
Abstract
Biofertilizer happens to be a promising alternative of chemical fertilizer in the establishment of sustainable agricultural practices. Following this observation, several nitrogen-fixing bacteria were isolated from the soil in which an isolate (AKS7) was selected for further studies as AKS7 showed considerable competence in growth on nitrogen-free growth medium. Acetylene reduction assay confirmed that AKS7 can fix atmospheric nitrogen efficiently. The result of Kjeldahl assay revealed that the organism (AKS7) could fix nitrogen up to 12 mg in 8 days. A strong positive correlation (r = 0.987) was observed between microbial cell biomass and the amount of nitrogen fixed by AKS7 over a period of 8 days. The organism was identified as Enterobacter cloacae through molecular and biochemical tests. To investigate the in situ nitrogen fixation by E. cloacae AKS7, naturally attenuated (AKS7 not-inoculated) and bioaugmented (AKS7-inoculated) soil microcosms were prepared. The bioaugmented microcosm showed higher level of soil nitrogen content than naturally attenuated microcosm. A large number of heterotrophic as well as nitrogen-fixing microorganisms were counted in bioaugmented microcosm than naturally attenuated microcosm. Results of the carbon source utilization patterns of BiOLOG ECO plate revealed that bioaugmented microcosm exhibited higher level of functional richness and evenness that lead to the exhibition of higher level of microbial functional-diversity in bioaugmented microcosm than the naturally attenuated microcosm. Taken together, the results indicated that augmentation of E. cloacae AKS7 into soil enhanced the nitrogen content and soil microbial functional-diversity considerably.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
At present epoch, the toughest challenge is to overcome the exponentially increasing food demand to support the ever-rising human population of almost 7 billion people in a sustainable way (Glick 2012). The enormous uses of chemical fertilizers in agriculture enhance the productivity of crop significantly to support the massive food demand. However, the application of chemical fertilizer causes eutrophication that leads to affect the ecosystem of water-bodies considerably (Mahanty et al. 2017). Additionally, the chemical fertilizers can also alter the water holding capacity of the soil, increase soil salinity and make the soil inconsistent with nutrients (Jetiyanon and Pliabanchang 2012). Moreover, some existing literature reported that in long run, application of chemical fertilizers to the soil for enhancing crop productivity is not recommended (Jetiyanon and Pliabanchang 2012; Mahanty et al. 2017). Considering all the consequences that could happen by the application of chemical fertilizers, biofertilizer seems to be a good alternative to boost soil fertility as they are environment-friendly and cost-effective (Bardi and Malusà 2012). Biofertilizers are mainly living microorganisms which when added to the soil improve plant growth considerably by promoting the supply of nutrients to the host plant (Bardi and Malusà 2012; Malusa and Vassilev 2014). It was documented in a literature that usage of biofertilizer promotes crop production up to 10–40% by raising the contents of proteins, vitamins, amino acids and fixing atmospheric nitrogen markedly (Bhardwaj et al. 2014). Above all, biofertilizer improves the fertility of soil considerably as they exhibit diverse functions including the fixation of aerial nitrogen to the soil (Mazid and Khan 2015). Atmospheric nitrogen (N2) is relatively inert and does not generally react with other molecules to form new compounds. Soil microbes play a pivotal role in nitrogen fixation as they convert atmospheric nitrogen into ammonia (NH3) using nitrogenase enzyme (Hoffman et al. 2009). In this way, the fixed nitrogen is used for the biosynthesis of the building blocks like nucleotides, amino acids and coenzymes that help the plants to grow comfortably. Though biofertilizers have been used quite frequently, certain factors need to be examined to ensure the safe usage of biofertilizers in agriculture. Biofertilizer which is to be introduced into soil may damage the pre-existing microbial community of soil or the introduced biofertilizer may be killed by the pre-existing microbial community of soil as well. Thus, biofertilizer following either option should not be recommended safe to use in agriculture (Mahanty et al. 2017). Therefore, biofertilizers that enhance soil fertility as well as stabilize the pre-existing microbial ecological balance in soil could be selected for sustainable agriculture development (Mahanty et al. 2017). In this current report, we have screened out an efficient nitrogen-fixing organism from soil based on its promising nitrogen-fixing potential. The organism was identified as Enterobacter cloacae AKS7. It was observed that after applying the organism (E. cloacae AKS7) into the soil, the nitrogen content as well as microbial functional-diversity of the augmented soil got increased substantially.
Methods
Sample collection
In the present study, the soil sample was collected from 5 cm below the surface of an agriculture field located in the District of South 24 Parganas, West Bengal, India. The collected soil samples were kept in sterile sealed plastic bags. Plastic bags carrying soil samples were kept at 4 °C until the required analysis was started. At first, gravels and hazardous materials were removed from the collected soil samples. After that, soils were homogenized and passed through a 2-mm pore size filter. Then, the filtered soil samples were air-dried at 37 °C.
