
Abstract
This study investigated the impacts of crude oil, diesel, and gasoline on the diversity of indigenous microbial communities as well as culturable microorganisms in the studied soil. Oil contamination led to shifts in the diversity of the soil’s microbial communities, regardless of the contaminant applied. Unpolluted soils were more diverse and evenly distributed than contaminated samples. The domain Bacteria accounted for 65.15% of the whole microbial community. The bacterial phylum Proteobacteria dominated in all samples, followed by Actinobacteria and Acidobacteria. Pseudomonas with 28.15% of reads dominated in Proteobacteria, while Rhodococcus (3.07%) dominated in Actinobacteria, and Blastocatella (2.53%) dominated in Acidobacteria. The dominant fungal phyla across all samples were Ascomycota dominated by Penicillium (50.48% of sequences), and Zygomycota dominated by Mortierella (16.87%). Sequences similar to the archaeal phyla, Euryarchaeota and Thaumarchaeota, were also detected. The number of culturable microorganisms increased following the contamination and was higher in contaminated samples than in clean samples. Oil contamination also resulted in the enrichment of oil-degrading strains. Two bacteria, Serratia marcescens strain PL and Raoultella ornithinolytica PS, which were isolated from crude oil-contaminated soil, exhibited strong crude oil degradation ability. Strain PL was the most efficient strain and degraded 75.10% of crude oil, while strain PL degraded 65.48%, after 20 days of incubation. However, the mixed culture of the two strains was more effective than single strain and could achieve up to 96.83% of crude oil degradation, with a complete abatement of straight-chain hydrocarbons (from C12 to C25), and more than 91% removal of highly branched hydrocarbons, phytane and pristane, which are known to be more recalcitrant to biodegradation. Strains PS and PL are two newly isolated crude oil degraders that are not among the most prominent crude oil-degrading strains referenced in the literature. Therefore, their high degradation capacity makes them perfect candidates for the bioremediation of petroleum hydrocarbon contaminated environments.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Petroleum (Crude oil) is a primary and essential source of energy that is widely used in various fields of human activity. However, there is growing concern about the rate of global environmental pollution because of the increasing production and use of products made from crude oil. Crude oil drilling, transportation, and refining can lead to accidental discharges of pollutants into the environment. Likewise, diesel and gasoline can also be released into the environment during transportation or dispensing at the gas stations.
Crude oil, diesel, and gasoline are mixtures of different hydrocarbons including alkanes, cycloalkanes, and polycyclic aromatic hydrocarbons (Ameen et al. 2016; Avanzi et al. 2015; Hazen et al. 2016). The release of hydrocarbons, from crude oil, diesel or gasoline, into the environment has numerous adverse effects on all living creatures including humans, animals, and even microorganisms (Borowik et al. 2017; Hassanshahian et al. 2012).
Microbial populations in soil play a critical role in ecosystem stability, plant nutrition, and organic matter transfer (Kirk et al. 2004). However, petroleum pollutants can change the composition and function of soil microbial communities due to their long-term toxic effects into the environments and their resistance to biodegradation (Hassanshahian et al. 2012; Mahjoubi et al. 2017; Sutton et al. 2013). Different methods and strategies have been adopted to clean up petroleum contaminants from the environments. Bioremediation, as one of the eco-friendly and cost-effective techniques, has received great attention over the past decade (Alegbeleye et al. 2017). Microorganisms, including bacteria, yeasts, archaea, and fungi possess the ability to adapt to petroleum-contaminated environments and utilize petroleum pollutants as carbon and energy sources for growth. However, no single microbial species can break down all components of crude or refined oils (Rahman et al. 2002; Sathishkumar et al. 2008). A consortium composed of many microbial populations with full enzymatic capacities is often used to improve the effectiveness of the biodegradation process (Sathishkumar et al. 2008). Nevertheless, the degrading rate based on the use of consortia is still not satificatory (Van Hamme et al. 2003; Crisafi et al. 2016; Patowary et al. 2016). Therefore, there is still a need for new strains that are highly effective in degrading all the compounds present in crude oil.
Given that microbial communities in soils include an extensive range of different kinds of organisms with distinct metabolisms, functions, and capabilities, it is essential to gain a better understanding of how these communities may respond to the environmental stress caused by petroleum pollutants. Several studies have reported the impacts of diesel oil or crude oil contamination on the microbial diversity in soils (Borowik et al. 2017; Sutton et al. 2013; Wu et al. 2017). However, simultaneous comparison of the effects of gasoline, diesel, and crude oil is lacking. Thus, the present study aimed to complement previous works by providing further insights into the overall effects of oil contamination on microbial communities in soils. This was achieved using a culture-independent approach based on Illumina sequencing of the bacterial 16S rRNA and the fungal ITS genes regions, and a traditional culture technique based on plate counts (Kirk et al. 2004). From the crude oil-impacted communities, we isolated microbial strains with crude oil degradation abilities. Raoultella ornithinolytica strain PS and Serratia marcescens strain PL, as well as their mixed culture, exhibited a great potential for crude oil degradation and could degrade all compounds in crude oil, as well as the most recalcitrant hydrocarbons, phytane and pristane. Therefore, these strains, especially as a mixed culture, can be successfully applied on bioremediation of petroleum polluted soil.
Materials and methods
Soil sampling and chemicals
The crude oil used in this study was obtained from an oil company located in Ningbo city, China. It was dark brown with a density of 0.845 g cm−3. 0# diesel and 90# gasoline with a density of 0.773 and 0.726 g cm−3, respectively, were purchased from a Sinopec gas station in Hangzhou, China.
For soil contamination experiment, a control of unpolluted soils were collected in an urban area within the Zhaohui campus (30° 17′ 40″ N–120 °9′ 38″ E) of the Zhejiang University of Technology (Hangzhou, China), at about 16 cm depth. The soil was dried and sieved at 2 mm, then analyzed for various parameters. Soil properties were determined according to methods previously described (Roy et al. 2014).
Soil contamination process
After removing large particles like roots, cobbles, and pebbles, 200 g of the fresh soil was distributed into four different 500 mL beakers, namely S1 (control on day 1), S3 (soil contaminated with crude oil), S4 (soil contaminated with diesel), and S5 (oil contaminated with gasoline). S3, S4, and S5 were contaminated with 28 mL of the corresponding oil, at weekly intervals during 22 days as follows:
On day 1, 4 mL of crude oil, diesel, and gasoline were added into S3, S4, and S5, respectively. The oil was thoroughly mixed with soil and then all beakers including S1 were covered with plastic wrap and incubated at 18 ± 2 °C for 7 days. On day 8, 8 mL of crude oil, diesel, and gasoline was added into each corresponding beaker. Then, all beakers were incubated under the same conditions as on day 1. On day 15, the volume of oil added was increased to 16 mL and the incubation conditions were as described above. On the last day of the experiment (day 22), no additional oil was added, and soil samples were taken from each treatment (S2, S3, S4, and S5) for analysis. S2 is the control on day 22. The process of soil contamination and analyses is shown in Fig. 1.

