, the absorbance of supernatant was taken at 660 nm (Gambhire et al. 2009). The percentage inhibition of protein denaturation was determined by using following formula:
Measurement of anti-inflammatory activity by nitrite quantification in J774A.1 cells
J774A.1 murine macrophage cells were cultured in DMEM media with 10% FBS and 1% pencillin-streptomycin. After 90% confluency, cells were trypsinized and seeded in 12 well plates (105 cells/well). After overnight incubation, cells were treated with 100 ng/ml LPS for 2 h followed D. oil (10 μg/ml) incubation at 37 °C under 5% CO2. After 2 h, 12 h and 24 h, cell-free supernatants were taken out and stored in -80 °C deep freezer. Nitric oxide (NO) quantification was performed by using griess reaction (Green et al. 1990). An aliquot (100 μl) of cell free supernatants were mixed with 50 μl of 1% sulphanilamide and 100 μl of 0.1% N-1-naphthylethylenediamine dihydrochloride in 2% (v/v) H3PO4 in 96 well plates at room temperature for 20 min. Untreated cells were taken as negative control. The presence of nitrite was then measured at an absorbance of 540 nm and was quantified by percentage inhibition.
Cell cytotoxicity
The cytotoxic effect of D. oil on J774A.1 cells was determined by MTT assay (Kumari et al. 2019). In summary, J774A.1 (2.5 × 104 cells/ml) were seeded in a 96-well plates and treated with D. oil (25–200 μg/ml) for 24 and 48 h followed by MTT assay to assess the cell viability. Cell viability was analyzed by the formula given below:
Immunomodulatory activity of D. oil
Isolation of human peripheral blood mononuclear cells (PBMC)
For isolation of human PBMC, blood was collected from healthy human donors in compliance with the ethical committee guidelines (Ref No. SUBMS/IEC/14/21) of Shoolini University, Solan, India. The blood was diluted with the equal volume of PBS (1:1) and then layered on lymphocytes separation media. The mixture was centrifuged at 2000 rpm for 20 min and then the undisturbed PBMC layer was isolated, washed twice with PBS and re-suspended in RPMI-1640 media supplemented with 10% Fetal bovine serum (FBS) and antibiotics (100 µg/ml penicillin and streptomycin). To analyze the cell viability, the cells were counted with a hemocytometer and viability was tested by trypan blue dye exclusion assay.
PBMC Proliferation Assay
PBMC were seeded (1 × 104 cells/well) in 10% RPMI-1640 medium in a 96-well plates. The cells were treated with different concentrations of D. oil (10–80 μg/ml) for 24 and 48 h. Concanavalin A (Con A, 10 μg/ml) treated PBMC cells was used as positive control and the untreated PBMC or DMSO treated cells were used as negative controls. The cells were incubated in 37 °C for 48 h in 5% CO2. Later the MTT assay was performed as described above.
Cytokine analysis
The isolated PBMC were seeded (106 cells/well) in six well plate. These cells were treated with D. oil (10 μg/ml) for 24 h or 48 h. After this, the secretion of cytokines (IFN-γ, IL-2 and TNF-α) was analyzed in culture supernatant using ELISA kits (Biolegend, USA) as per the manufacturer’s protocol.
Immunophenotyping of human lymphocytes
The PBMC (105 cells/well in six well plates) were stained (4 °C for 20 min) with FITC-labeled anti-CD8, perCP-Cy5-5-A-labeled anti-CD3, PE-A-labeled anti-CD56 (BD OncomarkTM CD8/CD56/CD3) after treating with D. oil at concentration of 10 μg/ml for 48 h at 37 °C. Inactivated PBMC and Con A treated PBMC were used as negative and positive controls, respectively. After staining, the cells were washed twice with PBS and were detected by flow cytometry (BD FACS CANTO II) using FACS Diva Version 6.1.3. Each test sample was run in duplicates (Hira et al. 2018).
Analysis of granulysin expression in D. oil activated PBMC
PBMC (105 cells) were treated with D. oil (10 μg/ml) for 48 or 72 h. The cells were centrifuged and washed with ice cold PBS and, then fixed with 4% paraformaldehyde for 20 min. After this, the cells were washed twice with ice cold Perm-wash buffer (0.1% Triton, 1% FBS in PBS) and then the primary antibody (rabbit anti-granulysin primary antibody, Santa Cruz) was added at a dilution of 1:100. After 30 min, cells were washed with cold PBS and treated with secondary antibody (anti-rabbit IgG allophycocyanin) at 1:400 dilution in dark for 30 min. Thereafter the cells were washed again twice with PBS and granulysin expression was analyzed by Flow cytometry (Saini et al. 2011).