Isolation of nitrogen-fixing bacteria
To obtain nitrogen-fixing microorganisms from the soil, 1 g soil was collected from soil and suspended in 9 mL of sterile 0.85% NaCl (Merck). Thereafter, a series of dilutions were accordingly prepared. A 0.1 mL aliquot was collected from appropriate dilution and aseptically spread onto Burk’s medium supplemented with agar (Merck). Burk’s medium has been found to be used in previous reports to isolate nitrogen-fixing bacteria from soil (Park et al. 2005; Kayasth et al. 2014; Chakraborty and Tribedi 2019). The composition of Burk’s medium was as follows-MgSO4 (200 mg/L), K2HPO4 (800 mg/L), KH2PO4 (200 mg/L), CaSO4 (130 mg/L), FeCl3 (1.45 mg/L), NaMoO3 (0.253 mg/L), sucrose (20 g/L). All the ingredients required for the preparation of Burk’s medium was purchased from Merck. The pH of the Burk’s medium was adjusted to 7.0. Agar (18 g/L) was added to make solidified medium. The plates were incubated at 30 °C for several days. Organisms that showed prominent colonies on Burk’s medium were considered as nitrogen-fixing organisms.
DNA extraction, polymerase chain reaction (PCR) of 16S rRNA gene and sequence analysis
Genomic DNA was extracted from the organism AKS7 by following the protocol as described by Ausubel et al. (1994). To extract the genomic DNA from bacteria, 1 mL overnight saturated culture of AKS7 [1 × 108 colony forming unit (CFU)] was centrifuged at 10,000 rpm for 2 min. The obtained pellet was re-suspended in 567 µL of Tris–EDTA buffer (Merck). Then, the cell suspension was further re-suspended with 3 µL of 10% SDS (Merck) and 30 µL of Proteinase K (SRL) and subsequently incubated at 37 °C for 1 h. After the incubation got over, 100 µL of 5 M NaCl solution was added and mixed thoroughly by inverting the Eppendorf (Tarson). Then, 80 µL of CTAB/NaCl (SRL) mixture was added, mixed thoroughly and further incubated at 65 °C for 10 min in a water bath. Thereafter, an equal volume (768 µL) of chloroform (Merck) was added to it and further centrifuged at 15,000 rpm for 5 min. Then, the supernatant was transferred carefully to a sterile eppendorf where an equal volume of phenol/chloroform/isoamyl alcohol (Hi media) in a ratio of 25:24:1 was added and mixed properly. Then, the mixture was centrifuged at 15,000 rpm for 5 min. To precipitate the genomic DNA, the supernatant was further transferred to a sterile eppendorf and incubated for 15 min at room temperature in presence of 0.6 mL of isopropanol (SRL). The mixture was then centrifuged at 15,000 rpm for 10 min. The precipitate was then washed three times with 100 µL of 70% ethanol (Merck). It was further centrifuged at 15,000 rpm for 10 min. After that, the supernatant was discarded and the eppendorf was dried in speed-vac for 20 min until the fragrance of ethanol got removed. Finally, the genomic DNA was re-suspended in 50 µL of Tris–EDTA buffer (Merck) and kept at 37 °C for future use.
Partial amplification of the 16S rRNA gene of our isolated organism AKS7 was carried out through PCR by following the protocol as described by Tribedi et al. (2012). The extracted genomic DNA from the bacteria (AKS7) was used as a template DNA and 16S rRNA gene-specific universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-TACGGYTACCTTGTTACGACTT-3′) was used as the primers for the PCR reaction. After the PCR got over, the amplified product of 16S rRNA gene was run through an agarose gel. The desired PCR product was then purified from the gel using the QIAGEN spin column gel extraction kit by following the manufacturer’s protocol. The purified PCR product was subsequently sequenced using Big Dye Terminator Cycle Sequencing Kit V3.1 (Applied Biosystems, USA) following the manufacturer’s protocol. The partial 16S rRNA gene sequence of the organism was subjected to BLAST search analysis (http://www.blast.ncbi.nlm.nih.gov) to identify the organism. The partial nucleotide sequence of the 16S rRNA gene of the organism was then deposited in the GenBank (http://www.ncbi.nlm.nih.gov/genbank) to get the accession number. To construct the phylogenetic tree, the partial 16S rRNA gene sequence of the isolate (AKS7) and various related 16S rRNA gene sequences of other microorganisms were aligned using MUSCLE tool of MEGA 7 (Kumar et al. 2016). A phylogenetic tree was then constructed from the aligned sequences using the maximum parsimony algorithm of MEGA 7.