Process of soil contamination with diesel, gasoline, and crude oil at weekly intervals during 22 days. S1 (control on day1), S2 (control after 22 days), S3 (crude oil–contaminated soil), S4 (diesel-contaminated soil), S5 (gasoline-contaminated soil)
Microbial diversity analysis: culture-dependent method based on plates counts
The plate counting method was carried out by using Luria–Bertani (LB) agar for bacterial counts, and Sabouraud agar (SAB) for fungal counts (Hare 2013). At each analysis period (days 1, 8, 15, and 22), 1 g of soil was collected from each sample and decimal dilutions of the soil were prepared with sterile distilled water. The procedure was performed in triplicate. Then, 0.1 mL of the highest dilutions was spread onto LB and SAB plates. All plates were incubated at 30 °C for 24–48 h. After incubation, the average number of colony-forming units per gram (CFU g−1) was determined. These plates were further used for the isolation of oil-degrading microorganisms.
Microbial diversity analysis: Illumina-based analysis
Soil DNA extraction
Total genomic DNA of soil was first extracted from S1 on day 1, and then on day 22 from S2, S3, S4, and S5. For each sample, DNA was extracted from 0.5 g of soil, in triplicate, using the Power Soil DNA Isolation Kit (MO BIO, Cat. no. 12888) according to the manufacturer’s instructions. The extracted DNA was run on a 1% agarose gel and then quantified using the Qubit® 2.0 Fluorometer (Life Technologies Inc., Cat. no. Q32866).
PCR amplification
The genomic DNA was amplified by a two-step PCR procedure. The first-round PCR was performed using modified primers containing barcodes and Illumina adapter sequences. For bacterial communities, the 16S rRNA genes (about 200–500 base pairs) flanking the V3–V4 hyper-variable regions were amplified using the modified primers 341F and 805R (Liu et al. 2015; Wu et al. 2017). For the fungal community, the internal transcribed spacer regions (200–400 base pairs) of the fungal genome were amplified using the modified primers ITS1 and ITS2. The PCR reaction was done in 50 μL volumes containing 20–30 ng of template DNA, 0.5 μL of primer F (50 µM), 0.5 µL of primer R (50 μM), 0.5 μL dNTP (10 mM each), 5 μL of 10 × PCR buffer, 0.5 μL Platinum Taq (5 U µL−1), and sterile distilled water to make up to a total volume of 50 µL.
The reaction mixtures were subjected to an initial step of 3 min at 94 °C, followed by 5 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 45 °C, and extension for 30 s at 65 °C. This step was followed by 20 cycles of denaturation for 30 s at 94 °C, annealing for 20 s at 55 °C, primer extension for 30 s min at 72 °C, and a final extension at 72 °C for 5 min.
PCR products were visualized on 1% agarose gels and then purified using the Agencourt AMPure XP PCR Purification kit. The amplicons were quantified on a Qubit 2.0 Fluorometer using the Qubit™ ssDNA Assay Kit (Life Technologies Inc., Cat. no. Q10212).
The second-round PCR was performed using the Illumina bridge PCR compatible primers. The PCR mixture and conditions were the same as with the first amplification. PCR products were analyzed, purified, and quantified as described above. Triplicate reactions for each sample were pooled and used for the sequencing. The sequences of the primers and barcodes used in this study are available in the Supplementary Table S1.
Illumina sequencing
Illumina sequencing libraries were prepared using the NEBNext® Ultra™ DNA Library Prep Kit according to the manufacturer’s instructions. Paired-end Illumina sequencing was performed on Illumina MiSeq (PE300) platform using the MiSeq Reagent kit 600-cycle V3 Standard Flow Cell. PCR amplification and the Illumina sequencing were carried out at the Institute of Microbiology (Zhejiang Tianke, Hangzhou, China).
Analysis of the Illumina sequencing dataset
Sequences of both bacteria and fungi were analyzed using Pre.cluster in Mothur. Quality control and data preprocessing were performed using Prinseq (Schmieder and Edwards 2011). Barcodes and low-quality sequences with less than 50 bp were removed. Then, all chimeras in the sequences were removed using chimera.uchime. After the quality check, sequences were clustered into operational taxonomic units (OTUs) at 97% similarity using Uclust (Sutton et al. 2013). Taxonomy was assigned to each OTU using RDP classifier at a confidence threshold of 80%. Sequences that were not assigned to known microbial phyla were designated as “unclassified”. The number of reads was normalized to 36,820 and 15,587 reads per sample for bacterial and fungal communities, respectively. Mothur was used to generate alpha diversity indexes. The evenness was calculated as previously described (Wilsey and Stirling 2007).
Sequence metadata of indigenous bacterial and fungal communities from all samples are available in the NCBI Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra) under accession number SRP104096.
Isolation of crude oil-degrading strains
Crude oil-degrading strains were isolated from the sample S3 (soil contaminated with crude oil) on day 22 using a Minimal Salt Medium (MSM) supplemented with crude oil. The MSM medium contained the following: K2HPO4·3H2O 1.3 g, MgSO4.7·H2O 1.0 g, CaCl2 0.005 g, Na2SO4 2.0 g, NH4Cl 1.0 g, ddH2O 1000 mL, pH 6.0, and autoclaving at 115 °C for 30 min.
Microbial inocula were prepared by mixing colonies that grew on LB agar (plates used for bacterial counts on day 22) corresponding to sample S3, with 10 mL of sterile MSM. Then, 5 mL of the solution obtained was inoculated into 250 mL Erlenmeyer flasks containing 100 mL of sterile MSM medium, and 1 mL of sterile crude oil (autoclaved at 115 °C for 30 min). Control flasks (uninoculated) were prepared under the same conditions. All flasks were sealed with silicone stoppers and then incubated for 10 days at 30 °C on a rotary shaker at 180 rpm. After a series of three subcultures in MSM flasks, a 1-mL aliquot from the flasks was spread onto sterile LB agar plates, and the plates were incubated at 30 °C for 1–5 days. Colonies with different phenotypes were picked and cultured onto crude oil-MSM agar plates to which 0.5 mL of crude oil was added to the lid of the plate. Control agar plates (uninoculated) were prepared in parallel. All plates were wrapped with Parafilm laboratory film and incubated inverted at 30 °C for 2–5 days. The procedure was repeated three times. Among the selected colonies, two isolates, PS and PL, showed continuous growth on MSM agar supplemented with crude oil. These isolates were stored for further characterization.
Identification of the most efficient crude oil degraders
The morphology of the cells was observed under a transmission electron microscope (JEOL JEM-1230). Different biochemical tests were performed as previously described (Stoica and Sorescu 2017).
