
Abstract
The repertoire and functions of MADS-box family transcription factors (TFs) largely remains unexplored with respect to floral organogenesis of Momordica dioica Roxb. Degenerative PCR followed by rapid amplification of cDNA ends was employed in the present study to clone and characterize 17 MADS-box genes (designated as MdMADS01 to MdMADS17) from the floral buds of M. dioica. The cloned genes were clustered into three subgroups (11 MIKCC, 4 MIKC* and 2 Mα) based on phylogenetic relationships with the MADS-box genes from Cucumis sativus, Cucumis melo and Arabidopsis thaliana. Southern hybridization showed that all the isolated genes were represented by single copy locus in the M. dioica genome. Gene structure analysis revealed 1–8 exons in MdMADS-box genes with the number of exons in MIKC greatly exceeding from that in M-type genes. Motif elicitation of the MdMADS-box genes indicated the presence of additional domains with MIKC type, suggesting that they had more complex structures. Expression analysis of MdMADS genes in six M. dioica transcriptome suggested that, 11 MIKCC—type genes are associated with floral homeotic functions, 4 MIKC*-type genes (MdMADS12 to MdMADS15) controlled the growth of male gametophyte, while the two M-type genes (MdMADS16 and MdMADS17) played significant role in female gametogenesis and seed development. Overall, these are the first set of MADS-box genes from M. dioica exhibiting a differential expression pattern during floral development. The results from this study will provide valuable information for further functional studies of candidate MADS-box genes in the sexual dimorphism of this economically important dioecious cucurbit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sexual systems in plants are characterized by multiple combinations of unisexual and hermaphrodite flowers. Interestingly, only six percent of the 240,000 species of the flowering plants have male and female functions in separate individuals (Renner and Ricklefs 1995). Multiple evidences indicate that separate sex types or dioecy has evolved independently and several times in land plants with a view to realizing simultaneous sex determination in different life forms (Vyskot and Hobza 2004). However, the variation in sexual behavior has greater evolutionary significance to maintain high genetic variability and adaptability across plant types. In the gourd family (Cucurbitaceae), flowers are represented by all the three major sexual patterns—monoecy, dioecy and hermaphrodite representing a distinct sexual dimorphism and evolution among the angiosperms. Although, major Cucurbitaceous genus such as Bryonia, Coccinia and Trichoxanthes have been well-documented to understand the genetics of sex determination (Volz and Renner 2008; Ming et al. 2011; Bhowmick et al. 2016; Mohanty et al. 2017), comparatively little or no interest has been attributed to the sexual development in Momordica, a genus with multiple perennial species that has undergone seven transitions from dioecy to monoecy (Schaefer and Renner 2010). Momordica dioica (spiny gourd) is a prominent dioecious member of this genus with a small genome size and an incipient set of homomorphic sex chromosomes that could be used as a sculpt for studying sexual phenotypes. Sex expression and inheritance have shown that M. dioica is governed by a unique factor for sexual variability representing heterozygous males and homozygous recessive females (Hossain et al. 1996; Baratakke and Patil 2009). More recently, we have shown that, a Copia-like retrotransposable element MdRE1 is exclusively found in the male genome suggesting a prominent Y-chromosome specific expression in the development of the male sex (Mohanty et al. 2016). However, the details of flowering biology and the genes involved in floral development of M. dioica are not yet known. Moreover, considering the fact that the plant has distinct culinary and medicinal attributes that are highly gender specific, characterization of the floral identity genes can provide a great opportunity for trapping the economic significance of M. dioica.
Sex differentiation is a complex phenomenon in the angiosperms with the involvement of several genes that are differentially expressed in diverse tissues and developmental phases (Charlesworth and Mank 2010). The most prominent is the genes encoding the MADS-box family transcription factors (TFs) that play important roles in many aspects of plant growth with crucial involvement in floral organ speciation and reproductive development (Smaczniak et al. 2012). These proteins are characterized by the presence of a 58–60 amino acids long conserved MADS-box DNA binding domain at the N-terminus that dimerize to specific DNA sequences called ‘CArG-boxes’ (Theiben and Gramzow 2016). Based on protein domain structures, the MADS-box genes have been divided into two lineages: Type I and Type II. The type I or M-type gene forms a heterogeneous group with small DNA sequences (≈ 180 bp) encoding only the MADS domain and are classified as Mα, Mβ and Mγ based on phylogeny (Parenicova et al. 2003). Although they constitute the major chunk of MADS-box genes in many plants, their functional attributes have only been characterized recently (Masiero et al. 2011). The type II or MIKC genes are characterized by the presence of additional domains, including an intervening (I) domain, a keratin-like (K) domain and a C-terminal (C) domain. They are classified as canonical (MIKCC) or star type (MIKC*) depending on the alteration of their motif structure. Additionally, MIKCC is further divided into 14 clades based on phylogenetic relationships and distinct sequence motifs in their C-terminal domains (Becker and Theissen 2003). Alteration in the C-terminal motif results in transcriptional activation of specific DNA sequences through formation of multimeric MADS-box protein complexes (Smaczniak et al. 2012). The MIKC subfamilies are often conserved and exhibit similar functions in different flowering plants. Several reports have shown that MIKCC genes are fundamental to the plant growth as well as reproductive and vegetative speciation such as in the differentiation of floral meristem [APETALA 1 (AP1), FRUITFUL (FUL) and CAULIFLOWER)], development of floral organs [AP1, APETALA3 (AP3), PISTILLATA (PI), AGAMOUS (AG) and SEPALLATA 1-3 (SEP1-3)] regulation of flowering time [SUPPRESSOR OF OVEREXPRESSION OF CONSTANT 1(SOC1), FLOWERING LOCUS C (FLC), SHORT VEGETATIVE PHAGE (SVP), AGAMOUS-LIKE 24 (AGL24)], fruit maturation [SHATTERPROOF 1-2 (SHP1-2)], embryonic development [TRANSPARENT TESTA 16 (TT16)] and root growth [AGAMOUS-LIKE 17 (AGL17)] (reviewed in Smaczniak et al. 2012; Theiben et al. 2016). The key function of MIKCC genes is classified into five classes-A, B, C, D and E each with multiple MADS-box TFs directly involved in the development of floral quartet model (Theiben et al. 2001, 2016). The ABCDE model regulates the floral organogenesis such that, the combination of A + E genes identifies the sepals, A + B + E specify petals, B + C + E denote stamens, C + E give carpels and C + D + E specify ovules. On the other hand, MIKC* genes have been implicated in floral transition and gametophytic development (Smaczniak et al. 2012).