Biochemical tests
Conventional biochemical tests such as gram character, motility, catalase activity and endo-spore formation were carried out by following the protocol as described previously by Dubey and Maheshwari (2012). However, urease (urea hydrolysis) production, phenylalanine deaminase assay and malonate utilisation tests of the organism were also performed by following the manufacture’s (SRL) protocol.
Nitrogen fixation assay
To examine the nitrogen-fixing ability of the isolated bacteria (AKS7), acetylene reduction assay was performed as described by Kessler and Leigh (1999). Briefly, 15 mL vials containing 5 mL of 8 days culture suspensions were incubated with acetylene gas (concentration of 10%). Such vials were then gently shaken and incubated at 30 °C for 8 days. 1 mL gas mixture was carefully taken out from the glass vial with an airtight syringe at a regular time interval. Thereafter, the collected gas mixture (1 mL) was injected into a specific column of Bruker Gas Chromatograph model GC 650. The column temperature was maintained at 100 °C. However, the injector and detector temperature were set at 110 °C. Generation of ethylene from acetylene with time confirms the nitrogen-fixing ability of the microorganism.
The amount of nitrogen fixed by the isolated organism (AKS7) was further estimated by following the protocol of the Kjeldahl method (APHA 2005). An overnight saturated culture of the isolated organism (AKS7) (1 × 108 CFU) was inoculated into 1.0 L of sterile Burk’s medium and incubated at 30 °C for 8 days. After the incubation got over, the growth medium was centrifuged at 12,000 rpm for 10 min to remove the bacterial cells. Then, cell-free supernatant was collected to determine the amount of nitrogen fixed by AKS7 through the Kjeldahl method. In parallel, the control set was also prepared in which AKS7 was not inoculated into Burk’s medium. Each growth medium was incubated at 30 °C for 8 days.
Microcosm studies
Preparation of microcosm
To examine the in situ nitrogen fixation by E. cloacae AKS7, a microcosm based study was carried out. For the making of soil microcosm, 450 g air-dried soil was put in the sterile glass beaker (Borosil). In the present study, two soil microcosms were prepared. The first one was considered as naturally attenuated microcosm in which E. cloacae strain AKS7 was not added. However, in the case of the bioaugmented microcosm, the soil was treated with E. cloacae strain AKS7. For the preparation of bioaugmented microcosm, 2 g microbial biomass of E. cloacae strain AKS7 was added to 450 g of soil. All the microcosms were separately covered with perforated aluminium foil and further incubated for 10 days at 30 °C. Sterile double distilled water was added at regular interval to make the soil microcosm moistened. Three replicates had been used for each type of microcosm. After the incubation got over, soil samples were collected from each microcosm and subsequently analyzed for several experiments.
Estimation of nitrogen content in soil microcosm
The total nitrogen content of soil microcosm was determined by the Kjeldahl method as described previously (Alef and Nannipieri 1995). In brief, soil samples collected from each soil microcosm were digested with concentrated H2SO4 (Merck) at 420 °C in presence of catalysts K2SO4 (Merck) and CuSO4 (Merck) followed by distillation in 10 N NaOH (Merck) and subsequent absorption in boric acid (Merck). The total nitrogen content of soil microcosm was determined using 0.1 N HCl (Merck) through titration.
Determination of heterotrophic microorganisms in soil microcosm
1 g soil was collected from different microcosms after 10 days of incubation and added into 9 mL of 0.85% NaCl in sterile test tubes. Thereafter, a series of dilution from 10−2 to 10−6 were prepared in 0.85% NaCl. For heterotrophic microbial count measurement, an aliquot (0.1 mL) of the appropriate dilution from each microcosm was spread aseptically onto Luria agar (LA) (Hi Media) plates for the enumeration of heterotrophic organisms. LA plates were incubated at 30 °C for 2 days.
Determination of nitrogen-fixing microorganisms in soil microcosm
To determine nitrogen-fixing microorganisms in soil microcosm, 1 g soil was collected from each microcosm after 10 days of incubation and suspended in 9 mL of 0.85% NaCl solution. Thereafter, a series of dilutions were accordingly prepared in sterile 0.85% NaCl solution. 0.1 mL aliquot of the appropriate dilution from each microcosm was aseptically spread onto Burk’s medium to obtain nitrogen-fixing bacteria. The plates were then incubated at 30 °C for 8 days.
Measurement of enzyme activities in soil microcosms
The dehydrogenase activity of soil microcosm was assessed by a modified protocol as suggested by Singh and Singh (2005). 5 g soil sample was taken from each microcosm and subsequently placed in a sterile 50-mL polypropylene tube. Then, the soil was mixed with 5 mL of 0.5% 2,3,5-triphenyl tetrazolium chloride (TTC)-Tris solution. Tubes were then incubated at 30 °C for 24 h in dark. After the incubation got over, TTC was reduced to triphenyl formazan (TPF) which was then extracted with 100 mL methanol (Merck). The concentration of TPF was then determined spectrophotometrically by recording the absorbance at 485 nm.