For the molecular identification, a colony PCR was used to amplify the 16S rRNA gene sequence from the genomic DNA, by using the universal primers 27 F and 1492R as previously described by Obi et al. (2016). PCR reactions consisted of a 50-μL amplification mix containing 2 µL of the template DNA, 1 µL of each primer (100 µM), 25 µL of PremixTaq DNA Polymerase (Takara), and 21 µL of sterile distilled water. The amplification reaction was performed in a Thermal Cycler (Bio-Rad T100™) and the conditions were as follows: 94 °C for 10 min, 30 cycles of 94 °C for 30 s, 56 °C for 30 s, 72 °C for 3 min, and a final extension at 72 °C for 5 min. The reaction was held at 16 °C for 5 min. The amplicons were then analyzed on a 1% agarose gel, and the gel was visualized using a Molecular Imager Gel Doc (Bio-Rad). PCR amplicons for each isolate were sent to Shanghai Invitrogen Biotechnology Co., Ltd for purification and sequencing. The amplified 16S rRNA gene sequences were submitted to GenBank and analyzed using the BLAST algorithm (www.ncbi.nlm.nih.gov/blast). The phylogenetic tree was constructed using the Maximum Composite Likelihood method (Tamura et al. 2004), and the bootstrap consensus tree was inferred from 1000 replicates. The phylogenetic tree was constructed in MEGA X (Kumar et al. 2018).
Biodegradation of crude oil by individual strain and mixed cultures
A set of 250 mL Erlenmeyer flasks containing 50 mL of MSM medium and 0.2 g of crude oil was prepared and autoclaved at 115 °C for 30 min. Then, 5 mL of pure isolates (strains PL and PS) were inoculated individually into flasks. All flasks including the control were sealed with silicone stoppers and incubated aerobically at 30 °C, 180 rpm for 20 days. Another set of 250 mL flasks was prepared as described above with and without yeast extract (2 mg mL−1). Yeast extract was used to stimulate the biodegradation ability of isolated strains. Then, 5 mL of a mixed culture of strains PS and PL (2.5 mL of inoculum from each strain) was inoculated into flasks in triplicate. The flasks without microorganisms were maintained as controls. All flasks were then incubated under the same conditions as described above. The extent of crude oil degradation in the medium was estimated by Gas chromatography–Mass spectrometry (GC–MS).
Inoculum preparation
Strains PS and PL were cultured individually in LB medium for 24 h. Then, the pellets, after centrifugation, were washed and re-suspended in MSM medium. The concentration of each inoculum obtained was determined by plate count technique. The concentration of the inoculum was 2.1 × 107 CFU mL−1 for strain PS and 2.3 × 107 CFU mL−1 for strain PL.
Estimation of the extent of biodegradation by GC–MS
After the incubation period, the whole culture broth in flasks was used to extract the remaining crude oil in the control and inoculated flasks. The extraction was performed at room temperature in a separatory funnel with 20 mL of n-hexane. This procedure was repeated three times. The top layer containing n-hexane and residual crude oil was recovered for analysis. Hydrocarbons in crude oil were analyzed by GC–MS using an Agilent Technologies 7890A GC system/ 5975C inert MSD with Triple-Axis-Detector. The injection volume was 1 μL. The analysis was performed as described by Yan et al. (2013).
Components of crude oil were identified by matching the retention time and the mass spectrum of each molecule with the reference data in the mass spectral library. The efficiency of biodegradation or crude oil degradation, expressed as a percentage, was calculated based on the difference between the total area of peaks in the treatments and the control, using the following formula: P (%) = 100 − (TAS × 100/TAC), where P is the amount of crude oil degraded, TAS is the total area of peaks in the sample and TAC is the total area of peaks in the control (Santisi et al. 2015).
Statistical analysis
All experiments were performed in triplicate. A two-way ANOVA in excel was used to compare the difference between means degradation with individual strains PS and PL, and the consortium of the two strains in the presence and absence of yeast extract after 20 days. The F-ratio and the associated probability value (p value) were calculated. When the p value was < 0.05, the statistically significant differences observed between treatments means were determined using the Student’s T test.
Results and discussion
General proprieties of the studied soil
The unpolluted soil sample used was moderately acidic with a pH of 5.98. Organic carbon and organic matter content were less than 0.3 and 0.5%, respectively. The soil contained 2.0% of CaCO3, 30.0% of moisture, 36.0% of clay, 35.0% of sand, and 29.0% of silt. The soil texture was clay loam.
Responses of the microbial community to crude oil, diesel, and gasoline contamination
Clustering of the reads at 97% similarity threshold resulted in 30,238 bacterial OTUs representing a total of 184,100 sequence reads with an average length of 409 bp, and 1365 fungal OTUs representing 77,935 sequences reads with an average length of 225 bp. The sequences of these OTUs were classified into the domain Bacteria (65.15% of the total data set) with 26 phyla, the domain Fungi (34.84%) with 4 phyla, and the domain Archaea (0.01%) with 2 phyla. Oil contamination substantially changed the composition and structure of bacterial and fungal communities in the tested soil.
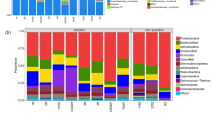
For the bacterial community, the highest number of bacterial OTUs was observed in S1 (8559 OTUs) and S2 (7495 OTUs) (Table 1). This number decreased significantly following the contamination in all contaminated samples (Table 1 and Supplementary Fig.S1). The community in S1 and S2 was more diverse and evenly distributed than that in S3, S4, and S5 as showed by the high values of Shannon diversity and evenness indices (Table 1). The coverage ranged from 86.6 to 93.0% (90.2 ± 2.62%), indicating that between 7 and 11 [1/(1- ‘Good's coverage')] additional reads would need to be sequenced before detecting a new bacterial OTU (Table 1). The highest Simpson's index was observed in S3. This value indicated the predominance of a few phyla (Proteobacteria) in this sample (Table 1; Fig. 2a). Proteobacteria dominated in all samples, but more importantly in polluted sample S3 in which it accounted for 95.61% of reads (Fig. 2a). This phylum accounted for 77.82% and 66.15% of reads in S5, and S4, and for 48.06% and 45.68% in clean samples S1 and S2 (Fig. 2a). Proteobacteria has been reported as the dominant phylum in contaminated soils in several previous studies (Sutton et al. 2013; Salam et al. 2018). The second most abundant phylum was Actinobacteria with average reads higher in polluted samples (S3, S4, and S5) than in clean samples (S1 and S2). On the other hand, oil contamination adversely affected phyla such as Acidobacteria, Bacteroidetes, Chloroflexi, Gemmatimonadetes, Planctomycetes, and Verrucomicrobia as their abundances significantly decreased following the contamination (Fig. 2a).

Abundance distribution of predominant phyla and genera in S1 (control on day 1), S2 (control after 22 days), S3 (crude oil-contaminated soil on day 22), S4 (diesel-contaminated soil on day 22), and S5 (gasoline-contaminated soil on day 22) determined by Illumina sequencing of 16S rRNA and ITS genes in the studied soil. a Predominant phyla of the bacterial community; b predominant phyla of the fungal community. Unclassified correspond to sequences that could not be assigned to known microbial phyla and Other corresponds to phyla with less than 1% and 0.5% relative abundance
For the fungal community, the highest number of OTUs was observed in S1 (315 OTUs). This number decreased following the incubation time and the contamination (Table 1 and Supplementary Fig.S2). S1 (4.42) and S5 (4.19), as well as S2 (3.22) and S4 (3.27), had a comparable Shannon index, and the coverage was 99.0% in all five samples (Table 1). The level of coverage indicated that most fungal OTUs present in these samples were identified in this study. The phylum Ascomycota predominated in polluted samples S4 (70.47%) and S5 (65.39%), but more significantly in S3 (80.80%), as also indicated by the highest Simpson's index observed in this sample (0.52) (Table 1; Fig. 2b). Zygomycota was the second well-represented phylum with dominance in S1 (28.08% of reads) and S2 (27.16% of reads) against 14.64%, 10.21%, and 10.77% of reads in S3, S4, and S5 (Fig. 2b). Likewise, Basidiomycota was more abundant in S1 (8.86%) and S2 (7.78%) than in S3, S4, and S5 in which it accounted for an average of only 1.07% of reads (Fig. 2b). The environmental stress caused by petroleum pollutants usually results in a natural selection of microbial population with the ability to survive and thrive under such conditions (Borowik et al. 2017; Yang et al. 2012).