Genome-wide analyses have led to the identification of multiple MADS-box genes in major cucurbitaceous plants such as melon and cucumber (Hu and Liu 2012; Hao et al. 2016). Furthermore, the expression analyses of these sequences have shown that they are involved in various aspects of the physiological and developmental processes relating to floral development and organogenesis. However, such studies have not yet been attempted in M. dioica due to the unavailability of its complete genome sequence. Therefore, to unearth the molecular mechanism controlling floral development and homeotic changes in M. dioica, we made an effort to clone and characterize the MADS-box genes by use of degenerative primer-based PCR approach. Degenerate primers are a mixture of similar but unidentical bases added together based on the coding ability of the same amino acids by different codons developed through variable bases. Considering that the MADS-box genes are highly conserved across plant species, we designed degenerative primers by aligning sequences found in the GenBank database to clone similar gene sequences from M. dioica without screening a cDNA library. In the present study, we describe the cloning and structural characterization of 17 M. dioica MADS-box genes followed by expression profiling of the isolated sequences in the male and female floral pattern development.
Materials and methods
Plant material
Male (M) and female (F) plants of M. dioica were grown and maintained in the experimental greenhouse of Centre for Biotechnology, Siksha O Anusandhan University, Bhubaneswar, India (20° 17′ 4.5852″ N, 85° 46′ 30.8496″ E). To avoid stochastic error, 20 male and 20 female plants grown at the same time were taken for experimental work. Sampling was done during the flowering season in July and the samples were floral buds, leaves and stems. Each tissue type was collected from multiple plants and pooled together to form male and female pools. In addition to this, male and female floral buds were collected separately from three developmental stages (early stage M1 & F1: 3 days after initiation; mid stage M2 & F2: 6 days after initiation and late stage M3 & F3: 9 days after initiation) for gene expression assays using qRT-PCR.
DNA and RNA isolation
DNA isolation was performed using the standard CTAB procedure of Doyle and Doyle (1990). Briefly, the frozen tissues were grounded with liquid nitrogen, added with extraction buffer (0.2 M Tris–HCl pH 7.5, 0.25 M NaCl, 25 mM EDTA pH 8.0, 0.5% SDS), and incubated at 65 °C for 10 min. Each sample was thrice treated with phenol: chloroform: isoamyl alcohol (25:24:1) for removal of non-nucleic acid compounds. DNA was precipitated using isopropanol and resuspended in 100 µL of 10 mM Tris, pH 8.0 with 10 µg RNaseA. The quantity and purity of the DNA was determined with UV–vis spectrophotometer (Thermo-Fischer Scientific, Waltham, USA) and 0.8% agarose gel electrophoresis.
Frozen plant material pulverized in a mortar and pestle with liquid nitrogen was used to isolate RNA using the TRIZOL reagent (Invitrogen, Darmstadt, Germany) according to the manufacturer’s instructions. RNA concentration and quality were determined with 1% formaldehyde denatured agarose gel electrophoresis and NanoDrop ND-1000 spectrophotometer (Thermo-Fischer Scientific, Waltham, USA). RNA samples with 260/280 nm ratio between 2.0 and 2.1 were used for further analysis.
Amplification and cloning of MADS-box partial cDNAs
Two microgram of RNA was used to synthesize the first strand cDNA using the high capacity cDNA synthesis kit (Life Technologies, Burlington, ON). The first strand cDNA was diluted tenfold and used as template for PCR using degenerative primers MB-F (corresponding to the IIKREIN motif) and MB-R (corresponding to the VLCDAEV motif) of the MADS-box genes. PCR was performed in a Veriti Thermal Cycler (Life Technologies, Burlington, ON, Canada) with 50 µL reaction mixture containing 200 µM dNTP mix, 10 pM each of forward and reverse primer, 10× PCR buffer and 1 unit of Taq polymerase. A gradient amplification was performed using the following temperature conditions: initial denaturation at 94 °C for 5 min followed by 36 cycles of 94 °C for 1 min, gradient annealing at 52 °C/54 °C/56 °C/58 °C for 1 min, extension at 72 °C for 1 min with a final extension at 72 °C for 10 min. The M. dioica Actin1 (MdActin1) gene was used as positive control, while RNA instead of cDNA was used as template for negative control. The amplified products were gel-purified using the Wizard SV gel and PCR cleanup system (Promega, USA) and subsequently cloned into the pGEM-T easy vector system (Promega, USA). The cloned products were sequenced using the BigDye Terminator v 3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) on an ABI Prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Isolation of full-length cDNA and genomic sequence
The full-length sequence of MADS-box cDNAs were obtained through 5′- and 3′-rapid amplification of cDNA ends (RACE) using the 5′/3′ RACE-PCR kit (Life Technologies, Burlington, ON, Canada). Gene-specific primers designed from the partial cDNA sequences and used in 5′ and 3′ RACE reactions are listed in Table S1. For 3′ RACE, 2 µg of RNA was reverse transcribed with 3′ RACE Adaptor Oligo-dT primer using Superscript II reverse transcriptase as per manufacturer’s instructions. Diluted cDNA was used as a template to amplify the 3′ region using gene-specific forward primer 1 and 3′ RACE Adaptor primer in a 50 µL reaction containing 1.5 mM MgCl2, 200 µM dNTP mix, 1× PCR buffer and 3 units of Taq polymerase. The PCR product was diluted and used as template in a second round of nested PCR using gene-specific forward primer 2 and 3′ RACE abridged universal amplification primer. The PCR conditions were: 3 min of initial denaturation at 94 °C followed by 35 cycles of 30 s at 94 °C, 30 s at 60 °C and 1 m 30 s at 72 °C. The cDNA synthesis for 5′ RACE was performed in the same manner as 3′ RACE, except that the Adaptor Oligo-dT primer was replaced with gene-specific reverse primer 1. Following the addition of a dC tail to the 3′ end of the first stand cDNA, it was used as a template to amplify 5′ region using gene-specific reverse primer and nested reverse primer in combination with 5′ RACE Abridged anchor primer and 5′ RACE Abridged Universal Amplification Primer, respectively. The amplified products of the 3′ and 5′ RACE were subsequently cloned and sequenced as mentioned in the previous section. Gene-specific primers designed based on the sequence alignment of the multiple clones from the 5′ and 3′ RACE products were used to amplify the full-length cDNA of MADS-box genes including the 5′ and 3′ untranslated regions (UTRs). PCR was performed in a 50 µL reaction mixture consisting of 200 μM dNTP mix, 1× PCR buffer, and 3 units of Taq DNA polymerase and 100 ng cDNA as template. The temperature conditions were 94 °C for 3 min followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 2 min.