Co-culture MTT micro-cytotoxicity assay
HCT-116 (human colorectal cancer) and SW620 (human colon cancer) were used as target cells, and were maintained in DMEM supplemented with penicillin (100 units/ml), streptomycin (100 mg/ml) and 10% FBS. Cells were incubated at 37 °C in a humidified, 5% CO2 atmosphere. PBMC pretreated with D. oil (10 μg/ml for 48 h) were used as effectors. Then, the untreated PBMC (as a negative control) and the activated PBMC were cultured with the cancer cells at different effector: target (E: T) ratios of 2.5:1, 5:1, 10:1 and 20:1 for 24 h in a 96 well plate. After the co-culture, 180 μl media and the suspended cells were discarded and washed with 200 μl PBS and MTT assay was carried out to analyze cancer cell death as described above. The percentage cell death was calculated by using the following formula:
The Abscontrol is cancer cells alone.
ROS generation via activated lymphocytes
PBMC were activated with D. oil (10 μg/ml for 48 h) and co-incubated with target cells (HCT-116 and SW620) at E:T ratio of 10:1 for 4 h at 37 °C in 5% CO2. Later 20 μM DCFDA was added for 30 min. The fluorescence was measured (emission at 538 nm and excitation at 485 nm) by using Varioskan Multiskan spectrophotometer (Kumar et al. 2020). ROS levels (% fold increase) were analyzed by the formula given below:
The Abscontrol was fluorescence of cells alone.
Mitochondrial membrane perturbation (MMP)
For mitochondrial membrane perturbation assay, target cells, (HCT-116 and SW620) were labelled with 20 μM cell trace far red (CTFR) dye and then co-cultured with D. oil (10 μg/ml for 48 h) pretreated PBMC at 37 °C in an atmosphere of 5% CO2. The effector and target cells were co-incubated at E: T ratio of 10:1 for 12 h. The untreated PBMC or Con A treated PBMC were used as negative and positive controls, respectively. Co-cultured cells were washed twice with PBS, trypsinized and then incubated with Rh 123 (50 μM) fluorescent probe in dark for 20 min. Thereafter, the cells were resuspended in PBS and analyzed for mitochondrial membrane depolarization (ΔΨm) by using two-color flow cytometry.
Statistical analysis
All the results were expressed as mean ± Standard Error. The significance of differences among mean values were determined by one-way ANNOVA and Student’s t-test by using GraphPad prism 5 software. A p value ≤ 0.05 was considered significant.
Results and discussion
Characterization of D. oil
Three repetitions of the extraction from 1 kg leaves of D. stramonium yielded 5 ml of D. oil (0.5%) which was stored at 4 °C. D. oil was analyzed by GC-MS and GC-FID. A total of 153 components of leaves were identified which covered 100% of total mass. The components with more than 1% of total mass are given in Table 1. The major components of D. oil were neophytadiene (Phytol acetate) (10.76%), β-damascenone (9.67%) and, β-eudesmol (7.2%). Previously, phytol had been identified as major constituent present in the leaves oil of D. stramonium (Aboluwodi et al. 2017). β-damascenone and β-eudesmol were also found earlier in the fruit of D. metel (Essien et al. 2010).
Antioxidant activity of D. oil
The antioxidant activity of D. oil was analyzed by DPPH and ABTS assay at a concentration range from 10 to 80 μg/ml. We found an increase in the free radicals scavenging as the D. oil concentration was increased (Fig. 1a, b). The IC50 values for DPPH and ABTS assays were 71.35 ±1.06 μg/ml and 61.01 ± 1.07 μg/ml, respectively. Previously, antioxidant potential of the leaves and seeds extracts of D. stramonium have been reported (Ananth and Rajan 2015; Iqbal et al. 2017). Antioxidant potential of the D. oil could be attributed to the active constituents like β-eudesmol and Neophytadiene. β eudesmol has been reported for scavenging intracellular ROS (Kim, 2018). Neophytadiene is also known for its anti-inflammatory and antioxidant compound (Santos et al. 2013).

