
Abstract
Finger millet is a rich source of seed storage proteins (SSPs). Various regulatory genes play an important role to maintain the quality and accumulation of SSPs in crop seeds. In the present study, nine regulatory genes of EAAs metabolic pathway, i.e., aspartate kinase, homoserine dehydrogenase, threonine synthase, threonine dehydratase, dihydrodipicolinate synthase, cystathionine γ synthase, anthranilate synthase, acetolactate synthase and lysine 2-oxoglutarato reductase/saccharopine dehydrogenase (LOR/SD) were identified from the transcriptomic data of developing spikes of two finger millet genotypes, i.e., GP-45 and GP-1. Results of sequence alignment search and motif/domain analysis showed high similarity of nucleotide sequences of identified regulatory genes with their respective homologs in rice. Results of promoter analysis revealed the presence of various cis-regulatory elements, like nitrogen responsive cis-elements (O2-site and GCN4), light responsive cis-elements, and stress responsive cis-elements. The presence of nine regulatory genes identified from the transcriptomic data of GP-45 and GP-1 was further confirmed by real time expression analysis in high and low protein containing genotypes, i.e., GE-3885 and GE-1437. Results of real time expression analysis showed significantly higher expression (p ≤ 0.01) of regulatory genes in GE-3885 rather than GE-1437 under control and treatment condition. Crude protein content of GE-3885 was found to be significantly higher (p ≤ 0.01) in comparison to GE-1437 under control condition, while under treatment condition GE-1437 was found to be more responsive to KNO3 treatment rather than GE-3885.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Millets are small seeded annual cereals, which rank 6th in the world cereal grain production (Verma and Patel 2012; Changmei and Dorothy 2014). Millets grow in tropical and arid climate and are known as climate resilient crops. They contain enormous minerals (Ca, Fe, Zn and others), vitamins, polyphenols, fiber, antioxidants and quality seed proteins that are good source of essential amino acids; and, therefore, are considered as nutricereals. Among the minor millets finger millet (Eleusine coracana) commonly known as madua (ragi), is an ethnic crop of Uttarakhand, India.
Since finger millet capitalizes on low nitrogen inputs, it could be considered as highly nitrogen use efficient crop (Gupta et al. 2012). Despite grown under low or limiting nitrogen conditions, the protein content of finger millet grain is comparable to that of the major cereals like wheat and rice, which consumes large amount of nitrogen fertilizers. Probably finger millet has developed unique mechanisms of protein accumulation under low nitrogen conditions in contrast to rice and wheat. Seed storage proteins (SSPs) are predominantly synthesized and stably accumulated in maturing endosperm tissue in cereals. Endosperm storage proteins present in cereal crops can be classified as albumins, globulins, prolamins, and glutelins based on their solubility (Landry et al. 2000; Osborne and Mendel 1914). The major storage proteins of finger millet are prolamins. During seed development, the expression of prolamin genes is regulated by concerted or combinatorial action of cis-elements/motifs of their promoters and seed-specific TFs, namely, Dof-PBF and bZIP TFs in conferring endosperm specificity (Shewry and Tatham 1995; Albani et al. 1997; Mena et al. 1998). Endosperm box, a conserved cis-element, has been found in many cereal prolamin genes (Forde et al. 1985). It is composed of P-box (TGTAAAG) and GCN4 motif [TGA(G/C)TCA]. The P-box has been reported to be recognized by a Dof (DNA binding with one finger)-type transcription factor (Vicente-Carbajosa et al. 1997). The Dof proteins are found to play an important role in activation of SSP genes expression and their accumulation, specifically of prolamin (Vicente-Carbajosas et al. 1997; Diaz et al. 2002; Yanagisawa et al. 2004; Gupta et al. 2018; Kumar et al. 2018).
Millets are known for their nutraceutical value and quality proteins (Kumar et al. 2015, 2016). Some of the SSPs are nutritionally superior having rich source of essential amino acids (Gaur et al. 2018). This may be due to the presence of common structural feature, i.e., amino acid sequences consisting of repeated blocks based on one or more short peptide motifs, or enriched specific amino acid residues, such as methionine. These features are responsible for high proportion of histidine, methionine, phenylalanine, and some other essential amino acids in some group of prolamin (Shewry and Halford 2002). Some regulatory enzymes found to play important role in biosynthesis of the essential amino acids using aspartate/pyruvate as common precursors (Wallsgrove et al. 1983; Azevedo et al. 1997; Ravanel et al. 1998). The detailed information about the gene sequence of these regulatory enzymes needs to be explored. The availability of the nucleotide sequence provides an opportunity to identify and characterize the regulatory genes and their regulatory element. These regulatory genes may increase the biosynthesis and accumulation of EAAs and, therefore, the quality of SSPs. The information obtained from the in silico analysis of regulatory genes will surely open up the clues about their regulatory mechanism. This will further lead to decipher the role of regulatory genes in EAAs metabolism, interaction with other regulatory elements and TFs, and accumulation of SSPs. This can further be exploited for crop engineering/crop improvement to develop biofortified crops with high quality seed proteins by altering the metabolism of EAAs, so that higher content of essential amino acids could be available to human and animal diets (Chakraborty et al. 2000; Molvig et al. 1997; Tabe et al. 1995).
Nucleotide sequences of several genes encoding seed storage proteins from important cereals are now available in the public databases and the list is increasing with the sequencing of cereal genomes. Identification and characterization of the SSPs and other regulatory genes involved in protein quality improvement has been done in the genome of rice (http://rice.plantbiology.msu.edu/), and studies are going on in Setaria italica and some other millets also (Foley et al. 2011; Gaur et al. 2018; Ponzoni et al. 2018). In this sequence, the present study is conducted for identification and characterization of regulatory genes involved in EAAs metabolism in finger millet.