The genomic sequences of M. dioica MADS-box genes were determined through genome walking technology using the Universal Genome walker 2.0 kit (Clontech Laboratories, CA, USA). Briefly, genomic DNA was digested with EcoRV, DraI, PvuII and StuI and ligated with GenomeWalker adaptors. Ligated DNA from the four genome walker libraries was used as template for PCR amplification using gene-specific primer and primary adaptor primer AP1 (Table S1). The diluted PCR product was used in a second round of PCR using nested adaptor primer AP2 and nested gene-specific primer. The reaction mixtures and temperature conditions used in GenomeWalker PCR were as described previously (Mohanty et al. 2016). Amplified fragments were purified, sub-cloned and sequenced as mentioned above.
Analysis of MADS-box cDNA and protein sequence
Sequence similarity searches for the full-length cDNA sequences were performed using BLASTn and BLASTp (http://www.ncbi.nlm.nih.gov). Reverse complementation and protein structure prediction was performed using the ExPaSy genomics and proteomics resource tools from SIB Bioinformatics resource portal (http://www.expasy.org). Multiple sequence alignments of the predicted MADS-box protein sequences were performed using CLUSTAL Omega (http://www.ebi.ac.uk) with default parameters. Phylogenetic tree was constructed from the aligned protein sequences by employing the neighbor joining method with Poisson correction, 1000 bootstrap replicates and pairwise deletion using the Molecular Evolutionary Genetic Analysis (MEGA v 6) package (Tamura et al. 2013). Motif structures of the predicted protein were analyzed using Multiple Expectation Maximization for motif Elicitation (MEME) tool (Bailey et al. 2006).
Southern blot analysis
Southern hybridization was performed as described previously (Mohanty et al. 2017). Briefly, the genomic DNA (10 µg) isolated from M. dioica was digested with restriction endonucleases EcoRI, XbaI and HindIII. Digested DNA samples were resolved on 1% agarose gel, transferred onto a Hybond-N+ nylon membrane (Amersham Pharmacia Biotech) by a vacuum transfer system and baked for 2 h at 80 °C. Partial sequences amplified from the C-terminal region and the 3′ untranslated regions of individual MADS-box genes were used as probes as listed in the Table S2. Digoxigenin (DIG) labeled probes were designed using a DIG DNA labeling and detection kit (Roche Diagnostics, Switzerland). The baked nylon membrane was blocked with DIG Easy Hyb for 1 h at 60 °C followed by hybridization with DIG labeled probes at 65 °C for 15 h. Probe-target hybridization was detected as per manufacturer’s instructions.
Semi-quantitative and quantitative RT-PCR
Semi-quantitative reverse transcription PCR (RT-PCR) and quantitative RT-PCR (qRT-PCR) was performed to determine the expression profiles of M. dioica MADS-box genes. The first strand cDNA was synthesized as described above and diluted ten times before use. For RT-PCR analyses, gene-specific primers amplifying the entire coding region were used (Table S3). The amplification was performed with an initial denaturation at 94 °C for 2 min followed by 34 cycles of 20 s at 95 °C, 40 s at 55 °C and 1 min at 72 °C, and final extension for 10 min at 72 °C. The equivalence of cDNA in different samples was verified using the RT-PCR product of M. dioica Actin gene. The PCR products were resolved on 1.2% agarose gel and visualized by staining with ethidium bromide.
The qRT-PCR assays were performed using the Kappa Biosystem’s FASTSYBR Green mix (D Mark, Toronto, ON) and unique sets of gene-specific primers amplifying a fragment of 125 bp (Table S4). Each real time RT-PCR reaction contained 5 ng of reverse transcribed cDNA, 5 µL of FASTSYBR Green mix, 2 µM concentration of forward and reverse primer and 2 µL of nuclease free water. The reactions were amplified on a StepOne Plus real time PCR system (Life Technologies, Burlington, ON) with following cycling conditions: 95 °C for 1 min (initial denaturation) followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s. The Ct values for each sample were determined from nine reactions, including three biological replicates with each biological replicates having three technical replicates. MdActin1 gene was used as endogenous control. The relative gene expression levels were computed using the 2−ΔΔ Ct method (Livak and Schmittgen 2001). A two-way analysis of variance (ANOVA) and multiple comparisons using uncorrected Fischer’s LSD test was performed to determine the statistical significance of the qRT-PCR results. Significant differences were scored at P < 0.05.
Results
Isolation of MADS-box genes from M. dioica
For the isolation of M. dioica MADS-box cDNAs related to floral differentiation, total RNA was extracted from the floral buds of both male and female flowers. RT-PCR amplification using degenerative primers designed from the conserved MADS-box domain resulted in several fragments between 150 and 250 bp. After sub-cloning, 108 clones were sequenced. A GenBank BLASTp and BLASTn search analysis revealed highest similarity of 17 partial sequences with known MADS-box genes from other plant species. Following 5′ and 3′ RACE analyses, we obtained the complete full-length cDNA sequences for the 17 exclusive MAD-box clones (Table 1). The 17 full-length cDNAs were designated as M. dioica MADS-box genes, MdMADS01 to MdMADS17 and the sequences were deposited in the NCBI GenBank under accession numbers MG491289-MG491305.