a–e Antioxidant effect of D. oil as measured by ABTS assay (a) and DPPH assay (b) using various concentrations (10-80 µg/ml). The anti-inflammatory effect of D. oil analyzed by RBC membrane stabilization assay (c) and BSA denaturation (d) done at different concentrations (10–80 µg/ml). NO production in J774. A macrophage cells (e) was determined after treating with D. oil (10 µg/ml) for 2–24 h. The results were expressed as the percentage inhibition of mean ± S.E.M. (n = 3), *p < 0.05
D. oil exhibit anti-inflammatory activity
In vitro anti-inflammatory activity of D. oil was analyzed by RBC membrane stabilization and BSA denaturation method. The RBC membrane stabilization has been widely used to study the in vitro anti-inflammatory activity as erythrocyte membrane is analogous to the lysosomal membrane and its stabilization implies that the tested drug is capable of stabilizing lysosomal membranes (Anosike et al. 2012). D. oil was found to stabilize RBC membrane in a concentration dependent manner (IC50 68.92 ± 1.08 μg/ml) (Fig. 1c) Additionally, D. oil was capable of inhibiting the albumin denaturation (IC50 91.30 ± 1.14 μg/ml) (Fig. 1d). Earlier, essential oils (leaves and seeds) of D. stramonium have shown significant anti-inflammatory properties in the rat model possibly due to the presence of phytol as one of the major components in leaves essential oil (Aboluwodi et al. 2017). Furthermore, the anti-inflammatory effects of eudesmol have also been reported previously (Seo et al. 2011).
The production of NO by immune cells has been used as an indicator for the presence and extent of inflammation in various diseases like arteriosclerosis, ischemic reperfusion, hypertension and septic shock (Pacher et al. 2007). We found that D. oil inhibited NO production by LPS-activated macrophage cells (Fig. 1e). The maximum NO production inhibition (52.43%) by D. oil was observed after 24 h of incubation. One of the components of essential oil, damascenone and its derivatives have been reported to inhibit LPS-induced nitric oxide synthase activity in Raw 264.7 macrophages (Gerhauser et al. 2009). Moreover, D. oil displayed nontoxic effects on J774A.1 macrophages at a concentration range of 25–200 μg/ml. The results revealed that even at the highest concentration (200 μg/ml) of the D. oil, 90% macrophages were viable after 24 and 48 h of incubation (data not shown). Taken together, our results show that D. oil has strong anti-inflammatory potential which can be attributed to the phytoconstituents present in the oil.
Immunomodulatory activity of D. oil
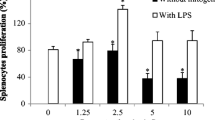
The immunopotentiating effect of D. oil was evaluated on human PBMC. The microscopic images (Fig. 2a) of PBMC treated with D. oil clearly indicated an increased cell number and activation as compared to the untreated cells and Con A treated immune cells. The lymphocyte proliferation assay provided evidence to the immunostimulatory potential of the D. oil (EC50 10.73 ± 1.49 μg/ml after 24 h) (Fig. 2b). This is the first report describing the immunoenhancing potential of the D. oil which could be due to the bioactive molecules present in the essential oil. Previously, it had been shown that D. stramonium leaves extract exhibited immunostimulatory potential on human lymphocytes (Gupta et al. 2016).

Microscopic investigation (20 ×) of immuno-proliferation activity of 10 µg/ml D. oil on human PBMC after 24 h of treatment (a). Untreated PBMC and Con A treated PBMC were taken as negative and positive controls, respectively. Effect of D. oil (10–80 µg/ml) on proliferation of human PBMC after 24 and 48 h was analyzed by MTT assay. The values were expressed as mean ± S.E.M of three independent observations done in triplicates, *p < 0.05 was considered as significant value
Cytokine analysis
The production of IL-2, IFN-γ, and TNF-α were analyzed after treating human PBMC with D. oil. The results revealed that D. oil significantly enhanced the secretion of IL-2, IFN-γ and TNF-α after 24 and 48 h of incubation as compared to the untreated PBMC (Fig. 3). Cytokines are major regulators of immune systems which control proliferation, differentiation, effector functions, and survival of leukocytes. IL-2 promotes the clonal expansion of antigen-activated CT8+ T cells and acts as growth factor for CD4+ T cells as well as NK cell proliferation (Boyman and Sprent 2012). IFN-ℽ and TNF-α play a vital role in the anti-tumor responses mediated through the activation of T-effector cells, macrophages and NK cells (Valencia et al. 2006).