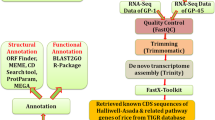
Materials and methods
Transcriptome wide identification of regulatory genes involved in EAAs metabolic pathway in finger millet
To identify regulatory genes of EAAs metabolic pathway from finger millet transcriptome, nucleotide and protein sequences of all regulatory genes, i.e., aspartate kinase (AK), homoserine dehydrogenase (HD), threonine synthase (TS), threonine dehydratase (TD), dihydrodipicolinate synthase (DHDPS), cysthathionine-y-synthase (CS), anthranilate synthase (AS), acetolactate synthase (ALS), lysine 2-oxoglutarato reductase/saccharopine dehydrogenase (LOR/SD) bifunctional and chorismate mutase (CM), were retrieved from NCBI and TIGR (Rice Genome Annotation Project). Transcriptome data of developing spikes of two finger millet genotypes, i.e., GP-45 and GP-1 was used as database (Kumar et al. 2015) and CDS of all regulatory genes of EAAs metabolic pathway from rice were used as query sequences for Blastn. Number of contigs from both the strands (i.e., sense and antisense) with different identity and score were obtained for every particular gene as a result of Blastn. Reverse complimentary tool was used to convert all the antisense sequences (complimentary sequence) into sense sequence (coding sequence) for all the genes. Among all, a single contig was selected for every gene on the basis of lower e value and highest score, which was indicating a better alignment and comparision.
ORF prediction
An online tool, ORF finder (http://www.ncbi.nlm.nih.gov/projects/gorf/) was used to find out the open reading frame of selected contigs. Among all, the longest frame was selected to retrieve nucleotide and protein sequences.
Phylogenetic analysis of regulatory genes of EAAs biosynthesis
Regulatory genes of EAAs biosynthesis pathway, i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS, and LOR/SD were retrieved from the transcriptome data of two finger millet genotypes (Kumar et al. 2015) followed by their sequence alignment with respective homologous genes of rice. Phylogenetic tree was constructed using MEGA5 standalone tool, and the evolutionary history was inferred using the Neighbor-Joining method (Tamura et al. 2011; Saitou and Nei 1987). EBI online software was used to check the percent similarity between protein sequences of finger millet and rice regulatory genes (http://www.ebi.ac.uk/Tools/psa/emboss_water/protein.html).
Domain and motif analysis of regulatory genes of EAAs biosynthesis
Domain analysis was done by Batch CD Search in NCBI for searching the Conserved Domain Database with protein/nucleotide query sequences (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). ScanProsite was used for structural and functional verification of the regulatory genes, which detected PROSITE signature matches and ProRule associated structural and functional residues in proteins (http://prosite.expasy.org/scanprosite/). The default parameters were used (with minimum width 6 and maximum width 50 amino acid) for analysis. Motif analysis of EAAs biosynthesis regulatory genes was done using MEME (Multiple EM for Motif Elicitation) version 4.11.3 program (Bailey et al. 2009).
Physicochemical properties of regulatory genes involved in EAAs biosynthesis
Analysis of protein sequences of EAAs biosynthesis regulatory genes, i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS and LOR/SD was done by Protein Identification and Analysis Tool (Protparam) on the ExPASy Server (http://web.expasy.org/protparam/) to explore physicochemical properties. Sub-cellular localization was done by TargetP 1.1 server (http://www.cbs.dtu.dk/services/TargetP/).
Promoter analysis of regulatory genes of EAAs biosynthesis
To conduct promoter analysis, upstream sequence (up to 1500 ntd) from transcription start site was taken for all the regulatory genes, i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS and LOR/SD. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences was used for promoter analysis (Lescot et al. 2002).
Expression analysis of differentially expressed regulatory genes of EAAs biosynthesis pathway
In the present study, sequencing reads were mapped to each gene, i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS, and LOR/SD and presented as FPKM (Fragments per Kilo base of exon per Million fragments mapped) value for transcriptome based expression analysis using Bed Tool (Trapnell et al. 2010). FPKM values of each contig were calculated and compared for transcriptome of both genotypes of finger millet followed by development of heat map using R package (R Development Core Team 2012).
Validation of gene expression through quantitative PCR
Validation of the regulatory genes of EAAs biosynthesis, i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS and LOR/SD was done by their expression analysis in 4 developmental stages of spikes by real-time PCR. 4 stages of developing spikes were S1—spike emergence; S2—pollination stage; S3—dough stage and S4—maturation stage. Two genotypes of finger millet, i.e., GE-1437 and GE-3885 were grown in polyhouse under control and treatment condition. In control condition, seeds were soaked overnight in water; while in treatment condition, seeds were soaked overnight in KNO3 solution before sowing. Total RNA extraction was done using RNA isolation iRIS solution (from IHBT Palampur) followed by DNase treatment (DNase I, 30 min at 37 °C + EDTA, 5 min at 70 °C) to remove residual DNA contamination. 2 μg of RNA was used for the first-strand cDNA synthesis using synthesis kit (Fermentas, Germany). Primers were designed for respective genes according to their nucleotide sequences (Supplementary table I) using Primer blast tool. The tubulin gene was used as an internal control to normalize the expression level of the gene of interest (GOI). To perform the Real-time PCR, each reaction contained Cyber Select Master Mix (applied biosystems), primers, and cDNA. For each sample, reaction was performed in triplicates and the relative expression of all genes was calculated by ΔΔCT method. Two factor ANNOVA was done for statistical analysis of data.
Construction of the EAAs biosynthesis pathway
All the regulatory genes involved in EAAs biosynthesis pathway, i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS and LOR/SD were identified from various sources, and the pathway was constructed accordingly on the basis of in silico analysis of regulatory genes. The network of EAAs biosynthesis pathway was created using Cell Designer 4.4 (http://celldesigner.org/) software, that enabled user to describe molecular interactions and biological networks using a well-defined and consistent graphical representation (Kitano et al. 2005). Cell Designer was used for understanding the logic and dynamics of gene regulatory and biochemical networks using system biology approach.
Determination of total nitrogen and crude protein content by micro-kjeldahl method
Micro-kjeldahl method was used for determination of total seed protein content, which followed 3 steps, i.e., digestion, distillation, and titration. 200 mg of seed sample was taken for digestion with concentrated H2SO4 + 1 gm of catalytic mixture at 150 °C temperature for initial 45 min, and then increases to 350 °C. Digestion resulted in clear solution was further proceeded for distillation. 4% boric acid, 40%NaOH, and mixed indicator were used or distillation. Mixed indicator was prepared by addition of 20 ml of mixture (0.2% methyl red + 0.2% methylene blue, in ethanol) in 4% boric acid.