Phylogenetic analysis of MADS-box genes in M. dioica
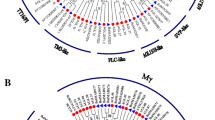
An unrooted neighbor joining phylogenetic tree was built from the protein sequence alignment of the deduced M. dioica MADS-box genes with those from closely related Cucumis sativus (14 genes) and Cucumis melo (18 genes) along with Arabidopsis thaliana (18 genes) as the outcrop species. Our results showed that, the 17 MADS-box genes were classified into two major groups: type I and type II (Fig. 1). The type I included two M-type MADS-box transcription factors (TFs) MdMADS16 and MdMADS17 from M. dioica with the closest similarity to M-type Arabidopsis thaliana AGL23 (68.3%) and AtAGL62 (77.8%), respectively (Fig. 1a; Table S5). The type II consisted of 15 MdMADS-box TFs including 11 MIKCC type and 4 MIKC* type genes (Fig. 1b; Table S5). Further analysis revealed that, the 11 MIKCC—type genes could be grouped into five subfamilies. MdMADS01 and MdMADS02 exhibited 82% amino acid identity with each other and were grouped with class-A MADS-box genes identified as APETALA 1/FRUITFUL (AP1/FUL) subfamily. AP1 and FUL are the two A-class MADS-box lineage genes found in core dicot species (Litt and Irish 2003). While MdMADS01 exhibited 95% identity with C. sativus agamous-like 7 (CsAGL7) and 68% identity with AtAP1, MdMADS02 showed 76% similarity with AtAGL8, a homolog of FUL in A. thaliana. Four cloned sequences- MdMADS03 to MdMADS06 clustered to form the B-class MADS-box genes. The B-class lineage consisted of three subgroups: AP3/DEFECIENS (DEF), TM6 and PI/GLOBOSA (GLO) (Theiben et al. 2001). MdMADS03 and MdMADS04 were grouped into the AP3/DEF subgroup, while MdMADS05 and MdMADS06 were categorized into the PI/GLO subgroup. No clones belonging to the TM6 subgroup could be held with the degenerate PCR. The deduced amino acid sequence of MdMADS03 exhibited 62.3, 51.2 and 44.6% with those of MdMADS04, MdMAD05 and MdMADS06, respectively. MdMADS03 exhibited 59.8, 64.7 and 68.2% identities, while MdMADS04 exhibited 47.7, 86.8 and 84.3% identities with Arabidopsis AP3, Cucumis melo DEF (MELO03C003778P1) and Cucumis sativus DEF (Csa017887), respectively. Similarly, MdMADS05 showed 61.2, 81.6, 82.5% and MdMADS06 exhibited 58.6, 79.2, 82.6% and identities with Arabidopsis PI1, Cucumis melo PI (MELO3C010515P1) and Cucumis sativus PI (Csa011135), respectively. The deduced amino acid sequence of MdMADS07 showed 69.3% identity with that of MdMADS08 and both were grouped under the AGAMOUS/SEADSTEAK (AG/STK) subfamily. Whereas, MdMADS07 was identified as a C-class MADS-box ortholog from the AG/FAR subgroup, MdMAD08 was closely related to Arabidopsis SEADSTEAK/Agamous-like 11 (STK/AGL11) protein (80.7%), encoded by a D-class MADS-box gene. The SEPALLATA (SEP) sub family integrated the two M. dioica genes, MdMAD09 and MdMADS10 with an amino acid identity of 66.9% among each other. MdMAD09 showed high sequence similarity with CsSEP2 (96.3%, Csa004117), CmSEP2 (92.8%, MELO3C026300P), AtSEP2 (76.2%) and AtAGL4 (76.4%), while MdMADS10 was identified as an ortholog of AtSEP3 (77.6%) and an isoform of CmAGL9 (91.6%, MELO3C022316P) and CsAGL9 (92.8%, Csa008448), all of which represented the E-class MADS-box genes. MdMADS11 was uniquely placed in a separate clade with 80.2% and 67.9% identities with Arabidopsis AtAGL17 and AtAGL21, respectively. Among the four M. doica MADS-box genes identified as orthologs of MIKC* subgroup, MdMAD12 and MdMAd14 exhibited 78.3–79.2% identities with AtAGL66, MdMADS13 was most similar to AtAGL65 (68.2%), while MdMAD15 showed high sequence similarity to AtAGL104 (81.7%). The predicted amino acid sequences when subjected to BlastP search against the non-redundant protein sequence database of NCBI revealed significant homology with MADS-box genes (Table S6) and corroborated with the groups as identified through phylogenetic analysis.

Phylogenetic tree of 17 MADS box proteins from Momordica dioica with their homologs in Arabidopsis thaliana, cucumber and melon. Phylogenetic analysis was performed by the neighbor joining method using ClustalW and visualized using MEGA 6. The solid dots represent the MADS-box protein from M. dioica identified in the present study. The letters in red represent the name of the subfamilies categorizing the clusters. a Type II MADS-box, b type I MADS-box proteins
Analysis of conserved motifs in MADS-box protein of M. dioica
Conserved motif analysis using multiple sequence alignment of the predicted proteins together with Multiple Expectation Maximization for Motif Elicitation (MEME) search tool resulted in the identification of ten conserved motifs from 17 candidate MADS-box genes of M. dioica (Fig. 2, S1; Table S7). Motif 1 (RQVTFSKRRNGLLKKAYELSVLCDA), 2 (EVALIVFSSSGKLYEYSSPS) and 3 (MGRGKIEIKRIENTT) representing the essentially conserved MADS-box domain was found in the N-terminus of all the 17 M. dioica MADS-box proteins. Likewise, the conserved I domain represented by motif 5 (IEKILERYERYSY) was clearly present in all type II members and partially represented in type I sequences of MdMADS16 and MdMADS17. Motif 4 (LMGEDJRSLSMKELESLEKQLDDAL) signifying the highly conserved K domain was found exclusively in the 15 MIKC proteins and thus acted as an important criterion for distinguishing the type I and type II MADS-box genes. Motif 6 (TRINQRREHIMSNHLSSYEAPALQQ), 8 (GPFENDVVGGWLPENGPNETH) and 9 (MGECHVANHNGDMFSPWAQAYNS) were only found in the members of MIKC* subfamily. Similarly, motif 7 (WQQEYEKLKARJEKLQDNNRN) and motif 10 (KQIRSRKYQLLLDZIETLQKK) were only found in MIKCC type genes. Interestingly, MdMAD05 and MdMAD06 both of which belong to the PI subfamily differed by the presence of motif 7 and 10 in the former and absence in the latter. Motifs 1, 2, 3 and 5 showed a significantly high degree of domain conservation, while it was relatively low in the rest of the motifs.

Conserved motif analyses of M. doica MADS-box proteins using MEME 4 program. Each motif is represented by a number in the colored box. The box length corresponds to motif length. Details of the motifs are listed in Table S7
Genomic organization of MADS-box genes in M. dioica
Southern blot analysis was performed to determine the genomic representation of the isolated MADS-box genes in the M. dioica genome. Since the isolated MADS-box genes exhibited a high degree of sequence conservation and size similarity, specific small size probes were designed from the C-terminal and 3′ UTR regions to avoid cross-hybridization. Under high stringency conditions, the DNA samples digested with EcoRI, XbaI and HindIII resulted in solitary hybridization band for all the 17 MADS-box genes (Fig. 3). The banding pattern and hybridization signals suggested that the M. dioica MADS-box genes are apparently represented by single copy locus in the M. dioica genome.