Induction of cytokines (IL-2, IFN-γ and TNF-α) measured after 24 and 48 h treatment of human PBMC with expressed as mean ± SEM (n=2) *p < 0.05
Immune cells profiling (CD3, CD8, CD56)
The immunoenhancing potential of D. oil was further validated by analyzing expression of cell surface markers on activated PBMC. D. oil significantly up-regulated the expression of CD3 (59.6%), CD8 (24%), and CD56 (11.5%) marker on PBMC at the concentration of 10 μg/ml as compared to untreated PBMC (Fig. 4). CD8+ T cells and CD 56+ NK cells are effective in eliminating self-cells especially those infected with a cytosolic pathogen and cancer cells (Taniguchi et al. 2003). These cells are known to eliminate target cells directly through cell-mediated cytotoxicity (Lanier 2008). Earlier, Datura withanolides had been reported to have immunostimulatory effects on murine model via CD4/CD8 cells activation and release of IL-2 and TNF- α (Bhat et al. 2005).

FACS analysis showing percentage of the cells expressing CD3, CD8 and CD56 markers on human PBMC after 48 h of treatment with D. oil at 10 μg/ml. Untreated PBMC (control) and Con A were taken as negative and positive controls, respectively. Data shown here is representative of three different experiments
Intracellular Granulysin expression
Granulysin is a protein present in cytotoxic granules of CTL and NK cells having anti-tumor and antimicrobial properties (Pena et al. 1997). Both biologic and clinical studies strongly suggested that enhanced granulysin expression act as an immunomarker for anti-tumor immune responses and cancer cell killing capacity of human lymphocytes (Clayberger and Krensky 2003). D. oil enhanced the intracellular granulysin expression (46.9%) of human lymphocytes after 48 h of treatment as compared to untreated PBMC (2.66%) (Fig. 5). After 72 h of treatment with D. oil, 26.2% of lymphocytes were granulysin positive as compared to 4.76% granulysin positive cells in untreated PBMC. When killer lymphocytes recognize infected or cancerous cells, they release these cytotoxic granules that contain cytolytic proteins like perforins and granzymes which are co-expressed with granulysin. The release of granule content accounts for the cytotoxic activity of CD8 effector T cells and NK cells which help in lysing the target cells by making pores in the lipid membrane (Gamen et al. 1998). These results show that the D. oil components are capable of modulating expression of granulysin, an immunomarker of activated human lymphocytes.

Intracellular granulysin expression analysis in human PBMC after 48 and 72 h of treatment with D. oil at 10 μg/ml. Untreated PBMC (control) and Con A treated PBMC were taken as negative and positive controls, respectively. Treated PBMC were fixed, permeabilized and probed with anti-granulysin antibody. Followed by treatment with secondary antibody (anti-rabbit IgG Allophycocyanin) and flow cytometry analysis. Results were expressed as percentage of granulysin positive cells
Co-culture of Human PBMC with cancer cells
To mimic in vivo conditions, cancer cells (target) were incubated with activated lymphocytes (effector) to analyze lymphocytes mediated cancer cell death. Two colon cancer cell lines (HCT-116 and SW620) were used as target cells and were co-cultured with D. oil activated lymphocytes at various E:T ratios for 48 h. The results revealed that with the increase in E:T ratios from 2.5:1 to 20:1, there was an increase in the target cell death (Fig. 7). The microscopic analysis also showed enhanced cancer cells death following co-incubation with D. oil treated PBMC as compared to the untreated PBMC or Con A treated PBMC (Fig. 6a–h). As shown in Fig. 7, D. oil pretreated PBL exhibited maximum cytotoxicity towards HCT-116 (51.09 ± 7.5%) and SW620 (48.57 ± 8.08%) at 20:1 ratio. The results confirmed that D. oil constituents were capable of activating the cytolytic properties of human lymphocytes towards colon cancer cells.