Fluorescein diacetate hydrolysis assay (FDAH) was performed in soil microcosms by following the modified protocol described by Adam and Duncan (2001). 5 g soil sample was taken from each microcosm and mixed with 15 mL of sterile sodium phosphate buffer (Merck) (pH 7.6) in a 50-mL Erlenmeyer flask. After that, 0.2 mL solution of fluorescein diacetate (1 mg mL−1) in acetone (Merck) was added to each sample. Flasks were then incubated for 20 min at 30 °C on a rotary shaker at 200 rpm. After the incubation got over, fluorescein was extracted from the soil with chloroform–methanol (2:1) mixture. Fluorescein concentration was thereafter measured spectrophotometrically by recording the absorbance at 490 nm.
Determination of microbial functional-diversity in soil microcosms
Previous literature documented that BiOLOG ECO plates could be used to compare the microbial functional-diversity between soil samples (Choi and Dobbs 1999; Tribedi and Sil 2013). BiOLOG ECO plate (Hayward, CA) contains Tetrazolium dye and triplicates of 31 different environmentally relevant carbon sources (Choi and Dobbs 1999). In brief, 10 g soil from each microcosm was added to 100 mL of sterile double distilled water in 250-mL sterile conical and shaken for 10 min. 150 μL of the soil suspension nearly containing ~ 3000 cfu were added to each well of the BiOLOG ECO plates. The plates were then incubated at 30 °C for 48 h. The carbon sources of BiOLOG ECO plate got utilized by the microorganisms present in each microcosm that resulted in the generation of purple colour. The intensity of the purple colour of BiOLOG ECO plate was then determined by measuring the Optical Density (OD) at 590 nm. The total biological activity of each BiOLOG ECO plate was expressed as average well colour development (AWCD). AWCD was determined by following the equation AWCD = ∑ODi/31, where ODi is the Optical Density value of each well. The Shannon–Weaver index (H), an indicator of functional-diversity, was measured using the following equation: H = − ∑pi ln pi, where pi is the ratio of the activity of each substrate (ODi) to the sum of activities of all substrates (ΣODi). The corresponding Lorenz curve was plotted for each microcosm. Thereafter, this curve was used to derive the Gini coefficient (G), which is a measure of inequality, using the formula:
Statistical analysis
Experimental results were subjected to statistical analysis of one-way analysis of variance (ANOVA) at 5% level. In the current manuscript, three replicates had been considered for each experimental set and each experiment was repeated three times.
Results and discussion
Isolation of soil bacterium that can fix aerial nitrogen efficiently
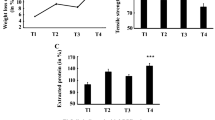
Though atmospheric nitrogen happens to be the most abundant gas (~ 78% N2) in the planet, living community except microorganisms cannot fix the atmospheric nitrogen directly. Only certain microorganisms are able to fix aerial nitrogen due to harbouring nitrogenase enzyme. These microorganisms can fix aerial nitrogen to ammonia which can be then easily assimilated by plants (Hoffman et al. 2009; Smith et al. 2013). Microorganisms that can fix atmospheric nitrogen to soil increase the fertility of soil considerably. To isolate nitrogen-fixing microorganisms from soil, Burk’s medium was selected as there was no external nitrogen source included in Burk’s medium (Park et al. 2005). Organisms that grow on Burk’s agar medium can fix atmospheric nitrogen and support their nitrogen demand on their own. In this context, we have been able to isolate 12 bacterial colonies on Burk’s medium that can fix atmospheric nitrogen. Among the isolated nitrogen-fixing organisms, the best growing bacterial isolate (AKS7) was selected for further studies by considering its colony morphology and growth efficiency. To demonstrate the nitrogen-fixing ability of the isolated bacterium AKS7, acetylene reduction assay was carried out. The acetylene reduction assay happens to be a popular method to detect the presence of nitrogenase enzyme in microorganisms as nitrogenase efficiently reduces acetylene to ethylene which can be quantified through gas chromatography (Dilworth 1966). To perform acetylene reduction assay, AKS7 was inoculated into the Burk’s liquid medium and the amount of ethylene produced by AKS7 was measured accordingly. The result showed that with time, the amount of ethylene production got increased considerably by AKS7 (Fig. 1). However, to gain further confidence, we had also examined the same in the control set wherein the Burk’s liquid medium was not inoculated with the organism (AKS7). After a similar length of incubation, we observed that ethylene was not produced from acetylene in the control set (data not shown). To determine the extent of nitrogen fixed by the microorganism (AKS7), the Kjeldahl method was followed. We had observed that with the progression of time, the amount of nitrogen in Burk’s liquid medium inoculated with AKS7 got increased continuously (Fig. 1). Over a period of 8 days, the isolated microorganism (AKS7) was found to fix approximately 12 mg of aerial nitrogen in 1.0 L of growth medium (Fig. 1). However, no aerial nitrogen was fixed in the control set in which Burk’s liquid medium was incubated in the absence of AKS7 (data not shown). Furthermore, with the progression of time, a continuous increase in cell