Southern blot analysis of 17 MADS-box genes in the M. dioica genome. DNA gel blots containing 30 μg of genomic DNA digested with EcoR I (a), Xba I (b) and HinD III (c) were hybridized under stringent conditions with probes from the 3′-specific region of the individual MADS-BOX genes. The sizes of DNA markers are shown at the left margin (kb). The probes used for southern blotting are represented in Table S2
To understand the genetic structures and evolutionary pattern of MADS-box genes, we determined the intron–exon organization by comparing the genomic DNA and coding regions of the 17 genes from M. dioica (Fig. S2). Sequence analysis revealed that all the MIKC type genes had complex genomic structures with an average of 6 introns and 7 exons. In contrast, the two Mα type genes MdMADS16 and MdMADS17 were comparatively smaller with only one intron and 2 exons. While the exons were mostly conserved and smaller in size (126–504 bp), the introns were highly variable with size ranging between 196 bp (intron 5, MdMADS08) to 5260 bp (intron 4, MdMADS11). Additionally, the MIKC* genes MdMADS12 and MdMADS13 had the highest number of 8 introns. Further analyses revealed that the majority of introns were highly conserved within the subfamily. For instance, all the six introns in the two AP3 subfamily members MdMADS03 and MdMADS04 were exactly similar. Likewise, in the four MIKC* genes, with the exception of introns 1 and 3 in MdMADS14 and introns 1 in MdMADS15, all other introns positions were highly conserved. All the introns had an elevated T content at the 5′ and 3′ splice junction representing a consensus di-nucleotide sequence of GT-AG, a typical structural characteristic found in majority of plants. Intron phase analyses indicated that, excepting a few centrally located introns with phase 0, majority of introns including the first and the last intron in all the MIKC type and the only intron in the Mα type genes were characterized by splicing site that occurred after the second nucleotide (phase 2) of the codon.
Spatial expression analysis of MADS-box genes in floral organ, leaves and stems
To explore the specificity of spatial expression patterns in the isolated MADS-box genes of M. dioica, we performed semi-quantitative RT-PCR in different tissues: stems, leaves and floral organs, including sepals, petals, stamens and pistils (Fig. 4). RT-PCR analyses revealed that, among the two class-A lineage genes, MdMADS01 belonging to the AP1 group was expressed in the sepals, petals and stem tissues, while MdMADS02 with similarity to FUL gene was additionally accumulated in the leaves. MdMADS02 was strongly expressed in petals and also in stem tissues. The expression of MdMADS03 and MdMADS04 was observed in all the whorls of the floral organs with high transcript accumulation in the petals and stamens. Transcripts of MdMADS03 and MdMADS04 were both detected in the stem tissues, whereas only MdMADS03 was detectable in the leaves. On the contrary, MdMADS05 and MdMADS06 genes were uniquely expressed only in the petal and stamen tissues. Thus, the expression of the genes from PI subgroup differed from AP3 subgroup in spite of the fact that they are categorized as class-B lineage genes. The C-class MADS-box gene, MdMADS07 was strongly accumulated in stamen and pistil with a minimal expression in the second whorl (petal) of the flower and no expression in the leaves or stems. Likewise, MdMADS08 gene with class-D lineage was expressed only in the fourth whorl (pistil) of the flower and was either least expressed or undetected in other whorls of the flower as well as in the vegetative tissues. The transcript accumulation of the two class-E MADS-box genes, MdMADS09 and MdMADS10 was realized in all the floral whorls but not in leaves and stems. The expression of MdMADS11 belonging to the AGL17 subfamily was detected in all the floral whorls albeit with a gradual decrease in the transcript levels from first whorl to the fourth whorl. However, the highest accumulation was observed in the stem tissue, while remaining undetected in the leaves. Among the four MIKC* genes, MdMADS12 expression was detected in all the four whorls, while the transcripts of MdMADS13, MdMADS14 and MdMADS15 were observed only in the first three whorls. The highest transcript levels for all the four genes were observed in the third whorl (stamen). In contrast, the expression was highly variable in the vegetative tissues. The transcripts of MdMADS15 were detected in both the stem and leaves, MdMADS12 and MdMADS13 were observed in only stems, whereas MdMADS14 transcripts were undetectable. The two Mα-type MADS-box genes, MdMADS16 and MdMADS17 were significantly expressed in the fourth floral whorl (pistil) but not in the leaves and stems. The transcript levels of MdMADS16 and MdMADS17 were marginally expressed in the second whorl, while the latter was also detected in the first whorl of the flower.

Spatial expression profiles of 17 MADS-box genes in different tissues of M. dioica. Semi-quantitative PCR was performed using total RNA isolated from sepals (Se), petals (Pe), stamens (st) and pistils (pi) of floral buds and from leaves (L) and stems (S). PCR products were separated on 1.5% agarose gel. MdActin that amplified a fragment of 156 bp served as the reference gene. The primers specific to each MdMADS gene are represented in Table S3
Differential expression of MADS-box genes in floral buds
To evaluate the potential roles of the isolated MADS-box genes in the development of floral buds and sex differentiation, independent qRT-PCR expression profiles were estimated from the small (MB1 & FB1), medium (MB2 & FB2) and large (MB3 & FB3) stages of the floral buds in M. dioica. Results revealed a differential expression patterns for all the 17 genes in the three bud stages (Fig. 5, Supplementary Fig. S3). Five genes including MdMADS01, MdMADS02 from AP1/FUL subfamily, MdMADS03 and MdMADS04 from AP3 subfamily and MdMADS11 from AGL17 subfamily showed a constitutive expression in both male and female buds with no significant difference across the bud developmental stages. Six genes (MdMADS05, MdMADS06 from the PI subfamily, MdMADS12, MdMADS13, MdMADS14 and MdMADS15 from MIKC* group) were significantly induced in the male buds and no difference in the expression could be observed in their female equivalents. While MdMADS05 and MdMADS06 demonstrated a gradual decrease in the accumulation of transcripts from MB1 (4.97 ± 0.13) to MB3 (1.78 ± 0.22), the four MIKC* genes showed a steady state up-regulation from the small (2.27 ± 0.17) to the large (6.01 ± 0.11) stages of the male bud. In contrast, three genes (MdMADS08, MdMADS16 and MdMADS17) showed a significant and rapid increase in transcript accumulation from FB1 to FB3, but were hardly detectable in the three stages of the male bud. MdMADS07 corresponding to the AG subfamily demonstrated an equal expression in both the male and female bud. As compared to the basal level, MdMADS07 transcripts were marginally increased (1.82 ± 0.23, FB; 1.89 ± 0.21) in the medium buds before significantly decreasing at the large bud stages. In contrast, MdMADS09 and MdMADS10 genes revealed a continuous accumulation of transcripts from the smaller to the larger stages of both the male and female buds.