Microscopic images (20 ×) depicting cytolytic activity of activated PBMC towards HCT-116 (a–d) and SW620 (e–h) cancer cells. Unactivated PBMC (PBL) or PBMC preincubated with Con A (10 μg/ml) and D. oil (10 μg/ml) for 48 h were co-cultured with cancer cells to analyse their cancer cells killing efficiency. Cancer cells alone (a and e) were used to evaluate the natural cell death

a, b Cytolytic activity of stimulated PBMC towards human cancer cells was analyzed by MTT assay. Untreated PBMC, Con A (10 μg/ml) and D. oil (10 μg/ml) treated PBMC, were co-cultured with HCT-116 (a) and SW620 cells (b) for 24 h at different E:T ratios (2.5:1 to 20:1). The results were expressed as mean ± SEM of percentage cell death (n = 3), *p < 0.05 was considered as significant
Co-culture of Human PBMC with cancer cells for ROS and MMP determination
Cytolytic activity requires direct cell–cell contact, which results in apoptosis of target cells. To elucidate the apoptotic pathways induced in target cells via D. oil activated lymphocytes, ROS generation and mitochondrial perturbation were analyzed in cancer cells. The intracellular ROS levels of cancer cells (HCT-116 and SW620 cells) was investigated after co-culturing these cells with D. oil activated lymphocytes for 4 h. At 10:1 (E:T) ratio, the ROS generation was found to be increased by fourfold in HCT-116 and threefold in SW620 cells as compared to target cells co-incubated with untreated lymphocytes (Fig. 8).

Effect of D. oil stimulated effector cells on the generation of ROS in HCT-116 (a) and SW620 (b) cells. Human PBMC were incubated with cancer cells at 10: 1 E:T ratio for 4 h. Cells were stained with DCFDA and fluorescence was measured. Untreated PBMC and Con A treated PBMC were used as negative and positive controls, respectively. Results were expressed as mean ± SEM from two similar experiments, *p < 0.05 is considered as significant
Mitochondrial membrane depolarization was examined in target cells after incubating with D. oil activated lymphocytes (Fig. 9). A significant decrease in Rh 123 fluorescence was found in HCT-116 and SW620 cells co-cultured with PBMC activated with D. oil depicting increased mitochondrial damage in these cells as compared to target cells co-cultured with untreated effector cells. Mitochondrial perturbation and hyperproduction of ROS have been shown to induce cell death in cancer cells. (Kroemer et al. 1995, Ricci et al. 2003). Taken together, the current data clearly demonstrated the immunoenhancing potential of D. oil which can stimulate cytolytic properties of human lymphocytes against colon cancer cells.

Analysis of the mitochondrial membrane depolarization in target cells (HCT-116 and SW620 cells). Target cells were co-cultured with D. oil (10 μg/ml) activated PBMC at 10: 1 (E:T ratio) for 12 h. Mitochondrial damage was analyzed by staining cancer cells with Rh 123 and analyzed by flow cytometry after gating CTFR+ target cells. Untreated PBMC and Con A pretreated PBMC were used as negative and positive controls, respectively. Representative flow diagrams are shown here from two similar experiments
Conclusion
GC-MS analysis revealed phytol acetate, β-damascenone and β-eudesmol as major compounds present in D. oil which might be responsible for free radicals scavenging, anti-inflammatory and immunostimulatory potential of D. oil. The D. oil was found to possess immunoenhancing activity which lead to an enhanced cell proliferation, increase in cytokine production, cell surface markers and cytolytic potential of peripheral blood lymphocytes. The activation of lymphocytes was also validated via heightened granulysin expression which is an immunomarker for the cancer killing capabilities of the lymphocytes. Moreover, D. oil activated immune cells are capable of inducing apoptosis in target cells by ROS generation and mitochondrial depolarization. In summary, D. oil could be explored as a potential new source of natural antioxidant, anti-inflammatory and immunomodulatory agent for cancer therapy.