biomass of AKS7 was also observed in Burk’s liquid medium inoculated with AKS7 (Fig. 1). Moreover, a strong positive correlation (r = 0.987) was noticed between cell biomass and microbial nitrogen fixation. Thus, the results of Fig. 1 confirmed that the isolated organism AKS7 can fix the atmospheric nitrogen for its own growth and survival. To determine the optimum temperature at which the organism (AKS7) grows and fixes maximum atmospheric nitrogen, similar numbers of organisms (AKS7) were separately inoculated in Burk’s liquid medium and incubated at different temperatures for 8 days. At regular time interval, cell biomass and ethylene production were measured in each growth medium to determine the microbial growth and extent of microbial nitrogen fixation, respectively. The result indicated that the organism (AKS7) was found to show maximum growth and could fix maximum aerial nitrogen at 30 °C (Supplementary Fig. 1A and 1B). Thus, 30 °C has been considered as the optimum temperature for obtaining the maximum growth of AKS7. Taken together, all the results showed that the nitrogen fixed by the microorganism (AKS7) might help the organism to grow considerably that resulted in the increase of cell biomass of AKS7.

Isolated organism (AKS7) showed efficient nitrogen fixing property. a Overnight saturated culture of AKS7 was incubated with acetylene gas in glass vial at 30 °C for 8 days. At different time interval, the cell-free supernatant was collected and the generation of ethylene was recorded through gas chromatography. 100% equates to 175 ppm ethylene (colored blue). b Overnight saturated culture of AKS7 was inoculated into sterile Burk’s medium and incubated at 30 °C for 8 days. The cell-free supernatant was collected at different time interval and the amount of nitrogen fixed by the organism was determined by the Kjeldahl method. 100% equates to 12 mg/L nitrogen (colored red). c Overnight saturated culture of AKS7 was inoculated into sterile Burk’s medium and incubated at 30 °C for 8 days. The weight of the microbial cell biomass was measured at different time interval during the course of incubation. 100% equates to 3.75 mg microbial cell biomass (colored green). Error bars indicated standard deviation (± SD). Mean values with different letters are significantly different from each other
Isolated organism is a new member of Enterobacter cloacae
The length of 16S rRNA gene of strain AKS7 was sequenced up to 1296 bp (accession number MK386888). BLAST analysis (http://www.blast.ncbi.nlm.nih.gov) of the obtained sequence showed that the closest relatives of strain AKS7 were members of E. cloacae (~ 99% similarity). However, BLAST analysis also revealed that the sequence of AKS7 showed considerable similarity (~ 98% similarity) with the 16S rRNA gene of different genera of Enterobacter, Klebsiella, Serratia and Escherichia which belongs to the family of Enterobacteriaceae. Based on the sequence similarity, a phylogenetic tree was constructed in which our isolated organism (AKS7) showed strong association with E. cloacae (Fig. 2). Besides, strain AKS7 could utilize malonate, was motile, non-sporulating, typically short rod-shaped gram-negative cells which were catalase and urease positive but oxidase and phenylalanine deaminase negative. Thus, all these results indicated that the isolated organism AKS7 was found to be a member of E. cloacae.

The phylogenetic tree was constructed from 16S rRNA gene sequences aligned with MUSCLE using MEGA 7. Enterobacter cloacae AKS7 used in this study (asterisked) is shown with its neighbors. Mesorhizobium loti LMG 17826t2 was used as an out-group. The scale bar represents 0.02 nucleotide exchanges per nucleotide and bootstrap values > 95% are shown at the internodes
The microcosm augmented with Enterobacter cloacae AKS7 showed higher nitrogen content and functional-diversity than naturally attenuated microcosm
To investigate the efficiency of AKS7 mediated nitrogen assimilation in the soil, we set up two different microcosms in triplicate form. In the first microcosm (naturally attenuated microcosm), the soil was not treated with AKS7 whereas in the second microcosm (bioaugmented), the soil was inoculated with AKS7. The result demonstrated that bioaugmented soil microcosm showed ~ 64% greater amount of nitrogen content compared to the nitrogen content of naturally attenuated soil microcosm (Fig. 3a). Thus, the result indicated that the highest level of nitrogen fixation took place in microcosm augmented with E. cloacae AKS7. The greater level of nitrogen content in bioaugmented microcosm could be attributed to the efficient abundance of nitrogen-fixing microorganisms in it. To verify this hypothesis, the abundance of heterotrophic as well as nitrogen-fixing microorganisms was counted in both bioaugmented and naturally attenuated soil microcosms. The result revealed that bioaugmented microcosm showed ~ 3 fold greater abundance of heterotrophic microorganisms than naturally attenuated microcosms (Fig. 3b). The number of nitrogen-fixing microorganisms present in the bioaugmented microcosm was found to be ~ 4.5 fold greater than the naturally attenuated microcosm (Fig. 3c). Thus, the results showed that higher numbers of nitrogen-fixing organisms were associated with bioaugmented microcosm that resulted in the enhanced assimilation of aerial nitrogen in bioaugmented soil.