Differential expression signatures of 17 MADS-box genes in different bud stages of male and female M. dioica. The results from the qRT-PCR analysis were analyzed using the MeV program. Color bar at the base represents the log2 expression values, thereby green color representing low level expression, black shows medium level expression and red signifies high level expression. Bud stages used for expression profiling are mentioned on top of each column. The relative expression level in the first stage female bud (FB1) was set to 1 for calibration of the qRT-PCR expressions. FB1, female bud stage 1 (3 days after initiation of bud); FB2, Female bud stage 2 (6 days after initiation of bud); FB3, Female bud stage 3 (9 days after initiation of bud); MB1, Male bud stage 1; MB2, Male bud stage 2; MB3, Male bud stage 3
Discussion
The trapping of nutritional attributes and economic values of M. dioica depends on effective breeding programs through identification of sex types as well as information on sex expression and inheritance. In a previous study, we demonstrated the association of a Copia-like retrotransposable element co-segregating with the male genotypes of M. dioica which could be possibly exploited for sexual dimorphism (Mohanty et al. 2016). However, for a dioecious plant like M. dioica, knowledge about the genes controlling the reproductive growth and floral development could be significant in designing of experimental systems for early identification of sexual phenotypes, which would be highly beneficial in breeding programs. In recent times, MADS-box genes have been shown to play major roles in floral initiation, differentiation of floral organs, development of seeds and fruits as well as stress responses (Smaczniak et al. 2012). In the present work, a degenerate primer-based RT-PCR approach was used for isolating MADS-box transcription factor genes that are possibly involved in floral development in M. dioica. We isolated 17 MADS-box genes expressed in the floral buds of M. dioica including two A-class genes (MdMADS01, MdMADS02), four B-class genes (MdMADS03 to MdMADS06), one C-class gene (MdMADS07), one D-class gene (MdMADS08), two E-class genes (MdMADS09, MdMAD10), four MIKC* genes (MdMADS11 to MdMADS15) and two type I MADS-box genes (MdMADS16 and MdMAD17). Interestingly, we could isolate only 11 MIKCC type genes in this study as against the presence of 29 genes in cucumber (Hu and Liu 2012) and 36 genes in melon (Hao et al. 2016). No genes could be isolated from subfamilies such as FLC, AGL12, BS, TM8, SOC and SVP from M. dioica. This may be ascribable to the fact that, all the genes might not have been amplified under the conditions set down by the degenerative PCR approach. Robust analysis through a genome-wide sequencing study could be attempted in future to characterize the genes that could not be trapped through this experimentation. However, three subfamilies FLC, AGL12 and BS were previously reported to be missing in the cucumber and melon genome (Hu and Liu 2012; Hao et al. 2016). FLC subfamily genes are known to control flowering through vernalization and associated pathways in Arabidopsis thaliana (De Lucia et al. 2008). The fact that M. dioica doesn’t require vernalization for flowering could be the reason for their absence in this dioecious plant. Arabidopsis AGL12 has been implicated in the proliferation of root meristem and the floral transition, while rice AGL12 gene promotes pigment accumulation and root development (Tapia-Lopez et al. 2008; Lee et al. 2008). Similarly, the BS family gene TT6 from Arabidopsis is involved in seed pigmentation and endothelial development (Nesi et al. 2002). The TM8 genes have a species-specific role thereby defining the flower development in tomato and grapevine while being absent in Arabidopsis (Heijmans et al. 2012). All the same, the SOC family genes act as activator, while SVP-related genes serve as a repressor of floral patterning and floral meristem determinacy in both monocots and dicots (Melzer et al. 2008; Liu et al. 2008; Tao et al. 2012). Therefore, the lack of these genes in M. dioica indicates that the molecular mechanism of flower transition is quite different in this species and it would be interesting to illustrate the possible mechanism fundamental to the absence of these genes in the M. dioica genome.
Genomic structure analyses of the isolated MADS-box genes revealed single cross-hybridizing signal for all the 17 sequences in the M. dioica genome. This suggest that the M. dioica MADS-box genes do not have duplicated loci as evident in other members of the Cucurbitaceae family (Hu and Liu 2012; Hao et al. 2016). A possible reason for this could be the presence of a diploid genome (2n = 28), asymmetric karyotypes and autopolyploid origin of the cultivated populations in M. dioica (Bhowmick and Jha 2015). Besides, a previously reported tetraploid nature of M. dioica genome has been recently negated due to misrepresentation of a related species (Bharathi et al. 2011). The intron–exon structure analysis showed that the two type I MADS-box genes were short and simpler, while the 15 type-II genes were complex and longer with multiple introns and exons. Genome-wide analysis of MADS-box genes in Arabidopsis (Parenicová et al. 2003), cucumber (Hu and Liu 2012), soybean (Shu et al. 2013) and more recently in melon (Hao et al. 2016) have shown the existence of a distinct bimodial pattern of intron distribution in which, MIKCC, MIKC* and Mδ have many introns, whereas Mα, Mβ and Mγ have no or a single intron. Moreover, the analysis of sequence alignment, 3 dimensional structure, conserved motifs and phylogenetics of the 17 isolated sequences reported significant similarity with those from cucumber, melon, Arabidopsis and rice. Taken together, these results suggest that the plant MADS-box gene family is relatively conserved across different species and are destined to play roles in a variety of developmental processes and floral transition.