Responses of bacterial genera to crude oil, diesel, and gasoline contamination
The most dominant bacterial genera in contaminated samples were Pseudomonas, followed by Serratia, Rhodococcus, and Sphingobium. Pseudomonas dominated in all three treatments, and especially in S3 (73.73%), while a significant increase of Rhodococcus (8.94% of reads) and Sphingobium (3.77% of reads) was observed in S4 and of Serratia (14.53%) and Gaiella (4.00%) in S5 (Fig. 3a and Supplementary Table S2). Their enrichment increased significantly following the contamination indicating that resistance to contaminants is widespread within these genera. Members of the Pseudomonas genus can adapt and grow under various environmental conditions, including petroleum contaminated sites. They are known to possess a versatile metabolism and many genes that allow them to use various carbon sources of energy (Das and Mukherjee 2007). Moreover, a great number of Pseudomonas spp. have already been reported as able to degrade crude oil (Das and Mukherjee 2007; Rahman et al. 2002; Sathishkumar et al. 2008), diesel oil, and gasoline (Avanzi et al. 2015; Lu et al. 2006).

Abundance distribution of predominant genera in S1 (control on day 1), S2 (control after 22 days), S3 (crude oil-contaminated soil on day 22), S4 (diesel-contaminated soil on day 22) and S5 (gasoline-contaminated soil on day 22) determined by pyrosequencing of 16S rRNA and ITS genes regions in the studied soil. a Predominant genera of the bacterial community; b predominant phyla of the fungal community. Unclassified correspond to sequences that could not be assigned to known microbial phyla and other corresponds to phyla with less than 1% and 0.5% relative abundance
The abundance of Rhodococcus in contaminated soils is correlated with their capacity to degrade petroleum hydrocarbons, which has already been reported in several studies (Lu et al. 2006). Similarly, Sphingobium species are also known for their natural ability to adjust to contaminated environments and use the contaminants as a growth and energy source (Waigi et al. 2015). The significant enrichment of the Serratia in gasoline-contaminated soil is consistent with the ability of members of this genus to use gasoline as a carbon and energy source for growth (Avanzi et al. 2015). On the other hand, there are no reports regarding the involvement of Gaiella in gasoline or hydrocarbon degradation. This may be because Gaiella is a novel genus that has recently been added to the phylum Actinobacteria (Albuquerque et al. 2011). Given that Gaiella was found in high abundance in gasoline-contaminated soils, the potential of members of this genus for petroleum hydrocarbon degradation could be worth investigating.
Responses of fungal genera to crude oil, diesel, and gasoline contamination
Following the contamination, Penicillium increased in abundance, whereas the abundance of Mortierella decreased. Penicillium dominated in contaminated samples with 78.38% of reads in S3, 60.97% in S4, and 48.78% in S5, while Mortierella, accounting for 28.02% in S1 and 27.15% in S2, dominated in control samples (Fig. 3b and Supplementary Table S3). The predominance of Penicillium in oil-contaminated soils could be associated with their ability to grow under different environmental conditions and use a wide range of carbon sources as previously reported (Govarthanan et al. 2017). Although Mortierella decreased in abundance after contamination, it was still the second most dominant genus in contaminated soils after Penicillium. A few members of this genus have been reported as able to degrade hydrocarbons (Hughes et al. 2007). However, overall, reports on the involvement of Mortierella spp. in petroleum hydrocarbons degradation are scarce.
It is worth noting the increased abundance of Didymosphaeria in S5 (5.84%) (Fig. 3b and Supplementary Table S3). Members of this genus could be potential candidate for the biodegradation of hydrocarbons contaminants. Minimedusa, Pluteus, and Podospora predominated in clean samples, and specifically in S1 accounting for 5.86%, 4.30%, and 3.14% of reads, respectively (Fig. 3b). However, their abundance decreased significantly to less than 0.1% following the contamination and the incubation time (Fig. 3b and Supplementary Table S3). Likewise, the abundances of bacterial genera such as Blastocatella, Sphingomonas, Escherichia-Shigella, Novosphingobium, Sphingopyxis, and Azospira decreased following the contamination (Fig. 3a and Supplementary Table S2). Crude oil, gasoline, and diesel consist primarily of complex mixtures of hydrocarbons including highly branched alkanes and polycyclic aromatic hydrocarbons. These compounds are known to have long-term toxic effects into the environments due to their resistance to biodegradation (Avanzi et al. 2015; Sathishkumar et al. 2008). Therefore, their presence in contaminated soil may have exerted adverse effects on the above species, resulting in the decreased of their abundances following the contamination.
Possible factors contributing to the microbial diversity shifts
Shifts in the microbial diversity observed between unpolluted and contaminated soils can most likely be attributed to the following main factors:
-
1.
Stress due to sudden changes in the environment following the contamination. In other words, the presence of crude oil, diesel, and gasoline in contaminated soils may have exerted adverse effects on some taxa, including Mortierella, Blastocatella and Azospira, while promoting the growth of others such as Pseudomonas, Rhodococcus, Gaiella, Sphingobium Serratia, Penicillium, and Didymosphaeria (Fig. 3a,b). The environmental stress caused by petroleum pollutants usually results in a natural selection of microbial population with the ability to survive and thrive under such conditions, leading to an increase in the number of microorganisms with the potential to degrade petroleum hydrocarbons (Borowik et al. 2017; Yang et al. 2012).
-
2.
Depletion of nutrients caused by the influx of carbon from the weekly spiking of the contaminants. Although diesel, gasoline, and crude oil are a substantial source of carbon, their high abundance may cause imbalanced carbons/nutrients ratios, resulting in competition for nutrients (Sutton et al. 2013). Nutrients, including nitrogen, phosphorus, and potassium, are essential for the metabolism and growth of indigenous microorganisms as well as for their biodegradation potential. The depletion of nutrients in the soil may have constrained the growth of certain groups of indigenous microorganisms such as Mortierella, Blastocatella, Azospira Minimedusa, Pluteus, and Podospora resulting in the decreased of their abundances after contamination (Fig. 3a, b)
-
3.