References
Aboluwodi AS, Avoseh NO, Lawal AO, Ogunwande IA, Giwa AA (2017) Chemical constituents and anti-inflammatory activity of essential oils of Datura stramonium L. J Med Plant Stud 5:21–25
Ananth A, Rajan S (2015) In-vitro antioxidant activity of Datura stramonium L. leaves. Adv Appl Sci Res 6:147–151
Anosike CA, Obidoa O, Ezeanyika LU (2012) Membrane stabilization as a mechanism of the anti-inflammatory activity of methanol extract of garden egg (Solanum aethiopicum). DARU J Pharm Sci 20:76
Berkov S, Zayed R, Doncheva T (2006) Alkaloid patterns in some varieties of Datura stramonium. Fitoterapia 77:179–182
Bhat BA, Dhar KL, Puri SC, Qurishi MA, Khajuria A, Gupta A, Qazi GN (2005) Isolation, characterization and biological evaluation of Datura lactones as potential immunomodulators. Bioorg Med Chem 13:6672–6677
Boyman O, Sprent J (2012) The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12:180–190
Brand-Williams W, Cuvelier M, Berset C (1995a) Use of free radical method to evaluate antioxidant activity. LWT Food Sci Technol 28:25–30
Brand-Williams W, Cuvelier M, Berset C (1995b) Use of free radical method to evaluate antioxidant activity. LWT Food Sci Technol 28:25–30
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424
Chahal A K, Chandan G, Kumar R, Chhillar AK, Saini AK, Saini RV (2019) Bioactive constituents of Emblica officinalis overcome oxidative stress in mammalian cells by inhibiting hyperoxidation of peroxiredoxins. J Food Biochem e13115
Clayberger C, Krensky AM (2003) Granulysin. Curr Opin Immunol 15:560–565
Essien EE, Walker TM, Ogunwande IA, Bansal A, Setzer WN, Ekundayo O (2010) Essential oil composition, cytotoxicity and antimicrobial activities of Datura metel L.from Nigeria. Int J Essent Oil Ther 4:69–72
Gambhire M, Juvekar A, Wankhede S (2009) Evaluation of the anti-inflammatory activity of methanol extract of Barleria cristata leaves by in vivo and in vitro methods. Int J Pharmacol 7:1–6
Gamen S, Hanson DA, Kaspar A, Naval J, Krensky AM, Anel A (1998) Granulysin-induced apoptosis. I. Involvement of at least two distinct pathways. J Immunol 161:1758–1764
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, Diaz LA (2019) Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 16:361–375
Gerhauser C, Klimo K, Hummer W, Holzer J, Petermann A, Garreta-Rufas A, Bhmer F-D, Schreier P (2009) Identification of 3-hydroxy-β-damascone and related carotenoid-derived aroma compounds as novel potent inducers of Nrf2-mediated phase 2 response with concomitant anti-inflammatory activity. Mol Nutr Food Res 53:1237–1244
Green SJ, Meltzer MS, Hibbs JB, Nacy CA (1990) Activated macrophages destroy intracellular Leishmania major amastigotes by an l-arginine-dependent killing mechanism. J Immunol 144:278–283
Guo R, Liu Y, Pan J, Guan W, Yang BY, Kuang HX (2019) A new sesquiterpenoid with cytotoxic and anti-inflammatory activity from the leaves of Datura metel L. Nat Prod Res 28:1–7
Gupta A, Kumar S, Mahindroo N, Saini RV (2016) Bioactive fraction from Datura stramonium linn. promotes human immune cells mediated cytotoxicity towards lung and breast cancer cells. Pharmacogn J 8:5435–5439
Hata T, Sakaguchi I, Mori M, Ikeda N, Kato Y, Minamino M, Watabe K (2003) Induction of apoptosis by Citrus paradisi essential oil in human leukemic (HL-60) cells. In Vivo (Athens, Greece) 17:553–559
Hira I, Kumar A, Kumari R, Saini AK, Saini RV (2018) Pectin-guar gum-zinc oxide nanocomposite enhances human lymphocytes cytotoxicity towards lung and breast carcinomas. Mater Sci Eng, C 90:494–503
Iqbal S, Sivaraj C, Gunasekaran K (2017) Antioxidant and Anticancer Activities of Methanol Extract of Seeds of Datura stramonium. Free Radic Antioxid 7:2
Iriti M, Colnaghi G, Chemat F, Smadja J, Faoro F, Visinoni FA (2006) Histo-cytochemistry and scanning electron microscopy of lavender glandular trichomes following conventional and microwave-assisted hydrodistillation of essential oils: a comparative study. Flavour Fragr J 21:704–712
Kim KY (2018) Anti-inflammatory and ECM gene expression modulations of β-eudesmol via NF-κB signaling pathway in normal human dermal fibroblasts. Biomed Dermatol 2(1):3
Kroemer G, Petit P, Zamzami N, Vayssiere JL, Mignotte B (1995) The biochemistry of programmed cell death. FASEB J Off Publ Fed Am Soc Exp Biol 9:1277–1287
Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ, Watanabe T (2015) Colorectal cancer. Nat Rev Dis Primers 5:15065
Kumar S, Saini RV, Mahindroo N (2017) Recent advances in cancer immunology and immunology-based anticancer therapies. Biomed Pharmacother 96:1491–1500
Kumar S, Gupta A, Saini RV, Kumar A, Dhar KL, Mahindroo N (2020) Immunomodulation-mediated anticancer activity of a novel compound from brugmansia suaveolens leaves. Bioorg Med Chem 28:115552
Kumari R, Saini AK, Kumar A, Saini RV (2019) Apoptosis induction in lung and prostate cancer cells through silver nanoparticles synthesized from Pinus roxburghii bioactive fraction. J Biol Inorg Chem 25:23–37
Lanier LL (2008) Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9:495–502
Lin SSC, Lu TM, Chao PC, Lai YY, Tsai HT, Chen CS, Lee YP, Chen SC, Chou MC, Yang CC (2011) In vivo cytokine modulatory effects of cinnamaldehyde, the major constituent of leaf essential oil from Cinnamomum osmophloeum Kaneh. Phytother Res 25:1511–1518
Ohshima H, Tatemichi M, Sawa T (2003) Chemical basis of inflammation-induced carcinogenesis. Arch Biochem Biophys 417:3–11
Orhan IE, Mesaik MA, Jabeen A, Kan Y (2016) Immunomodulatory properties of various natural compounds and essential oils through modulation of human cellular immune response. Ind Crop Prod 81:117–122
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424
Pathania AS, Guru SK, Verma MK, Sharma C, Abdullah ST, Malik F, Chandra S, Katoch M, Bhushan S (2013) Disruption of the PI3K/AKT/mTOR signaling cascade and induction of apoptosis in HL-60 cells by an essential oil from Monarda citriodora. Food Chem Toxicol 62:246–254
Pena SV, Hanson DA, Carr BA, Goralski TJ, Krensky AM (1997) Processing, subcellular localization, and function of 519 (granulysin), a human late T cell activation molecule with homology to small, lytic, granule proteins. J Immunol 158:2680–2688
Ricci JE, Gottlieb RA, Green DR (2003) Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol 160:65–75
Saini RV, Wilson C, Finn MW, Wang T, Krensky AM, Clayberger C (2011) Granulysin delivered by cytotoxic cells damages endoplasmic reticulum and activates caspase-7 in target cells. J Immunol 186:3497–3504
Santos CCDMP, Salvadori MS, Mota VG, Costa LM, de Almeida AAC, de Oliveira GAL, Costa JP, de Sousa DP, de Freitas RM, de Almeida RN (2013) Antinociceptive and antioxidant activities of phytol in vivo and in vitro models. J Neurosci 2013:949452
Seo MJ, Kim SJ, Kang TH, Rim HK, Jeong HJ, Um JY, Kim HM (2011) The regulatory mechanism of β-eudesmol is through the suppression of caspase-1 activation in mast cell–mediated inflammatory response. Immunopharm immunot 33:178–185
Sharma A, Goyal R, Sharma L (2015) Potential biological efficacy of Pinus plant species against oxidative, inflammatory and microbial disorders. BMC Complem Altern Med 16:35
Sveen A, Kopetz S, Lothe RA (2020) Biomarker-guided therapy for colorectal cancer: strength in complexity. Nat Rev Clin Oncol 17:11–32
Taniguchi M, Harada M, Kojo S, Nakayama T, Wakao H (2003) The regulatory role of Vα14 NKT cells in innate and acquired immune response. Annu Rev Immunol 21:483–513
Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE (2006) TNF downmodulates the function of human CD4+ CD25hi T-regulatory cells. Blood 108:253–261
Verma MK, Anand R, Chisti AM, Kitchlu S, Chandra S, Shawl AS, Khajuria RK (2010) Essential oil composition of Artemisia dracunculus L. (tarragon) growing in Kashmir-India. J Essent 13:31–335
Zuzarte M, Salgueiro L (2015) Essential oils chemistry. In: de Sousa DP (ed) Bioactive essential oils and cancer. Springer, Cham, pp 19–61
Acknowledgements
Authors are grateful to Director (CSIR-IIIM, Jammu), Central Research Cell (MMDU Mullana) and SUBMS for providing the facilities to conduct this research. The work was funded by “Pilot Project grant for Young Investigators in Cancer Biology”, Department of Biotechnology, Govt. of India. BT/PR9613/MED/30/1260/2013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chandan, G., Kumar, C., Verma, M.K. et al. Datura stramonium essential oil composition and it’s immunostimulatory potential against colon cancer cells. 3 Biotech 10, 451 (2020). https://doi.org/10.1007/s13205-020-02438-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-020-02438-4