Sex differentiation genes often get segregated as different functional alleles among the two different individuals of dioecious species (Diggle et al. 2011). Analysis of genetic and molecular basis of sexual development in many plants have revealed the involvement of a number of MADS-box genes with differential expression pattern in the development of male and female sexes (Gramzou and Theissen 2010). In the present study, we investigated the expressional changes of the isolated MADS-box genes to deduce their specific role in floral organ speciation in M. dioica. According to the ABCDE model of floral development, A specify the petals and sepals, B and C are conscientious to stamen development, while carpel is formed by the same C-class AG genes (Coen and Meyerowitz 1991). Our results also suggest that MdMADS01 and MdMADS02 are putative orthologs of AP1 and FUL and have petal and sepal specific expression in both male and female flowers of M. dioica (Fig. 4). Similarly, MdMADS03 to MdMADS06 were identified as close orthologs of AP3/PI subclass genes that demonstrated distinct expression in male reproductive organs but insignificant in the female reproductive system. The AP3/PI genes have been previously reported to act as switching factor in the activation of male and repression of female development (Wuest et al. 2012). Besides, the AP3 homologs have also been associated with the development of cucumber fruits, apple stems and leaves (Tian et al. 2015) justifying the expression of MdMADS03 and MdMADS04 in the vegetative tissues which might be playing a similar role in M. dioica. The unique expression of MdMADS08 in the carpel tissue is suggestive of the fact that it is highly homologous with the STK gene of Arabidopsis thaliana which along with SHATTERPROOF 1 and 2 (SHP1, 2) are involved in the development and differentiation of ovules (Matias-Hernandez et al. 2010). Furthermore, two close orthologs of SEP genes (MdMADS9 and MdMADS10) showed widespread expression in all the floral tissues suggesting their direct involvement in organ development. SEP genes are redundant sets of floral homeotic factors that form large complexes with other homeotic proteins to regulate the growth of floral meristem and specify the development of petal, stamen and carpel in the unisexual flowers (Pelaz et al. 2000; Rijpkema et al. 2009). AGL17-like genes have been primarily involved in root-related developmental processes (Parenicova et al. 2003). However, as it is difficult to isolate RNA from the roots of spiny gourd, we were unable to indicate the role of AGL17-like gene in M. dioica root development. Nevertheless, the uniform expression of the AGL17-like ortholog in all the floral organs as well as in the different stages of the male and female buds of M. dioica corroborate with the assumption that it is a positive regulator of flower development through photoperiodic induction of AP1 and LFY genes (Han et al. 2008). Further experimental evidences are required to prove this hypothesis in M. dioica.
Recent genetic studies have revealed that MIKC*-type MADS-box genes including AGL65, AGL66 and AGL104 are imperative to the development of male gametophytes (Smaczniak et al. 2012). Previous reports have shown that double and triple mutants of A. thaliana MIKC* genes, agl65, agl66 and agl104 results in pollen-affected phenotypes, abnormal growth of the pollen tube and delayed germination (Verelst et al. 2007a; Adamczyk and Fernandez 2009). Expression studies on the mutant phenotypes have further revealed that MIKC* type genes form a protein interaction complex and exhibit their redundant function by regulating transcriptome dynamics during pollen development (Verelst et al. 2007b). In the present study, the MIKC* homologs were predominantly expressed in the male reproductive organ and in the mature male buds of M. dioica. A recent report on transcriptome dynamics in the floral buds of a dioecious cucurbit, Coccinia grandis have shown that the genes encoding AGL65, AGL66 and AGL104 were significantly induced in the late stage of the male buds (Mohanty et al. 2017). Therefore, it is reasonable to suggest that the homologs of AGL65, AGL66 and GAL104 are critical in the speciation of stamen and development of pollen in the course of sexual dimorphism in the Cucurbitaceae family.
While the MIKC* genes predominantly regulate the male gametophytic development, type I MADS-box genes have been implicated in female gametogenesis and seed development (Masiero et al. 2011). MdMADS16 homologous to AGL62 and MdMADS17 homologous to AGL23 demonstrated significant expression in the late stage of the female buds. AGL62 in association with AGL80 and AGL61 facilitates endosperm development and suppresses premature formation of cell wall in the endosperm (Bemer et al. 2008; Kang et al. 2008). Likewise, AGL23 plays an important role in regulating the biogenesis of organelles during embryo sac development (Colombo et al. 2008). Mutant analysis of AGL23 in Arabidopsis thaliana has shown that agl23 mutant lines develop albino seeds that did not germinate into viable plants (Colombo et al. 2008). These reports together with our finding suggest that a broad class of type I MADS-box genes could be associated with female gametophyte, embryo sac and seed development in M. dioica. Although large sets of type I MADS-box genes have been identified from other cucurbitaceous species such as cucumber and melon, so far no direct functions have been attributed to them. Further experimentation through large-scale yeast two hybrid protein interaction screening and development of functional knockouts will throw open more insights into their role in sex differentiation.
Conclusion
In conclusion, we investigated the role of MADS-box TFs in the sex differentiation of M. dioica wherein we isolated, systemically analyzed and demonstrated the expression patterns of 17 MADS-box genes in different growth organs and floral buds. A comparison of phylogenetic relationship and expression studies between M. dioica MAD-box genes and those from other plant species suggest that even if the ABCDE model of flower development is conserved in M. dioica, representation of new functions and segregation of existing gene functions is essential for sexual dimorphism in this evolutionarily important dioecious cucurbit. The information generated through this study will pave way for the selection of appropriate candidate MADS-box genes for further functional characterization as required for growth, development and commercial exploitation of M. dioica.
References
Adamczyk BJ, Fernandez DE (2009) MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol 149:1713–1723
Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucl Acids Res 34:W369–W373
Baratakke RC, Patil CG (2009) Karyomorphological investigations in dioecious climber Momordica dioica Roxb. J Cytol Genet 11:91–96
Becker A, Theissen G (2003) The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29:464–489
Bemer M, Wolters-Arts M, Grossniklaus U, Angenent GC (2008) The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 20:2088–2101
Bharathi LK, Munshi AD, Vinod Chandrashekaran S, Behera TK, Das AB, John KJ, Nath V (2011) Cytotaxonomical analysis of Momordica L. (Cucurbitaceae) species of Indian occurrence. J Genet 90:21–30
Bhowmick BK, Jha S (2015) Dynamics of sex expression and chromosome diversity in Cucurbitaceae: a story in the making. J Genet 94:793–808