The properties of the studied soil may have also influenced the microbial composition and diversity in this study. Several phyla including Actinobacteria, Acidobacteria, Bacteroidetes, and Gemmatimonadetes were abundant in S1, but their abundances were reduced by half or more in S2 after 22 days (Fig. 2a). Likewise, the relative abundances of genera such as Sphingopyxis, Escherichia-Shigella, Sphingomonas, Gaiella, Blastocatella, and Rhodococcus, Minimedusa, Pluteus, and Podospora significantly decreased after 22 days (Fig. 3a, b). The soil investigated in this study was moderately acidic (pH = 5.98) with 30.0% of moisture. Given that no water was supplemented in the soil during the whole experimental period, the soil may have lost its initial moisture content over time leading to change in the soil pH, as the pH is known to often co-vary with different soil characteristics including moisture (Rousk et al. 2010). Several studies have reported the effect of pH on the relative abundances of Acidobacteria, Actinobacteria, and Bacteroidetes (Lauber et al. 2009; Sutton et al. 2013). The effects of soil moisture on microbial community composition were reported by Ma et al. (2015). The soil tested was a clay loam soil. This type of soil ensures a better transfer of water and air. The texture of the soil determines its ability to transmit air, water, and nutrients (Agbor et al. 2018). Therefore, the impacts of the soil structure on the composition and diversity of indigenous microbial community will depend entirely on the species of microorganisms present in these communities. Organic matter content is another important soil parameter that may influence microbial growth. Organic matter plays a crucial role in bioavailability of contaminants to microorganisms and their biodegradation potential (Mrozik and Piotrowska-Seget 2010). Many studies have reported that the rates of pollutant degradation were higher in low-organic-matter soil than in high-organic-matter soil because of lower rates of desorption and microbial growth (Mrozik and Piotrowska-Seget 2010). In this study, the tested soil contained low organic matter (0.5%). This could be the reason why well-known petroleum hydrocarbon-degrading genera, including Pseudomonas, Serratia, Rhodococcus, Blastocatella, and Penicillium dominated in this study (Fig. 3).
-
4.
Oxygen availability may have also played an important role in the shifts of soil microbial communities, and especially on the archaeal community. Archaeal phyla Thaumarchaeota and Euryarchaeota were found in very low abundance (less than 0.01%) and almost exclusively in clean samples, whereas in the study by Sutton et al. (2013), archaea have been found in higher abundant and more significantly in contaminated samples. However, it is worth noting that archaea are mostly active under anaerobic conditions (Siles and Margesin 2018). Therefore, the aerobic conditions under which the present study was carried out could be the main reason why archaea were observed at such a very low abundance.
Effects of crude oil, diesel, and gasoline contamination on culturable microbes
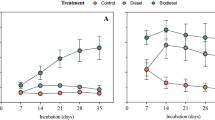
On day 1, the number of culturable bacteria was around 6.65 log CFU g−1 in all samples. This number increased reaching the maximum on day 8 in S5 (8.36 log CFU g−1) at a low concentration of gasoline (20 mL kg−1 of soil), and on day 15 in S3 (7.94 log CFU g−1) and S4 (8.24 log CFU g−1) at a relatively high concentration of diesel and crude oil (60 mL kg−1 of soil) (Fig. 4a).

The number of culturable microbes in S1 (control on day 1), S2 (control on day 22), S3 (crude oil-contaminated soil on day 22), S4 (diesel-contaminated soil on day 22), and S5 (gasoline-contaminated soil on day 22) determined at weekly intervals during 22 days. a Culturable bacteria; b culturable fungi. Error bars indicate the standard deviation of triplicate samples
Similarly, the number of culturable fungi decreased continuously in the control from day1 to day 22 (Fig. 4b). The number of culturable fungi reached the highest value in S3 (6.02 log CFU g−1) and S4 (7.49 log CFU g−1) on day 15, while in S5 the maximum value (6.51 log CFU g−1) was observed on day 8 (Fig. 4b). Overall, heterotrophic bacterial and fungal counts were more significant in contaminated soils than in clean samples. Gasoline, diesel, and crude oil contained compounds that are known to be highly toxic, and the toxicity of these compounds may have an inhibitory effect on soil microbial communities. However, many communities in the soil are able to adapt to these extreme conditions and metabolize the pollutants. This group of microorganisms usually represents less than 0.1% of the indigenous communities in non-contaminated environments (Mahjoubi et al. 2017; Yang et al. 2012). Their number can increase reaching 10% to even 100% of the whole microbial community after contamination (Mahjoubi et al. 2017), resulting in a higher number of culturable microorganisms in contaminated soils than in non-contaminated soils.
Isolation and identification of crude oil-degrading strains
After enrichment culture in flasks, two isolates (strains PL and PS) were selected from the sample S3 as the most efficient crude oil degraders based on their growth response on crude-oil-enriched media. The biodegradation efficacy of these strains was investigated in MSM medium individually, and as a consortium, after 20 days of incubation.
Colonies of strain PS, on LB agar, were 1 to 2 mm in diameter, circular, cream-white, glistening and moist with an entire margin (Fig. S3). This strain could grow on LB agar at 10 °C and 37 °C after 1–3 days. Colonies of isolate PL on LB agar slant were red, 1–3 mm in diameter, circular with an entire margin (Fig. S3). Isolate PS was rod-shaped non-motile with a size from 0.2 to 1 μm, while strain PL was a straight rod, motile bacterium, with a diameter between 0.5 and 1 μm (Fig. S3). Isolates PS and PL were gram-negative, oxidase negative, arginine dihydrolase negative, citrate positive, catalase positive, glucose positive, and non-sporulated (Supplementary Table S4). BLAST search based on the 16S rRNA gene sequences similarity indicated that isolate PS shared 99% identity with Raoultella ornithinolytica species, while isolate PL shared the highest degree of similarity with Serratia marcescens strains. The evolutionary tree of these isolates is shown in Fig. 5.

Phylogeny trees of the 16S rRNA sequences of strains PS and PL
By comparing the physiological, morphological, and biochemical characteristics of these two isolates (Supplementary Table S4), strain PL was identified as Serratia marcescens, while strain PS was identified as Raoultella ornithinolytica. The sequences of these two strains were submitted to GenBank under accession number KY464986 (strain PS) and KY652842 (strain PL). R. ornithinolytica strain PS, which is a novel strain, was deposited in the China Center for Type Culture Collection (CCTCC) under the collection number CCTCC M2018151.