Distilled samples were titrated with 0.04 N HCl. Total nitrogen was calculated by following formula (Humphries 1956). Total nitrogen content of seed sample was multiplied by factor 6.25 to obtain crude protein content in gm/100 gm of dried seed:
Results
Identification and characterization of regulatory genes involved in EAAs metabolism
To identify regulatory genes involved in EAAs metabolism in Finger millet, nucleotide sequences of such regulatory genes (i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS, LOR/SD, and CM) from rice were taken as reference. Rice sequences for the same ten regulatory genes were used as query for blastn; and numbers of homologous contigs were obtained for each regulatory gene (except CM) from the transcriptome data of finger millet. Best contig which showed maximum homology and best query cover among all was fetched out for each and every regulatory gene in finger millet. The identified genes were designated on the basis of their respective rice homologs. Nucleotide sequences of nine regulatory genes were submitted to NCBI. Table 1 presents the accession no. and percent nucleotide sequence similarity of identified regulatory genes with the regulatory genes of rice. All the regulatory genes of finger millet showed high sequence similarity with their rice homologs.
Phylogenetic analysis of regulatory genes
Nucleotide sequences of nine regulatory genes (i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS, and LOR/SD) from rice and finger millet were taken to construct the phylogenetic tree. MEGA7 software was used to construct the phylogenetic tree. Phylogenetic tree revealed the evolutionary relationship of regulatory genes of EAAs metabolism in rice and finger millet. Phylogenetic tree of regulatory genes grouped into 3 clusters A, B, and C (Fig. 1). TD, AS, TS, CS and ALS were included in cluster A; AK1, AK2 and HD in cluster B; and DHDPS, LOR/SD bifunctional and SD were in cluster C. AK1 was found to be more closely related to HD rather than AK2 in cluster B, while FM LOR/SD bifunctional and Os LOR/SD bifunctional were included in cluster C.

Phylogenetic tree showing the evolutionary relationship among regulatory genes of EAAs metabolism of rice and finger millet. Regulatory genes are grouped into 3 clusters A, B, and C
Domain and motif analysis
To reveal the structural and functional aspects of identified regulatory genes (i.e., AK1, AK2, HD, TS, TD, DHDPS, CS, AS, ALS, and LOR/SD) protein sequences of respective genes were subjected to domain analysis using CD search tool (Marchler-Bauer and Bryant 2004; Marchler-Bauer et al. 2015, 2011, 2017) and Protparam tool. Results indicated that there were 13 conserved domains for 10 regulatory genes. AK, DHDPS, TD, CS, AS, and ALS consisted two functional domains, while the other enzymes consisted single domain. Among all the domains, activity of most of the domains was known, while the functions of few others belonged to AK, HD, DHDPS, and TD was found to be unknown. The list of the domains with known functions and respective superfamily of enzymes to which they belong are mentioned in Table 2. One of the domains of AK which belonged to LysC superfamily was found to be responsible for amino acid transport and metabolism. Single domain of TD belonged to PALP superfamily was a pyridoxal-phosphate dependent enzyme. CS was also a pyridoxal-phosphate dependent enzyme, which consisted two domains belonged to AAT-I superfamily and Cys-Met-Meta-PP. AS also had two domains; one belonged to chorismate-binding superfamily and made the catalytic region of enzyme, while the other was a part of anthranilate synthase component I. Two domains of ALS belonged to ALS-ss-C- and ILvH superfamily. One domain was found to be involved in the catalysis of first step in branched-chain amino acid biosynthesis, and the other one was found to be a part of acetolactate synthase 3 regulatorysubunit. One of the domains of DHDPS belonged to ICL KPHMT superfamily was found to catalyze the formation and cleavage of either P–C or C–C bonds, while the function of another one wasunknown. A single domain present in LOR/SD was belonged to saccharopine dehydrogenase C superfamily and found to be involved in the catabolism of lysine. All the domains present in regulatory enzymes of EAAs metabolism of finger millet showed similar characteristics to their rice counterparts and belonged to the same superfamily as in rice.
Motif analysis revealed the structural similarity between the regulatory enzymes of rice and finger millet, and allowed identification of regulatory genes of EAAs metabolism retrieved from finger millet transcriptome. Figure 2 shows the results for motif analysis of regulatory genes of EAAs metabolic pathway using MEME program. OsAK1 and FMAK1 both the genes were found to commonly consist of motif 1, motif 2, motif 3, motif 5, motif 6, and motif 8, whereas motif 4 was found to be present only in OsAK1. OsAK2 and FMAK2 both the genes were found to commonly consist of motif 3, motif 5, motif 9, and motif 10, whereas motif 1 and motif 4 were present only in OsAK2. OsHD and FMHD were found to commonly consist of motif 3, motif 4, motif 5, and motif 10, whereas motif 1 was present only in OsHD. OsTS and FMTS were found to commonly consist of motif 1, motif 2, motif 4, motif 6, and motif 8. OsTD and FMTD both were found to commonly consist of motif 1, motif 2, motif 3, motif 5, and motif 8. OsCS and FMCS were found to commonly consist of motif 1, motif 7, motif 8, and motif 10, whereas motif 5 was present only in FMCS. OsAS and FMAS both were found to commonly consist of motif 1, motif 2, motif 3, motif 4, motif 9 and motif 10, whereas motif 8 was present in OsAS only. OsALS and FMALS both the genes were found to commonly consist of motif 2, motif 5 and motif 6, whereas motif 4 was present only in OsALS. OsDHDPS and FMDHDPS both the genes were found to commonly consist of motif 1, motif 2, motif 3, motif 6, and motif 7, whereas motif 4 and motif 8 were present only in FMDHDPS. OsLOR/SD and FMLOR/SD both the genes were found to consist of motif 3 only. Results of motif analysis showed that the regulatory genes of EAAs metabolism shared the common motifs between the genes of rice and finger millet. Supplementary table II shows the list of motif along with their width, occurrence among regulatory enzymes and best possible match along the database.