Bioaugmented microcosm showed higher amount of soil nitrogen content and increased microbial functional-diversity than naturally attenuated microcosm. a Soils were separately collected from each microcosm and nitrogen content of each microcosm was measured through the Kjeldahl method. b Soils were collected from each microcosm and heterotrophic microbial count was performed on LA plate. c The abundance of nitrogen-fixing microbial population in each microcosm was counted on Burk’s medium. d Soils were separately collected from both naturally attenuated and bioaugmented microcosm after an incubation of 10 days at 30 °C and dehydrogenase assay was performed accordingly in each microcosm to measure the viable cells population. e Fluorescein diacetate hydrolysis assay was carried out in each microcosm to measure the metabolic activity of microbial cells. f Soil samples were separately collected from each microcosm and were diluted accordingly. Diluted soil suspension was then separately added to BiOLOG ECO plate and then incubated at 30 °C for 3 days. The extent of utilization of different carbon sources was measured by recording OD at 590 nm. AWCD of each microcosm was measured by looking at the extent of utilization of carbon sources of BiOLOG ECO plate. g Microbial functional-diversity of each microcosm was determined by measuring Shannon–Weaver index as mentioned in the methods section. h Gini coefficient of each microcosm was determined from the Lorenz curve obtained from the carbon source utilization spectrum. Error bars indicated standard deviation (± SD). Mean values with different letters are significantly different from each other
Since microbial ecosystem is very sensitive to even small perturbation, it is very important to understand the microbial ecology of soil challenged with E. cloacae AKS7. In this regard, microbial activity was examined in both naturally attenuated and bioaugmented microcosm. Dehydrogenase happens to be an intracellular enzyme and every living microorganism possesses dehydrogenase enzyme for various functions including energy production through electron transport chain (Gu et al. 2009; Salazar et al. 2011). Thus, it was reported in literature that dehydrogenase enzyme could be considered as an efficient indicator of overall soil microbial activity (Gu et al. 2009; Salazar et al. 2011). The result of dehydrogenase activity indicated that bioaugmented microorganism showed ~ 75% greater dehydrogenase activity than naturally attenuated microcosm (Fig. 3d). Thus, the result showed that bioaugmented microcosm exhibited a greater number of viable microbial populations in comparison to naturally attenuated soil microcosm. Besides, microorganisms usually secrete various types of hydrolytic enzymes such as hydrolase, esterase and protease which can hydrolyze the ester bond of FDA resulting in the generation of fluorescein (Chakraborty and Tribedi 2019). It was reported in literature that FDA hydrolysis assay could be followed to measure the microbial metabolic activity of soil microcosms (Chakraborty and Tribedi 2019). Therefore, we have compared the metabolic activity between naturally attenuated and bioaugmented soil microcosm. The result indicated that bioaugmented microcosm showed ~ 2 fold higher metabolic activity than naturally attenuated soil microcosm (Fig. 3e). Thus, the results demonstrated that higher level of dehydrogenase and FDA hydrolysis activity were spotted in bioaugmented microcosm in comparison to naturally attenuated microcosm (see Fig. 3d, e). Taken together, the results showed that bioaugmented microcosm exhibited higher nitrogen content and microbial activity than naturally attenuated soil microcosm. It was reported in literature that the augmenting agent while exhibiting its functions in soil should not damage the pre-existing native ecological balance (Epelde et al. 2009; Tribedi and Sil 2013; Mahanty et al. 2017). In the present study, we already showed that the isolated organism E. cloacae AKS7 increased the nitrogen content of soil considerably. To study the ecological balance of ecosystem, in the present study, efforts were put together to measure the microbial metabolic potential and functional-diversity of soil microcosms as both of them influence the ecosystem stability notably (Mason et al. 2005; Epelde et al. 2009; Clark et al. 2012; Tribedi and Sil 2013). Therefore, to determine the metabolic potential, we determined the average well colour development (AWCD) of both naturally attenuated and bioaugmented soil microcosms from the carbon source utilization pattern of BiOLOG ECO plates. The result revealed that bioaugmented microcosm showed ~ 50% higher value of AWCD than naturally attenuated soil microcosm (Fig. 3f). Thus, the