Crude oil degradation by individual strains and the mixed culture
The extent of crude oil degradation in MSM medium by strain PS and strain PL was estimated by GC–MS after 20 days of incubation. Owing to the complex composition of crude oil, some of its constituents could not be readily resolved and identified by GC–MS chromatography. Compounds that could be identified in the crude oil used included linear chain n-alkanes from C12 to C25 and branched alkanes including 2,6,10-trimethyldodecane (iso-C15), 2,6,10-trimethyltridecane (iso-C16), norpristane (iso-C18), pristane (iso-C19), and phytane (iso-C20) (Table 2). After 20 days, both strains completely degraded short chain n-alkanes C12, C13, and iso-C15, but only partially degraded long-chain alkanes (Table 2). Nevertheless, more than 50% of aliphatic hydrocarbons such as C14, C15, C16, C17, C19, C20, C21, iso-C16, and iso-C19 were degraded by the two strains (Table 2).The overall biodegradation capacity of strain PS was greater than that of strain PL. Strain PS degraded 75.10% of crude oil, while strain PL degraded 65.48%, after 20 days. There was a statistically significant difference between crude oil degradation by strains PS and PL (p = 0.014). Isolate PL was identified as Serratia marcescens, members of which have been previously reported as petroleum hydrocarbon degraders (Alegbeleye et al. 2017; Morales-Guzmán et al. 2017). However, there are few reports regarding the involvement of Raoultella ornithinolytica in petroleum contaminants’ biodegradation. Only a few researchers have reported the use of Raoultella ornithinolytica to degrade petroleum hydrocarbons. For examples, Alegbeleye et al. (2017) reported a Raoultella ornithinolytica strain that could degrade acenaphthene and fluorene compounds in shake culture after 14 days. Likewise, Morales-Guzmán et al. (2017) reported a Raoultella ornithinolytica C5S3 that degraded less than 10.0% of diesel after 8 days of incubation. R. ornithinolytica strain PS, isolated in this study, showed high efficiency for crude oil degradation compared to the above-mentioned strains.
Strains PS and PL showed good crude oil degradation ability. They could simultaneously degrade linear and highly branched alkanes present in the crude oil, with a complete abatement of short-chain hydrocarbons. However, the microbial combination of the two strains significantly increased the biodegradation of crude oil compared to individual strains (p = 0.00001). As shown in Table 2, all alkanes from C12 to C25 were completely degraded by the mixed culture in the presence of yeast extract, except for pristane (iso-C19) and phytane (iso-C20). Peaks of these hydrocarbons completely disappeared from the chromatograms (Supplementary Fig.S4). Isoprenoid alkanes phytane and pristane were more recalcitrant to biodegradation. However, the mixed culture could degrade 91.15 and 97.30% of these compounds, after 20 days in the presence of yeast extract (Table 2). The relative abundances of their peaks on the GC–MS chromatograms were also significantly reduced compared to the control (Supplementary Fig. S4).
Overall, the mixed culture degraded up to 91.52 and 96.83% of crude oil in the absence and presence of yeast extract (Table 2). Yeast extract was used as biostimulant agent in supplying limiting nutrients, including nitrogen and phosphorus, to stimulate the biodegradation activity of the strains. Nutrients, whether organic or inorganic, are intensively used to improve and accelerate the biodegradation of pollutants (Santisi et al.2015). The biodegradation of crude oil by the mixed culture in the presence and absence of yeast extract was not statistically different (p = 0.113). Therefore, the presence of yeast extract did not contribute significantly in increasing the rate of crude oil degradation. Instead, it is the two strains together that could achieve the degradation observed, indicating that there was a synergetic interaction between strains PS and PL. Several studies have shown that synergetic interactions among members of a consortium would result in a degradation ratio higher than that observed with individual strains (Ameen et al. 2016).
Many studies have already reported the effectiveness of consortia of two or more strains in degrading crude oil. However, it is worth noting that when comparing the ability of one consortium to another, a conclusion should not be drawn based solely on the biodegradability of the strains used, as the overall performance of a consortium depends on many factors, including the temperature, the density and concentration of the crude oil used, the incubation time, which differ from one study to another (Table 3). For instance, Li et al. (2016) reported a consortium of Bacillus TCOB-4 and Castellaniella TCOB-5, that could degrade 51.87% of crude oil (2%, w/v) after 7 days of incubation at 30 °C. Xia et al. (2017) reported a consortium of five strains, instead, that degraded 85.26% of crude oil (1%, v/v) after 15 days of incubation. Likewise, Chen et al. (2017a) tested the ability of a mixed culture of five strains to degrade different concentrations of crude oil and reported that the highest crude oil degradation (75.10%) was observed at lowest concentration of crude oil (1%, w/v). Similarly, Yang et al. (2019) compared the ability of two consortia consisting of two and four bacterial strains. Their results showed that the consortium with four strains was more affective and degraded 73.00% of crude oil, whereas the mixed culture of two strains only degraded 53.00% of crude oil (Yang et al. 2019). In the study of Zhao et al. (2011), a consortium of seven strains degraded 52.10% of crude oil, while a microbial combination of six strains that was tested by Varjania et al. (2015) achieved 83.49% of crude oil degradation. In the present study, the mixed culture, which included only two strains (strain PL and PS), was as effective as many previous studies and achieved up to 96.83% of crude oil degradation after 20 days of incubation at 30 °C (Table 3). The mixed culture could degrade all compounds in crude oil, including pristane and phytane, with a complete abatement of linear chain hydrocarbons (Supplementary Fig. S4). Isoprenoid alkanes, pristane and phytane, are known to be more resistant to biodegradation due to their highly branched nature (Yang et al. 2019). However, the mixed culture tested could degrade more than 91% of these compounds (Table 2). Therefore, strains PS and PL can be used in degrading crude oil-contaminated soils. In our recent publication, a consortium of five strains containing strains PL and PS exhibited the best degradation rate of crude oil in highly contaminated soil compared to many previous studies (Bidja Abena et al. 2019).
Many hydrocarbons in crude oil were significantly degraded to some extent by strains PS and PL as well as by the mixed culture of the two strains in the presence and absence of yeast extract. Overall short- and medium-chain alkanes were degraded more quickly than long-chain alkanes. Alkanes, in particularly those with relatively shorter chains (< C12), are generally readily degraded due to their lower molecular weight and water solubility. Medium-length alkanes (C12–C16), and long-chain alkanes (> C16) with higher molecular weight are generally degraded at a relatively slow rate owing to their hydrophobic nature (Sathishkumar et al. 2008).
Moreover, the susceptibility of crude oil to microbial degradation depends on the intrinsic ability of the microorganism to degrade crude oil and activate the key enzyme for the first step of the biodegradation. The crude oil used in this study consisted primarily of medium- and long-chain alkanes. Therefore, the key enzymes that might have been involved in the biodegradation process were more likely to be soluble cytochrome P450s and integral membrane non-heme iron monooxygenase (AlkB) enzymes for the degradation of medium-chain alkanes (C8–C16), and alkane monooxygenases (AlmA and LadA) for the degradation of long-chain alkanes (Chen et al. 2017b; Hassanshahian et al. 2012). Researches in the past decade have gained many new insights into the mechanism of alkane degradation, including branched and linear alkanes, by microorganisms and the genes encoding for the enzymes involved in the process (Chen et al. 2017b). The general aerobic biodegradation pathway of alkanes by bacteria proceeds as follows: alkanes are first converted into the corresponding alcohols that are further oxidized by alcohol dehydrogenases and aldehydes dehydrogenases to fatty acids, which then enter β-oxidation (Van Hamme et al. 2003).
The specific metabolic pathway of hydrocarbons is still unclear for many bacterial species, including Serratia marcescens and Raoultella ornithinolytica species. Therefore, further investigations are still needed to understand the mechanism of hydrocarbons’ degradation in strain PS and PL and to identify the intermediates metabolites that are produced during the biodegradation process. Likewise, the potential of these strains remains to be investigated in crude oil-contaminated soils.