Motif analysis of regulatory genes of EAAs metabolic pathway using MEME program
Physicochemical properties of regulatory genes
To study physicochemical properties of identified regulatory genes of finger millet and its rice orthologs, protparam tool was used. Sub-cellular localization, stability, and instability index of these regulatory enzymes are given in Table 3. Three out of 10 enzymes, i.e., AK1, HD and CS were found to be stable, whereas two enzymes, i.e., TD and AS were found to be unstable, in both rice and finger millet. Among rest of the five enzymes, four enzymes, i.e., AK2, CS, ALS, and LOR/SD were found to be stable in finger millet, while DHDPS was found to be unstable in finger millet. The stability of the protein was found to be dependent upon the instability index. Whenever the value of instability index was less than 40, then it was found to be structurally stable and vice versa. Most of the regulatory enzymes were found to be structurally stable in finger millet; this might be a possible reason for high EAAs content in finger millet in comparison to rice. DHDPS (unstable) was found to be involved in lysine biosynthesis, while LOR/SD (stable) was found to be involved in its catabolism. This might be a probable reason for low lysine content in finger millet in comparison to other EAAs. Most of the enzymes were found to be localized in chloroplast, while few were found to be localized in mitochondria, and one was in secretary pathway. Localization of rest of the enzymes could not be predicted. Table 3 shows the stability, instability index, and localization of the regulatory enzymes of EAAs metabolism (i.e., AK1, AK2, HD, TS, TD, DHDPS, CS, AS, ALS and LOR/SD) in finger millet and rice.
Promoter analysis
Promoter analysis of regulatory genes (i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS, and LOR/SD) was done using PlantCARE software to found out different cis-elements involved in the regulation of regulatory genes of EAAs metabolism. The nucleotide sequence of ~ 1500 bp upstream from the transcription initiation site was selected for promoter analysis. Different cis-regulatory elements were found along with two common cis-regulatory elements, i.e., CAAT-box and TATA-box (Table 4). Two cis-elements, GCN4 motif and O2-site found to be involved in regulation of nitrogen/SSP metabolism were also obtained. GCN4 motif was uniquely present in AK1, while O2-site was found in AK1, AK2, AS, CS and HD genes. In addition, many light responsive cis-elements, i.e., AE-box, Box 4, GA-motif, G-Box, GATA-motif, GT1-motif, GTGGC-motif, I-box, L-Box, Sp1, TCT-motif, TCCC-motif, chs-Unit 1 m1, chs-CMA1a and 4 cl-CMA2b were also detected in regulatory genes. Some hormone responsive cis-elements, i.e., ABRE, TGA-element, AuxRR-core, GARE-motif, P-box, TGACG-motif, CGTCA-motif, TCA-element; and some stress responsive cis-elements like CCAAT-box, MBS, ARE and GC-motif were also present in regulatory genes. These cis-elements directly or indirectly may regulate the expression of regulatory genes of EAAs metabolism and in turn the synthesis of SSPs, accumulation of EAAs in SSPs, and SSP accumulation (Devoto et al. 2002; Catala et al. 2007; Yang et al. 2011).
Expression analysis of regulatory enzymes of EAAs biosynthetic pathway
Data obtained from RNA sequencing of developing spike of finger millet was used to calculate the FPKM value of regulatory genes (i.e., AK, HD, TS, TD, DHDPS, CS, AS, ALS, and LOR/SD). FPKM is defined as a quantification method for gene expression, which normalizes the total read length and the number of sequence reads (Mortazavi et al. 2008). FPKM values obtained from transcriptome data of finger millet spike was further used to generate the heat map (Fig. 3). The expression of all the regulatory genes of EAAs metabolism was found to be higher in GP-45 in comparison to GP-1 except TD and TS genes, which showed similar expression in both the genotypes of finger millet.

Transcriptome wide expression analysis of regulatory genes of EAAs metabolism in two genotypes (GP-1 and GP-45) of finger millet
Real time expression analysis was done for validation ofidentified regulatory genes (AK, HD, TS, TD, DHDPS, CS, AS, ALS and LOR/SD) in four developing stages of spikes of two finger millet genotypes, i.e., GE-1437 and GE-3885. Seeds were sown under control and treatment condition. In control, sowing was done without any pre-treatment of seeds, while in treatment, seeds were primed overnight with KNO3 solution before sowing. Both the genotypes, i.e., GE-1437 and GE-3885 showed similar response for all the genes, while the level of expression was found to be higher in GE-3885 in comparison to GE-1437. The expression of TS, CS, AS, and ALS regulatory genes was found to be higher under treatment condition, while the expression of TD, DHDPS and LOR/SD was found to be lower under treatment condition in comparison to control. Results of ANNOVA also showed the significant variation (p ≤ 0.01) between both the genotypes for all the genes. Both, the control and treatment conditions also showed highly significant variation (p = 0.00) for all the regulatory genes. The interaction of genotypes and treatments was also found to be significant (p ≤ 0.01) for all the genes except LOR and CS. Figure 4 shows the relative expression of both the genotypes under control and treatment conditions in four developmental stages of spikes (S1, S2, S3, and S4) of GE-1437 and GE-3885 for all the regulatory genes of EAAs metabolic pathway.

Expression profiling of regulatory enzymes of essential amino acid metabolic pathway through real time PCR in developing spikes of finger millet genotypes (GE-1437 and GE-3885) under control (C) and treatment (T) condition. S1, S2, S3, and S4 are the developmental stages of spikes. a Aspartate kinase (AK), b homoserine dehydrogenase (HD), c threonine synthase (TS), d cystathione γ synthase (CS), e anthranilate synthase (AS), f acetolactate synthase (ALS), g threonine dehydratase (TD), h dihydrodipicolinate synthase (DHDPS) and i lysine 2-oxoglutarate reductase–saccharopine dehydrogenase
Inter-relationship of protein accumulation with EAAs biosynthesis
To further establish the relationship between EAAs biosynthesis and protein accumulation, and to see the effect of nitrogen treatment on protein accumulation in mature seeds of finger millet; the total protein content was measured in mature seeds of GE-1437 and GE-3885 genotypes. Seeds of finger millet were grown under two different conditions, i.e., control (C), without any nitrogen treatment; and treatment (T), with KNO3 priming of seeds before sowing. Micro-kjeldahl method was used to measure the total protein content of each sample. Trends of protein accumulation in the mature seeds were observed through the determination of crude protein content. The estimated protein content in mature seeds of GE-1437 and GE-3885 for control (C) condition was found to be 7.25% and 10.40%, respectively, and for treatment conditions (T) it was 10.98% and 11.35%, respectively. Results of statistical analysis showed significant variation (p ≤ 0.01) between genotypes under control condition, while under treatment condition both the genotypes showed non-significant variation. GE-1437 showed significant increase (p ≤ 0.01) in seed protein content under treatment condition in comparison to control, while non-significant variations were obtained in seed protein content of GE-3885 under control and treatment condition. Results clearly indicated the higher accumulation of protein in GE-3885 under control condition (C) in comparison to GE-1437, while GE-1437 was found to be more responsive to KNO3 treatment for protein accumulation rather than GE-3885 (Fig. 5).