result indicated that higher metabolic potential was associated with bioaugmented microcosm compared to the metabolic potential of naturally attenuated soil microcosm. Microbial functional-diversity refers to the diverse functional traits that microorganisms can exhibit in a given ecosystem (Tribedi and Sil 2013; Chakraborty and Tribedi 2019). It plays an important role in overall ecosystem functioning and productivity (Mason et al. 2005; Clark et al. 2012). Thus, we have targeted our efforts to measure the functional-diversity of both bioaugmented and naturally attenuated soil microcosms. To determine the functional-diversity, Shannon–Weaver index was measured by utilizing the extent of intensities of the well colour of BiOLOG ECO plates. Shannon–Weaver index has been widely used in literature to determine the functional-diversity of a given ecosystem (Tribedi and Sil 2013; Chakraborty and Tribedi 2019). Our result showed that bioaugmented microcosm revealed ~ 15% greater value of Shannon–Weaver index than naturally attenuated microcosm (Fig. 3g). Thus, the result indicated that higher microbial functional-diversity was observed in a microcosm that was augmented with E. cloacae AKS7. To gain further confidence, functional evenness was compared between naturally attenuated and bioaugmented soil microcosm. Functional evenness refers the pattern of distribution of metabolic functions in a given ecosystem (Tribedi and Sil 2013; Chakraborty and Tribedi 2019). Existing literature reported that the Gini coefficient has been determined to understand the functional evenness in an ecosystem (Tribedi and Sil 2013; Chakraborty and Tribedi 2019). The Gini coefficient describes inequality in a population and it ranges from zero (when all individuals are identical) to a maximum of one (in which all individuals except one has a size of zero) (Tribedi and Sil 2013). Our result revealed that bioaugmented microcosm showed ~ 20% lesser value of Gini coefficient than naturally attenuated microcosm (Fig. 3h). The result demonstrated that a greater extent of functional homogeneity prevailed in the bioaugmented microcosm than naturally attenuated microcosm. Thus, our results showed that functional richness and evenness got significantly increased in bioaugmented microcosm suggesting higher degree of functional-diversity in bioaugmented microcosm than naturally attenuated soil microcosm. Taken together, the successful augmentation of E. cloacae AKS7 increased the nitrogen content of soil considerably without damaging the native ecological balance.
Conclusion
In conclusion, the current study showed that the exogenous addition of E. cloacae AKS7 to agricultural soil enhanced the soil nitrogen content as well as increased microbial functional-diversity of soil considerably. Thus, E. cloacae AKS7 could be considered as a promising nitrogen-fixing organism to enhance the fertility of the soil. However, further studies including the pathogenicity of the organism (if any) are required before recommending the organism (E. cloacae AKS7) to be used as a potential biofertilizer for sustainable agricultural practice. In this regard, we shall be putting our efforts in future to understand the epidemiology of our isolate E. cloacae AKS7 through multilocus sequence typing (MLST) using the sequences of internal fragments of multiple house-keeping genes. MLST could help us to analyze the genetic relations between isolates of E. cloacae with the aid of sequencing technology.
References
Adam G, Duncan H (2001) Development of a sensitive and rapid method for the measurement of total microbial activity using fluorescein diacetate (FDA) in a range of soils. Soil Biol Biochem 33:943–951
Alef K, Nannipieri P (1995) Methods in applied soil microbiology and biochemistry. Academic Press, London
APHA (2005) Standard methods for the examination of water and wastewater. American Public Health Association, Washington DC
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1994) Current protocols in molecular biology. Wiley, New York
Bardi L, Malusà E (2012) Drought and nutritional stresses in plant: alleviating role of rhizospheric microorganisms. Abiotic stress: new research. Nova Science Publishers Inc., Hauppauge, pp 1–57
Bhardwaj D, Ansari MW, Sahoo RK, Tuteja N (2014) Biofertilizers function as key player in sustainable agriculture by improving soil fertility, plant tolerance and crop productivity. Microb Cell Fact 13(1):66
Chakraborty P, Tribedi P (2019) Functional diversity performs a key role in the isolation of nitrogen-fixing and phosphate-solubilizing bacteria from soil. Folia Microbiol (Praha). https://doi.org/10.1007/s12223-018-00672-1 (Epub ahead of print)