Strains PS and PL can be applied as exogenous strains, in a process known as bioaugmentation, to accelerate the biodegradation of soils contaminated with crude oil. Bioaugmented strains along with indigenous microbial communities would then carry out the biodegradation of the contaminants. However, the process of bioaugmentation still encounters many environmental challenges that should be considered when setting up an in situ bioremediation. The survival of inoculated strains is one of the most challenging issues as the number of exogenous microorganisms generally decreased shortly after inoculation into the soil (Mrozik and Piotrowska-Seget 2010). Therefore, appropriate nutrients would have to be supplied to stimulate the activity of the introduced microorganisms. Nutrients, whether organic or inorganic, can be amended to a soil system. However, the use of organic wastes would not only provide all the necessary nutrients and appropriate conditions for the growth of microbial populations, but would also reduce the overall cost of the bioremediation process. Besides, the effectiveness of organic nutrient in enhancing the biodegradation of crude oil-polluted soils has already been reported in many studies (Agbor et al. 2018). Another important factor is the availability of pollutants to microorganisms. Biosurfactants can be used to reduce surface tension and increase the solubilization of hydrophobic hydrocarbons for their optimal utilization by microorganisms (Patowary et al. 2016). Other factors such as temperature, pH, and moisture content of the soil also determine the efficiency of bioaugmentation and should be taken into account when designing the experiment.
Conclusions
Oil contamination substantially changed the composition and structure of bacterial, archaeal, and fungal communities in the tested soil. Gasoline, diesel, and crude oil contamination all resulted in a significant decrease in the number of both bacterial and fungal OTUs. Proteobacteria was the most abundant phylum in the bacterial community, with Pseudomonas as the dominant genus, while Ascomycota dominated in the fungal community, with Penicillium as the dominant genus.
Crude oil contamination also resulted in the enrichment of indigenous strains with crude oil degradation potential. Serratia marcescens PL and Raoultella ornithinolytica PS exhibited high ability to degrade crude oil in liquid medium. However, the mixed culture of the two strains was more effective and could degrade all compounds in crude oil including highly branched hydrocarbons phytane and pristane. Strains PL and PS, especially as a mixed culture, are good candidates for in situ bioremediation of crude oil-contaminated soils.
References
Agbor RB, Antai SP, Nkanang AJ (2018) Microbial degradation of total petroleum hydrocarbon in crude oil polluted soil ameliorated with agro-wastes. Glo J Earth Environ Sci 3:1–7
Albuquerque L, França L, Rainey FA et al (2011) Gaiella occulta gen. nov., sp. nov., a novel representative of a deep branching phylogenetic lineage within the class Actinobacteria and proposal of Gaiellaceae fam. nov. and Gaiellales ord. nov. Syst Appl Microbiol 34:595–599. https://doi.org/10.1016/j.syapm.2011.07.001
Alegbeleye OO, Opeolu BO, Jackson V (2017) Bioremediation of polycyclic aromatic hydrocarbon (PAH) compounds: (acenaphthene and fluorene) in water using indigenous bacterial species isolated from the Diep and Plankenburg rivers, Western Cape, South Africa. Braz J Microbiol 48:314–325. https://doi.org/10.1016/j.bjm.2016.07.027
Ameen F, Moslem M, Hadi S et al (2016) Biodegradation of diesel fuel hydrocarbons by mangrove fungi from Red Sea Coast of Saudi Arabia Saudi. J Biol Sci 23:211–218. https://doi.org/10.1016/j.sjbs.2015.04.005
Avanzi I, Gracioso L, Baltazar M et al (2015) Aerobic Biodegradation of Gasoline Compounds by Bacteria Isolated from a Hydrocarbon-Contaminated Soil. Environ Eng Sci 32:990–997. https://doi.org/10.1089/ees.2015.0122
Bidja Abena MT, Li T, Shah MN, Zhong W (2019) Biodegradation of total petroleum hydrocarbons (TPH) in highly contaminated soils by natural attenuation and bioaugmentation. Chemosphere 234:864–874. https://doi.org/10.1016/j.chemosphere.2019.06.111
Borowik A, Wyszkowska J, Oszust K (2017) Functional diversity of fungal communities in soil contaminated with diesel oil. Front Microbiol 8:1862. https://doi.org/10.3389/fmicb.2017.01862
Chen Q, Li J, Liu M et al (2017a) Study on the biodegradation of crude oil by free and immobilized bacterial consortium in marine environment. PLoS ONE 12(3):e0174445. https://doi.org/10.1371/journal.pone.0174445
Chen W, Li J, Sun X et al (2017b) High efficiency degradation of alkanes and crude oil by a salt-tolerant bacterium Dietzia species CN-3. Int Biodeter Biodegr 118:110–118
Crisafi F, Genovese M, Smedile F, Russo D, Catalfamo M, Yakimov M, Giuliano L, Denaro R (2016) Bioremediation technologies for polluted seawater sampled after an oil-spill in Taranto Gulf (Italy): a comparison of biostimulation, bioaugmentation and use of a washing agent in microcosm studies. Mar Pollut Bull 106:119–126
Das K, Mukherjee AK (2007) Crude petroleum-oil biodegradation efficiency of Bacillus subtilis and Pseudomonas aeruginosa strains isolated from a petroleum-oil contaminated soil from North-East India. Bioresource Technol 98:1339–1345. https://doi.org/10.1016/j.biortech.2006.05.032
Govarthanan M, Fuzisawa S, Hosogai T et al (2017) Biodegradation of aliphatic and aromatic hydrocarbons using the filamentous fungus Penicillium sp. CHY-2 and characterization of its manganese peroxidase activity. RSC Adv 7:20716–20723. https://doi.org/10.1039/C6RA28687A
Hare JM (2013) Sabouraud agar for fungal growth. In: Gupta V, Tuohy M, Ayyachamy M (eds) Laboratory protocols in fungal biology. Springer, New York
Hassanshahian M, Emtiazi G, Cappello S (2012) Isolation and characterization of crude-oil-degrading bacteria from the Persian Gulf and the Caspian Sea. Mar Pollut Bull 64:7–12. https://doi.org/10.1016/j.marpolbul.2011.11.006
Hazen TC, Prince RC, Mahmoudi N (2016) Marine oil biodegradation. Environ Sci Technol 50:2121–2129. https://doi.org/10.1021/acs.est.5b03333
Hughes KA, Bridge P, Clark MS (2007) Tolerance of antarctic soil fungi to hydrocarbons. Sci Total Environ 372:539–548. https://doi.org/10.1016/j.scitotenv.2006.09.016
Kirk JL, Beaudette LA, Hart M et al (2004) Methods of studying soil microbial diversity. J Microbiol Meth 58:169–188. https://doi.org/10.1016/j.mimet.2004.04.006
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lauber CL, Hamady M, Knight R et al (2009) Pyrosequencing-based assessment of soil ph as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111. https://doi.org/10.1128/AEM.00335-09