Total protein content (%P) in mature seeds of two finger millet genotypes (GE-1437 and GE-3885) under control (C) and treatment conditions (T)
Deducing the metabolic pathway for description of EAAs metabolism
A model of EAAs metabolic pathway was created using Cell designer 4.4 for better understanding of the role of metabolic networks of EAAs accumulation in relation to storage protein content in finger millet seeds (Fig. 6). Aspartic acid and pyruvate were found to be the precursor molecules for biosynthesis of nine essential amino acids. In aspartate pathway, aspartate kinase catalyzed the conversion of aspartate into aspartic semialdehyde. Homoserine dehydrogenase catalyzed the conversion of aspartic semialdehyde into homoserine. Conversion of O-phosphohomoserine into cysthathionine was catalyzed by cysthathionine γ synthase, which further lead to synthesize methionine. Conversion of O-phosphohomoserine into threonine was catalyzed by threonine synthase. Threonine dehydratase catalyzed the conversion of threonine into 2-oxobutyrate, which further lead to synthesize isoleucine. Dihydrodipicolinate synthase was found to play a regulatory role in lysine biosynthesis and catalyzed the conversion of aspartic semialdehyde into 2, 3-dihydrodipicolinate; whereas saccharopine dehydrogenase was found to play a regulatory role in lysine catabolism, and catalyzed the conversion of saccharopine into 2-aminoadipate semialdehyde. In pyruvate pathway, acetolactate synthase was found to play a regulatory role in valine and leucine synthesis and catalyzed the conversion of pyruvate into hydroxyethyl-TPP. Anthranilate synthase was found to play catalytic role in the conversion of chorismate into anthranilate. Regulatory enzyme involved in the synthesis of tyrosine and phenylalanine was found to be unknown in finger millet. Some compounds of metabolic pathway were found to be responsible for feedback regulation of these regulatory enzymes. Aspartate kinase was found to be negatively regulated by threonine and S-adenosyl methionine. Aspartate kinase and homoserine dehydrogenase was found to be negatively regulated by threonine. Similarly, threonine dehydratase was found to be negatively regulated by isoleucine. S-adenosyl methionine was found to be a negative regulator of aspartate kinase and dihydrodipicolinate synthase, while it was found to be a positive regulator of cysthathionine γ synthase and threonine synthase. Acetolactate synthase was found to be negatively regulated by valine and leucine. Similarly, anthranilate synthase was found to be negatively regulated by tryptophan. An unknown regulatory enzyme was found to be positively regulated by tyrosine and phenylalanine in finger millet. Deduced pathway suggested that the accumulation of essential amino acid might be increased with increased activity of these regulatory enzymes.

EAAs metabolic pathway was created of using systems biology graphical notation (SBGN) by Cell Designer 4.4., showing regulatory enzymes of EAAs metabolism in Finger millet
Discussion
Identification of gene in any plant species whose genome is yet not annotated could be done using either domain features of the gene family or sequences characterized from related organisms as a query sequence. In this study, regulatory genes of EAAs metabolic pathway of rice were taken as query sequence for the identification of their homologs in transcriptome of finger millet spike. Among the cereals, rice was the one with fully annotated genome. Rice and finger millet both were cereal crops; and finger millet showed high levels of similarity with rice genome according to the studies of Dida et al. (2007). Transcriptomic approaches were used to identify and characterize, regulatory genes involved in EAAs metabolic pathway, which might play an important role in accumulation of EAAs in seed storage proteins in finger millet. All the regulatory genes were found to be present in the transcriptome of finger millet except CM, which was responsible for the regulation of tyrosine biosynthesis in rice. This indicated that synthesis of tyrosine in finger millet might be regulated by some other regulatory gene. Classification of gene families included grouping of genes or proteins into families, which revealed important insights to gene evolution and gene function (Wu et al. 2003; Demuth et al. 2006). The results found on the basis of in silico analysis showed that, the gene sequences identified in finger millet genotypes were homologs of regulatory genes in rice. Each regulatory gene of rice and finger millet was belonged to the same domain superfamily, and shared the similar motif structure and, therefore, should follow the same pattern as in rice.
Promoter analysis was done by PlantCARE; a database of cis-regulatory elements (Lescot et al. 2002). These cis-elements were found to play important role in nitrogen metabolism, SSP synthesis, light response, hormone response, or stress response and directly or indirectly may regulate EAAs biosynthesis. GCN4 motif and O2-site were found to be involved in the regulation of nitrogen metabolism, SSPs synthesis and amino acid metabolism (Diaz et al. 2002, 2005; Hinnebusch 2005; Kawakatsu and Takaiwa 2010). SSPs were found to be a rich source of EAAs, so there may be possibility that these regulatory elements might be involved in the regulation of EAAs biosynthesis. The light responsive cis-elements were also found to be present, which may directly be involved in the regulation of EAAs biosynthesis (Mills and Wilson 1978; Mills et al. 1980; Bi et al. 2005; Hacham et al. 2013). Some other light responsive elements like G-box and GATA-motif were also found to be involved in the nitrogen metabolism and SSP synthesis (De Pater et al. 1993; Kawagoe and Mura 1996; Rastogi et al. 1999; Chandrasekharan et al. 2003; Bi et al. 2005). Some hormone responsive cis-elements like ABRE, GARE-motif, and P-box were found to be involved in nitrogen metabolism and SSP synthesis and, therefore, might regulate the biosynthesis of EAAs indirectly (Finkelstein and Crouch 1986; Hobo et al. 1999; Chandrasekharan et al. 2003; Mönke et al. 2004; Naito et al. 2007; Ufaz et al. 2011). Expression of some TFs was found to be stimulated by plant hormones, which in turn might regulate the EAAs biosynthesis (Nambara et al. 1992; Brocard et al. 2002; Verdier and Thompson, 2008; Ruffel et al. 2011; Léran et al. 2015). Some stress responsive cis-elements like CCAAT-box, MBS, ARE, GC-motif was also found to be involved into control the expression of regulatory genes (Devoto et al. 2002; Catala et al. 2007; Yang et al. 2011).