Choi KH, Dobbs FC (1999) Comparison of two kinds of BiOLOG microplates (GN and ECO) in their ability to distinguish among aquatic microbial communities. J Microbiol Methods 36:203–213
Clark CM, Flynn DF, Butterfield BJ, Reich PB (2012) Testing the link between functional diversity and ecosystem functioning in a Minnesota grassland experiment. PLoS One 7:e52821
Dilworth MJ (1966) Acetylene reduction by nitrogen-fixing preparations from Clostridium pasteurianum. Biochim Biophys Acta 127:285–294
Dubey RC, Maheshwari DK (2012) Practical microbiology, 5th edn. S. Chand Publishing, New Delhi (ISBN: 9788121921534)
Epelde L, Mijangos I, Becerril JM, Garbisu C (2009) Soil microbial community as bioindicator of the recovery of soil functioning derived from metal phytoextraction with sorghum. Soil Biol Biochem 41:1788–1794
Glick BR (2012) Plant growth promoting bacteria: mechanisms and applications. Scientifica 2012:15
Gu Y, Wag P, Kong C (2009) Urease, invertase, dehydrogenase and polyphenoloxidase activities in paddy soils influenced by allelophatic rice variety. Eur J Soil Biol 45:436–441
Hoffman BM, Dean DR, Seefeldt LC (2009) Climbing nitrogenase: toward a mechanism of enzymatic nitrogen fixation. Acc Chem Res 42:609–619
Jetiyanon K, Pliabanchang P (2012) Potential of Bacillus cereus strain RS87 for partial replacement of chemical fertilisers in the production of Thai rice cultivars. J Sci Food Agric 92:1080–1085
Kayasth M, Gera R, Dudeja SS, Sharma PK, Kumar V (2014) Studies on salinization in Haryana soils on free-living nitrogen-fixing bacterial populations and their activity. J Basic Microbiol 54:170–179
Kessler PS, Leigh JA (1999) Genetics of nitrogen regulation in Methanococcus maripaludis. Genetics 152:1343–1351
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Mahanty T, Bhattacharjee S, Goswami M, Bhattacharyya P, Das B, Ghosh A, Tribedi P (2017) Biofertilizers: a potential approach for sustainable agriculture development. Environ Sci Pollut Res Int 24:3315–3335
Malusa E, Vassilev N (2014) A contribution to set a legal framework for biofertilisers. Appl Microbiol Biotechnol 98:6599–6607
Mason NW, Mouillot D, Lee WG, Wilson JB (2005) Functional richness, functional evenness and functional divergence: the primary components of functional diversity. Oikos 111:112–118
Mazid M, Khan TA (2015) Future of bio-fertilizers in Indian agriculture: an overview. Int J Agric Food Res 3:10–23
Park M, Kim C, Yang J, Lee H, Shin W, Kim S, Sa T (2005) Isolation and characterization of diazotrophic growth promoting bacteria from rhizosphere of agricultural crops of Korea. Microbiol Res 160:127–133
Salazar S, Sanchez L, Alvarez J, Valverde A, Galindo P, Igual J, Peix A, Santa-Regina I (2011) Correlation among soil enzyme activities under different forest system management practices. Ecol Eng 37:1123–1131
Singh J, Singh DK (2005) Dehydrogenase and phosphomonoesterase activities in groundnut (Arachis hypogaea L.) field after diazinon, imidacloprid and lindane treatments. Chemosphere 60:32–42
Smith BE, Richards RL, Newton WE (eds) (2013) Catalysts for nitrogen fixation: nitrogenases, relevant chemical models and commercial processes, vol 1. Springer, Berlin, p 340
Tribedi P, Sil AK (2013) Bioaugmentation of polyethylene succinate contaminated soil with Pseudomonas sp. AKS2 results in increased microbial activity and better polymer degradation. Environ Sci Pollut Res Int 20:1318–1326
Tribedi P, Sarkar S, Mukherjee K, Sil AK (2012) Isolation of a novel Pseudomonas sp. from soil that can efficiently degrade polyethylene succinate. Environ Sci Pollut Res Int 19:2115–2124
Acknowledgements
The authors’ would like to express sincere gratitude to the learned experts for their immense guidance and cooperation in improving the manuscript. We sincerely thank Ms. Rakshita Dave and Ms. Sutirtha Dutta for their help in correcting the language of the manuscript. This current work was supported by a grant in aid from the Department of Science and Engineering research board (SERB), DST, Govt. of India (Sanction number: YSS/2015/000387). Reagents like fluorescein diacetate (FDA) and 2,3,5-triphenyl tetrazolium chloride (TTC) were received as a kind gift from Dr. Alok Kumar Sil (Department of Microbiology, University of Calcutta) and Mr. Tapan Saha (Institute of Environmental Studies and Wetland Management, Kolkata), respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors’ declare that they do not have any conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chakraborty, P., Sarker, R.K., Roy, R. et al. Bioaugmentation of soil with Enterobacter cloacae AKS7 enhances soil nitrogen content and boosts soil microbial functional-diversity. 3 Biotech 9, 253 (2019). https://doi.org/10.1007/s13205-019-1791-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-019-1791-8