Li X, Zhao L, Adam M (2016) Biodegradation of marine crude oil pollution using a salt-tolerant bacterial consortium isolated from Bohai Bay, China. Mar Pollut Bull 105:43–50
Liu H, He H, Cheng C et al (2015) Diversity analysis of the bacterial community in tobacco waste extract during reconstituted tobacco process. Appl Microbiol Biotechnol 99(1):469–476. https://doi.org/10.1007/s00253-014-5960-8
Lu S-j, Wang H-q, Yao Z-h (2006) Isolation and characterization of gasoline-degrading bacteria from gas station leaking-contaminated soils. J Environ Sci 18:969–972. https://doi.org/10.1016/S1001-0742(06)60023-5
Ma L-N, Guo C-y, Lü X-T et al (2015) Soil moisture and land use are major determinants of soil microbial community composition and biomass at a regional scale in northeastern China. Biogeosciences 12:2585–2596. https://doi.org/10.5194/bg-12-2585-2015
Mahjoubi M, Cappello S, Souissi Y et al (2017) Microbial bioremediation of petroleum hydrocarbon—contaminated marine environments. In: Zoveidavianpoor M (ed) Recent insights in petroleum science and engineering. IntechOpen, London
Morales-Guzmán G, Ferrera-Cerrato R, Rivera-Cruz MDC et al (2017) Diesel degradation by emulsifying bacteria isolated from soils polluted with weathered petroleum hydrocarbons. Appl Soil Ecol 121:127–134. https://doi.org/10.1016/j.apsoil.2017.10.003
Mrozik A, Piotrowska-Seget Z (2010) Bioaugmentation as a strategy for cleaning up of soils contaminated with aromatic compounds. Microbiol Res 165:363–375
Obi LU, Atagana HI, Adeleke RA (2016) Isolation and characterisation of crude oil sludge degrading bacteria. SpringerPlus 5:1946–1946. https://doi.org/10.1186/s40064-016-3617-z
Patowary K, Patowary R, Kalita MC, Deka S (2016) Development of an efficient bacterial consortium for the potential remediation of hydrocarbons from contaminated sites. Front Microbiol 7:1092. https://doi.org/10.3389/fmicb.2016.01092
Rahman KS, Thahira-Rahman J, Lakshmanaperumalsamy P et al (2002) Towards efficient crude oil degradation by a mixed bacterial consortium. Bioresource Technol 85:257–261
Rousk J, Bååth E, Brookes PC et al (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. Isme J 4:1340. https://doi.org/10.1038/ismej.2010.58
Roy AS, Baruah R, Borah M et al (2014) Bioremediation potential of native hydrocarbon degrading bacterial strains in crude oil contaminated soil under microcosm study. Int Biodeter Biodegr 94:79–89. https://doi.org/10.1016/j.ibiod.2014.03.024
Salam LB, Ilori MO, Amund OO et al (2018) Characterization of bacterial community structure in a hydrocarbon-contaminated tropical African soil. Environ Technol 39:939–951. https://doi.org/10.1080/09593330.2017.1317838
Santisi S, Cappello S, Catalfamo M et al (2015) Biodegradation of crude oil by individual bacterial strains and a mixed bacterial consortium. Braz J Microbiol 46:377–387. https://doi.org/10.1590/s1517-838246120131276
Sathishkumar M, Binupriya AR, Baik S-H et al (2008) Biodegradation of crude oil by individual bacterial strains and a mixed bacterial consortium isolated from hydrocarbon contaminated areas. Clean: Soil, Air, Water Air Water 36:92–96. https://doi.org/10.1002/clen.200700042
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. https://doi.org/10.1093/bioinformatics/btr026
Siles JA, Margesin R (2018) Insights into microbial communities mediating the bioremediation of hydrocarbon-contaminated soil from an Alpine former military site. Appl Microbiol Biotechnol 102:4409–4421. https://doi.org/10.1007/s00253-018-8932-6
Stoica C, Sorescu I (2017) The great bacteria Book-ABIS online encyclopedia. Regnum Prokaryotae. https://tgw1916.net/ABIS/encyclopedia.html. Accessed on Mar 2017
Sutton NB, Maphosa F, Morillo JA et al (2013) Impact of long-term diesel contamination on soil microbial community structure. Appl Environ Microbiol 79:619–630. https://doi.org/10.1128/aem.02747-12
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101:11030–11035. https://doi.org/10.1073/pnas.0404206101
Van Hamme JD, Singh A, Ward OP (2003) Recent advances in petroleum microbiology. Microbiol Mol Biol R 67:503–549. https://doi.org/10.1128/MMBR.67.4.503-549.2003
Varjania SJ, Rana DP, Jain AK et al (2015) Synergistic ex-situ biodegradation of crude oil by halotolerant bacterial consortium of indigenous strains isolated from on shore sites of Gujarat, India. Int Biodeter Biodegr 103:116–124
Waigi MG, Kang F, Goikavi C et al (2015) Phenanthrene biodegradation by sphingomonads and its application in the contaminated soils and sediments: a review. Int Biodeter Biodegr 104:333–349. https://doi.org/10.1016/j.ibiod.2015.06.008
Wilsey B, Stirling G (2007) Species richness and evenness respond in a different manner to propagule density in developing prairie microcosm communities. Plant Ecol 190:259–273. https://doi.org/10.1007/s11258-006-9206-4
Wu M, Ye X, Chen K et al (2017) Bacterial community shift and hydrocarbon transformation during bioremediation of short-term petroleum-contaminated soil. Environ Pollut 223:657–664. https://doi.org/10.1016/j.envpol.2017.01.079
Xia M, Liu Y, Taylor AA et al (2017) Crude oil depletion by bacterial strains isolated from a petroleum hydrocarbon impacted solid waste management site in California. Int Biodeter Biodegr 123:70–77. https://doi.org/10.1016/j.ibiod.2017.06.003
Yan S, Wang Q, Qu L et al (2013) Characterization of oil-degrading bacteria from oil-contaminated soil and activity of their enzymes. Biotechnol Biotec Eq 27:3932–3938. https://doi.org/10.5504/BBEQ.2013.0050
Yang S, Wen X, Jin H et al (2012) Pyrosequencing investigation into the bacterial community in permafrost soils along the China–Russia Crude Oil Pipeline (CRCOP). PLoS ONE 7:e52730. https://doi.org/10.1371/journal.pone.0052730
Yang R, Zhang G, Li S et al (2019) Degradation of crude oil by mixed cultures of bacteria isolated from the Qinghai–Tibet plateau and comparative analysis of metabolic mechanisms. Environ Sci Pollut Res 26(2):1834–1847. https://doi.org/10.1007/s11356-018-3718-z
Zhao D, Liu C, Liu L et al (2011) Selection of functional consortium for crude oil-contaminated soil remediation. Int Biodeter Biodegr 65:1244–1248
Acknowledgements
This research was supported by the College of Biotechnology and Bioengineering at the Zhejiang University of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bidja Abena, M.T., Chen, G., Chen, Z. et al. Microbial diversity changes and enrichment of potential petroleum hydrocarbon degraders in crude oil-, diesel-, and gasoline-contaminated soil. 3 Biotech 10, 42 (2020). https://doi.org/10.1007/s13205-019-2027-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-019-2027-7