FPKM values of the regulatory genes were calculated to analyze the expression level of regulatory genes of EAAs metabolic pathway in transcriptome of finger millet spike. Validation of regulatory genes was done by real time expression analysis of developing stages of spikes in two finger millet genotypes grown under control and treatment conditions. The promoter analysis revealed that, there were some nitrogen responsive cis-elements present in the promoter of regulatory enzymes of EAAs metabolic pathway. Therefore, comparative expression analysis was done to establish the effect of nitrogen on expression of these regulatory enzymes in comparison to control. Regulatory genes having nitrogen responsive cis-elements, i.e., AK, HD, TS, CS, AS and ALS showed significant variation in their expression under treatment conditions in comparison to control in all the developmental stages of spikes. HD, TS, CS, AS, and ALS showed higher expression under treatment condition. This might be due to the positive response of nitrogen responsive cis-elements for nitrogen (KNO3) treatment (Devoto et al. 2002; Chandrasekharan et al. 2003; Mönke et al. 2004; Diaz et al. 2002, 2005; Hinnebusch, 2005; Bi et al. 2005; Naito et al. 2007; Catala et al. 2007; Kawakatsu and Takaiwa 2010; Ufaz et al. 2011; Yang et al. 2011). AK, TD, DHDPS, and LOR/SD showed lower expression in treatment condition as compared to control. The possible reason might be the presence of nitrogen responsive cis-elements like GCN4, which negatively regulate the expression of these genes in the presence of nitrogen (Diaz et al. 2002, 2005; Hinnebusch 2005, Kawakatsu and Takaiwa 2010). The results of FPKM value and real time expression analysis inferred that the accumulation of amino acids was genotype dependent and regulation of amino acids metabolism might influence the accumulation of protein as well in finger millet genotypes. Results of micro-kjeldahl indicated higher protein content in mature seeds of GE-3885 in comparison to GE-1437 under control condition. Barneix (2007)reported the correlation of free amino acid content with final grain protein content (GPC) during grain filling (Barneix and Guitman, 1993). It might, therefore, be possible to increase the amount of grain protein, if more available amino acids are produced during the late grain filling stage. In addition, accumulation of amino acids and protein might also be affected by fertilizer sources (Poulsson 1973; Timms et al. 1981; Cromwell et al. 1983; Tsai et al. 1984; Baxter and Rideout 2006 and Kaur et al. 2016). Both the genotype grown under KNO3 treated conditions showed high protein content in comparison to control, while GE-1437 was found to be significantly responsive to KNO3 treatment in comparison to GE-3885. The seed protein content of finger millet was found to be rich source of EAAs from the study and indicated the neutraceutical value of finger millet SSPs.
Conclusion
In this study nine regulatory genes of EAAs metabolic pathway had been identified from the transcriptome of finger millet spike, which revealed the regulatory pathway of EAAs metabolism in finger millet seeds. Higher expression of these regulatory genes in high protein content genotype indicated their important role in accumulation of storage proteins. Promoter analysis revealed the presence of various cis-regulatory elements in promoter region of these regulatory genes. In addition, higher expression of regulatory genes under KNO3 treatment also indicated the presence of some nitrogen responsive regulatory elements. Moreover, the total protein content of both genotypes suggested that the protein accumulation in finger millet seeds may be genotype dependent. Protein accumulation is a complex trait, which is affected by genetic, epigenetic, and environmental factors. Total protein content of both the genotypes increased with KNO3 primed seeds of finger millet, which indicated that protein accumulation also get affected with the supply and/or source of nitrogen. The information from this study can further be utilized for confirming the presence of nitrogen responsive and other regulatory cis-elements, which were found to be involved in the accumulation and quality improvement of SSPs. These improved quality proteins can be exploited to overcome the problem of protein malnutrition. Further, a minimum dose of nitrogen can be standardarize, at which these regulatory genes or regulatory elements would respond well enough for accumulation of EAAs and protein. This will minimize the excessive use of fertilizers and lead to sustainable environment and sustainable agriculture.
Abbreviations
- EAAs:
-
Essential amino acids
- SSPs:
-
Seed storage proteins
- TFs:
-
Transcription factors
- AK:
-
Aspartate kinase
- HD:
-
Homoserine dehydrogenase
- TS:
-
Threonine synthase
- TD:
-
Threonine dehydratase
- DHDPS:
-
Dihydrodipicolinate synthase
- CS:
-
Cystathione γ synthase
- AS:
-
Anthranilate synthase
- ALS:
-
Acetolactate synthase
- LOR/SD:
-
Lysine 2-oxoglutarato reductase/saccharopine dehydrogenase
- CM:
-
Chorismate mutase
- FPKM:
-
Fragments per kilobase of exon per million fragments mapped
References
Albani D, Hammond-Kosack MC, Smith C, Conlan S, Colot V, Holdsworth M, Bevan MW (1997) The wheat transcriptional activator SPA: a seed-specific bZIP protein that recognizes the GCN4-like motif in the bifactorial endosperm box of prolamin genes. Plant Cell 9:171–184
Azevedo R, Arruda P, Turner WL, Lea PJ (1997) The biosynthesis and metabolism of the aspartate derived amino acids in higher plants. Phytochemistry 46:395–419
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME suite: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208
Barneix AJ (2007) Physiology and biochemistry of source-regulated protein accumulation in the wheat grain. J Plant Physiol 164:581–590
Barneix AJ, Guitman MR (1993) Leaf regulation of the nitrogen concentration in the grain of wheat plants. J Exp Bot 44(10):1607–1612
Baxter P, Rideout L (2006) Decision making of 2nd year baccalaureate nursing students. J Nurs Educ 45(4):121–128
Bi YM, Zhang Y, Signorelli T, Zhao R, Zhu T, Rothstein S (2005) Genetic analysis of Arabidopsis GATA transcription factor gene family reveals a nitrate-inducible member important for chlorophyll synthesis and glucose sensitivity. Plant J 44:680–692
Brocard I, Lynch T, Finkelstein R (2002) Regulation and role of the Arabidopsis ABA-insensitive (ABI) 5 gene in ABA, sugar and stress response. Plant Physiol 129:1533–1543
Catala R, Ouyang J, Abreu IA, Hu Y, Seo H, Zhang X, Chua N (2007) The Arabidopsis E3 SUMO ligase SIZ1 regulates plant growth and drought responses. Plant Cell 19:2952–2966
Chakraborty S, Chakraborty N, Dutta A (2000) Increased nutritive value of transgenic potato by expressing a nonallergenic seed albumin gene from Amaranthus hypochondriacus. Proc Natl Acad Sci USA 97:3724–3729
Chandrasekharan MB, Bishop KJ, Hall TC (2003) Module-specific regulation of the beta-phaseolin promoter during embryogenesis. Plant J 33:853–866
Changmei S, Dorothy J (2014) Millet-the frugal grain. Int J Sci Res Rev 3:75–90
Cromwell GL, Bitzer MJ, Stahly TS, Johnson TH (1983) Effects of soil nitrogen fertility on the protein and lysine content and nutritional value of normal and opaque-2 corn. J Anim Sci 57(6):1345
De Pater S, Pham K, Chua NH, Memelink J, Kijne J (1993) A 22-bp fragment of the pea lectin promoter containing essential TGAC-like motifs confers seed-specific gene expression. Plant Cell 5:877–886
Demuth JP, De Bie T, Stajich JE, Cristianini N, Hahn MW (2006) The evolution of mammalian gene families. PLoS One 1(1):e85
Devoto A, Nieto-Rostro M, Xie D, Ellis C, Harmston R, Patrick E, Davis J, Sherratt L, Coleman M, Turner JG (2002) COI1 links jasmonate signalling and fertility to the SCF ubiquitin–ligase complex in Arabidopsis. Plant J 32:457–466
Diaz I, Vicente-Carbajosa J, Abraham Z, Martinez M, Isabel L, Moneda I, Carbonero P (2002) The GAMYB protein from barley interacts with the DOF transcription factor BPBF and activates endosperm-specific genes during seed development. Plant J 29:453–464
Diaz I, Martinez M, Isabel-LaMoneda I, Rubio-Somoza I, Carbonero P (2005) The DOF protein, SAD, interacts with GAMYB in plant nuclei and activates transcription of endosperm-specific genes during barley seed development. Plant J 42:652–662
Dida MM, Ramakrishnan S, Bennetzen JL, Gale MD, Devos KM (2007) The genetic map of finger millet, Eleusine coracana. Theor Appl Genet 114(2):321–332
Finkelstein RR, Crouch ML (1986) Rapeseed embryo development in culture on high osmoticum is similar to that in seeds. Plant Physiol 81:907–912
Foley RC, Gao L, Spriggs A et al (2011) Identification and characterisation of seed storage protein transcripts from Lupinus angustifolius. BMC Plant Biol 11:59. https://doi.org/10.1186/1471-2229-11-59
Forde BG, Heyworth A, Pywell AH, Kreis M (1985) Nucleotide sequence of a B1 hordein gene and the identification of possible upstream regulatory elements in endosperm storage protein genes from barley, wheat and maize. Nucleic Acids Res 13:7327–7339
Gaur VS, Sood S, Tiwari S, Kumar A (2018) Genome-wide identification and characterization of seed storage proteins (SSPs) of foxtail millet (Setaria italica (L.) P. Beauv.). 3 Biotech 8(10):415
Gupta N, Gupta AK, Gaur VS, Kumar A (2012) Relationship of nitrogen use efficiency with the activities of enzymes involved in nitrogen uptake and assimilation of finger millet genotypes grown under different nitrogen inputs. Sci World J 2012:625731. https://doi.org/10.1100/2012/625731
Gupta S, Pathak RK, Gupta SM, Gaur VS, Singh NK, Kumar A (2018) Identification and molecular characterization of Dof transcription factor gene family preferentially expressed in developing spikes of Eleusine coracana L. 3 Biotech 8(2):82
Hacham Y, Matityahu I, Amir R (2013) Light and sucrose up-regulate the expression level of Arabidopsis cystathionine gamma-synthase, the key enzyme of methionine biosynthesis pathway. Amino Acids 45:1179–1190
Hinnebusch AG (2005) Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59:407–450
Hobo T, Kowyama Y, Hattori T (1999) A bZIP factor, TRAB1, interacts with VP1 and mediates abscisic acid-induced transcription. Proc Natl Acad Sci 21:15348–15353
Humphries ES (1956) Mineral components and ash analysis. In: Paech K, Tracey MV (eds) Modern methods of plant analysis, vol 1. Springer, Berlin, pp 468–502
Kaur K, Gupta AK, Rajput A, Kumar M (2016) ge-CRISPR—an integrated pipeline for the prediction and analysis of sgRNAs genome editing efficiency for CRISPR/Cas system. Sci Rep 6:30870
Kawagoe Y, Mura N (1996) A novel basic region/helix-loop-helix protein binds to a G-box motif CACGTG of the bean seed storage protein b-phaseolin gene. Plant Sci 116:47–57
Kawakatsu T, Takaiwa F (2010) Cereal seed storage protein synthesis: fundamental processes for recombinant protein production in cereal grains. Plant Biotechnol J 8:939–953
Kitano H, Funahashi A, Matsuoka Y, Oda K (2005) Using process diagrams for the graphical representation of biological networks. Nat Biotechnol 23(8):961–966
Kumar A, Gaur VS, Goel A, Gupta AK (2015) De novo assembly and characterization of developing spikes transcriptome of finger millet (Eleusine coracana): a minor crop having nutraceutical properties. Plant Mol Biol Report 33:905–922
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kumar A, Jaiswal J, Sharma N, Gupta S, Kumar A (2018) Understanding the molecular basis of differential grain protein accumulation in wheat (Triticum aestivum L.) through expression profiling of transcription factors related to seed nutrients storage. 3 Biotech 8(2):112
Landry J et al (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci 97(11):5807–5811
Léran S, Edel KH, Pervent M, Hashimoto K, Corratgé-Faillie C, Offenborn JN et al (2015) Nitrate sensing and uptake in Arabidopsis are enhanced by ABI2, a phosphatase inactivated by the stress hormone abscisic acid. Sci Signal 8:ra43-ra
Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, Rouzé P, Rombauts S (2002) PlantCAREPlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30(1):325–327
Marchler-Bauer A, Bryant SH (2004) CD-Search: protein domain annotations on the fly. Nucleic Acids Res 32(W):327–331
Marchler-Bauer A et al (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39(D):225–229
Marchler-Bauer A et al (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43(D):222–226
Marchler-Bauer A et al (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45(D):200–203
Mena M, Vicente-Carbajosa J, Schmidt RJ, Carbonero P (1998) An endosperm-specific DOF protein from barley, highly conserved in wheat, binds to and activates transcription from the prolamin-box of a native B-hordein promoter in barley endosperm. Plant J 16:53–62
Mills WR, Wilson KG (1978) Amino acid biosynthesis in isolated chloroplast; metabolism of labelled aspartate and sulphate. FEBS Lett 92:129–132
Mills WR, Lea PJ, Miflin BJ (1980) Photosynthetic formation of the aspartate family of amino acids in isolated chloroplasts. Plant Physiol 65:1166–1172
Molvig L, Tabe LM, Eggum BO, Moore AE, Craig S, Spencer D, Higgins TJV (1997) Enhanced methionine levels and increased nutritive value of seeds of transgenic lupins (Lupinus angustifolius L.) expressing a sunflower seed albumin gene. Proc Natl Acad Sci 94:8393–8398
Mönke G, Altschmied L, Tewes A, Reidt W, Mock H, Bäumlein H, Conrad U (2004) Seed-specific transcription factors ABI3 and FUS3: molecular interaction with DNA. Planta 219(1):158–166
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7):621–628
Naito T, Kiba T, Koizumi N, Yamashino T, Mizuno T (2007) Characterization of a unique GATA family gene that responds to both light and cytokinin in Arabidopsis thaliana. Biosci Biotechnol Biochem 71:1557–1560
Nambara E, Satoshi N, Peter M (1992) A mutant of Arabidopsis which is defective in seed development and storage protein accumulation is a new abi3 allele. Plant J 2(4):435–441
Osborne TB, Mendel LB (1914) Nutritive properties of the maize kernel. J Biol Chem 18:1–16
Ponzoni E, Brambilla IM, Galasso I (2018) Genome-wide identification and organization of seed storage protein genes of Cannabis sativa. Biol Plant 62(4):693–702. https://doi.org/10.1007/s10535-018-0810-7
Poulsson E (1973) Amino acid analyses of wheat grain and gain fractions. Sci Rep Agric Univ Norway 52:1–18
Rastogi RP, Kumar A, Tyagi MB, Sinha RP (1999) Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010:592980
Ravanel S, Gakiere B, Job D, Douce R (1998) The specific features of methionine biosynthesis and metabolism in plants. Proc Natl Acad Sci 95:7805–7812
R Development Core Team (2012) R: A language and environment for statistical computing. R foundation for Statistical Computing, Vienna. http://www.R-project.org/
Ruffel S, Krouk G, Ristova D, Shasha D, Birnbaum KD, Coruzzin GM (2011) Nitrogen economics of root foraging: transitive closure of the nitrate-cytokinin relay and distinct systemic signaling for N supply vs. demand. Proc Natl Acad Sci 108:18524–18529
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Shewry PR, Halford NG (2002) Cereal seed storage proteins: structures, properties and role in grain utilization. J Exp Bot 370:947–958
Shewry PR, Tatham AS (1995) Seed storage proteins: structure and biosynthesis. Plant Cell 7:945–956
Tabe LM, Wardley-Richardson T, Ceriotti A, Aryan A, McNabb W, Moore A, Higgins TJV (1995) A biotechnological approach to improving the nutritive value of alfalfa. J Anim Sci 73:2752–2759
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Timms MF, Bottomley RC, Ellis JRS, Schofield JD (1981) The baking quality and protein characteristics of a winter wheat grown at different levels of nitrogen fertilisation. J Sci Food Agric 32:684–698
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat Biotechnol 28(5):511
Tsai CY, Huber DM, Glover DV, Warren HL (1984) Relationship of N deposition to grain yield and N response of three maize hybrids. Crop Sci 24:277–281
Ufaz S, Shukla V, Soloveichik Y, Golan Y, Breuer F, Koncz Z, Galili G, Koncz C, Zilberstein A (2011) A Transcriptional control of aspartate kinase expression during darkness and sugar depletion in Arabidopsis: involvement of bZIP transcription factors. Planta 233(5):1025–1040
Verdier J, Thompson RD (2008) Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant Cell Physiol 49(9):1263–1271
Verma V, Patel S (2012) Nutritional security and value added products from finger millets (ragi). J Appl Chem 1:485–489
Vicente-Carbajosa J, Moose SP, Parsons RL, Schmidt RJ (1997) A maize zinc-finger protein binds the prolamin box in zein gene promoters and interacts with the basic leucine zipper transcriptional activator Opaque2. Proc Natl Acad Sci 94:7685–7690
Wallsgrove RM, Lea PJ, Miflin BJ (1983) Intracellular localization of aspartate kinase and the enzyme of threonine and methionine biosynthesis in green leaves. Plant Physiol 71:780–784
Wu CH, Huang H, Yeh LSL, Barker WC (2003) Protein family classification and functional annotation. Comput Biol Chem 27(1):37–47
Yanagisawa S, Schmidt RJ (1999) Diversity and similarity among recognition sequences of Dof transcription factors. Plant J 17(2):209–214
Yanagisawa S, Akiyama A, Kisaka H, Uchimiya H, Miwa T (2004) Metabolic engineering with Dof1transcription factor in plants: improved nitrogen assimilation and growth under low-nitrogen conditions. Proc Natl Acad Sci 101:7833–7838
Yang X et al (2011) Gene expression biomarkers provide sensitive indicators of in plant a nitrogen status in maize. Plant Physiol 157:1841–1852
Acknowledgements
The authors wish to acknowledge University Grant Commission, Govt. of India, New Delhi for providing fellowship. Author is also thankful to the SUB-DIC Bioinformatics GBPUA&T, Pantnagar for providing valuable support.
Author information
Authors and Affiliations
Contributions
AK, DP, and KG conceptualized the manuscript. KG, AT, and AA conducted the experiments. KG and AA wrote the manuscript. AK, DP, and SG assisted and edited the manuscript. KG and AT has statistically analyzed the data. AK and DP contributed in critically revising the draft and updating the manuscript for publication.
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gururani, K., Kumar, A., Tiwari, A. et al. Transcriptome wide identification and characterization of regulatory genes involved in EAA metabolism and validation through expression analysis in different developmental stages of finger millet spikes. 3 Biotech 10, 347 (2020). https://doi.org/10.1007/s13205-020-02337-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-020-02337-8