Bhowmick BK, Yamamoto M, Jha S (2016) Chromosomal localization of 45S rDNA, sex specific C values and heterochromatin distribution in Coccinia grandis (L.) Voigt. Protoplasma 253:201–209
Charlesworth D, Mank JE (2010) The birds and the bees and the flowers and the trees: lessons from genetic mapping of sex determination in plants and animals. Genetics 186:9–31
Coen ES, Meyerowitz EM (1991) The war of the whorls: genetic interactions controlling flower development. Nature 353:31–37
Colombo M, Masiero S, Vanzulli S, Lardelli P, Kater MM, Colombo L (2008) AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J 54:1037–1048
De Lucia F, Crevillen P, Am Jones, Greb T, Dean C (2008) A PHD-polycomb repressive complex 2 triggers the epigenetic silencing of FLC during vernalization. Proc Natl Acad Sci USA 105:16831–16836
Diggle PK, Di Stilio VS, Gschwend AR, Golenberg EM, Moore RC, Russell JR, Sinclair JP (2011) Multiple developmental processes underlie sex differentiation in angiosperms. Trends Genet 27:368–376
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:1241–1251
Gramzow L, Theißen G (2010) A hitchhiker’s guide to the MADS world of plants. Genome Biol 11:214
Han P, García-Ponce B, Fonseca-Salazar G, Alvarez-Buylla ER, Yu H (2008) AGAMOUS-LIKE 17, a novel flowering promoter, acts in a FT independent photoperiod pathway. Plant J 55:253–265
Hao X, Fu Y, Zhao W, Liu L, Bade R, Hasi A, Hao J (2016) Genome-wide identification and analysis of the MADS-box gene family in melon. J Am Soc Hort Sci 141:507–519
Heijmans K, Morel P, Vandenbussche M (2012) MADS-box genes and floral development: the dark side. J Expt Bot 63:5397–5454
Hossain MA, Islam M, Ali M (1996) Sexual crossing between two genetically female plants and sex genetics of kakrol (Momordica dioica Roxb.). Euphytica 90:121–125
Hu L, Liu S (2012) Genome-wide analysis of the MADS-box gene family in cucumber. Genome 55:245–256
Kang IH, Steffen JG, Portereiko MF, Lloyd A, Drews GN (2008) The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 20:635–647
Lee S, Woo YM, Ryu S, Shin YD, Kim WT, Park KY, Lee IJ, An G (2008) Further characterization of a rice AGL12 group MADS-box gene, OsMADS26. Plant Physiol 147:156–168
Litt A, Irish V (2003) Duplication and diversification in the APETALA/FRUITFUL floral homeotic gene lineage: implications for the evolution of floral development. Genetics 165:821–833
Liu C, Chen H, Er HL, Soo HM, Kumar PP, Han JH, Liou YC, Yu H (2008) Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development 135:1481–1491
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C (T)) method. Methods 25:402–408
Masiero S, Colombo L, Grini PE, Schnittger A, Kater MM (2011) The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23:865–872
Matias-Hernandez L, Battaglia R, Galbiati F, Rubes M, Eichenberger C, Grossniklaus U, Kater MM, Colombo L (2010) VERDANDI is a direct target of the MADS domain ovule identity complex and affects embryo sac differentiation in Arabidopsis. Plant Cell 22:1702–1715
Melzer S, Lens F, Gennen J, Vanneste S, Rohde A, Beeckman T (2008) Flowering-time genes modulate meristem determinacy and growth form in Arabidopsis thaliana. Nat Genet 40:1489–1492
Ming R, Bendahmane A, Renner SS (2011) Sex chromosomes in land plants. Annu Rev Plant Biol 62:485–514
Mohanty JN, Nayak S, Jha S, Joshi RK (2016) A sequence tagged site (STS) marker encoding Copia-like retrotransposable element is associated with male specific sex expression in Momordiac dioica Roxb. Sci Hort 201:265–270
Mohanty JN, Nayak S, Jha S, Joshi RK (2017) Transcriptome profiling of the floral buds and discovery of genes related to sex-differentiation in the dioecious cucurbit Coccinia grandis (L.) Voigt. Gene 626:395–406
Nesi N, Debeaujon I, Jond C, Stewart AJ, Jenkins GI, Caboche M, Lepiniec L (2002) The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell 14:2463–2479
Parenicová L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J, Cook HE, Ingram RM, Kater MM, Davies B, Angenent GC, Colombo L (2003) Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15:1538–1551
Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF (2000) B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405:200–203
Renner SS, Ricklefs RE (1995) Dioecy and its correlations in the flowering plants. Am J Bot 82:596–606
Rijpkema AS, Zethof J, Gerats T, Vandenbussche M (2009) The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J 60:1–9
Schaefer H, Renner SS (2010) A three genome phylogeny of Momordica (Cucurbitaceae) suggests seven returns from dioecy to monoecy and recent long-distance dispersal to Asia. Mol Phylogenet Evol 54:553–560
Shu Y, Yu D, Wang D, Guo D, Guo C (2013) Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol Biol Rep 40:3901–3911
Smaczniak C, Immink RGH, Angenent GC, Kaufmann K (2012) Developmental and evolutionary diversity of plant MADS-box domain factors: insights from recent studies. Development 139:3081–3098
Tamura K, Stecher G, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tao Z, Shen L, Liu C, Liu L, Yan Y, Yu H (2012) Genome-wide identification of SOC1 and SVP targets during the floral transition in Arabidopsis. Plant J 70:549–561
Tapia-López R, García-Ponce B, Dubrovsky JG, Garay-Arroyo A, Pérez- Ruíz RV, Kim SH, Acevedo F, Pelaz S, Alvarez-Buylla ER (2008) An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol 146:1182–1192
Theiben G, Melzer R, Rumpler F (2016) MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development 143:3259–3271
Theißen G (2001) Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol 4:75–85
Theißen G, Gramzow L (2016) Structure and evolution of plant MADS domain transcription factors. In: Gonzalez DH (ed) Plant transcription factors: evolutionary, structural and functional aspects. Elsevier, Philadelphia, pp 127–138
Tian Y, Dong Q, Ji Z, Chi F, Cong P, Zhou Z (2015) Genome-wide identification and analysis of the MADS-box gene family in apple. Gene 555:277–290
Verelst W, Saedler H, Münster T (2007a) MIKC* MADS-protein complexes bind motifs enriched in the proximal region of late pollen-specific Arabidopsis promoters. Plant Physiol 143:447–460
Verelst W, Twell D, de Folter S, Immink R, Saedler H, Münster T (2007b) MADS-complexes regulate transcriptome dynamics during pollen maturation. Genome Biol 8:R249
Volz SM, Renner SS (2008) Hybridization, polyploidy and evolutionary transitions between monoecy and dioecy in Bryonia (Cucurbitaceae). Am J Bot 95:1297–1306
Vyskot B, Hobza R (2004) Gender in plants: sex chromosomes are emerging from the fog. Trends Genet 20:432–438
Wuest SE, O’Maoileidigh DS, Rae L, Kwasniewska K, Raganelli A, Hanczaryk K, Lohan AJ, Loftus B, Graciet E, Wellmer F (2012) Molecular basis for the specification of floral organs by APETALA3 and PISTILLATA. Proc Natl Acad Sci USA 109:13452–13457
Acknowledgement
The study is funded by a research grant (BT/PR3919/PBD/16/959/2011) from the Department of Biotechnology, Government of India. JNM is grateful to DBT, India for financial support in the form of Senior Research Fellowship. The authors are thankful to Prof. Manoj Ranjan Nayak, President, Siksha O Anusandhan University for his guidance and support. We also thank Prof. Sumita Jha, Centre for Advanced Study, Dept. of Botany, Calcutta University and Prof. Sanghamitra Nayak, Head, Centre of Biotechnology, Siksha O Anusandhan University for their able guidance and support. We also thank DST-FIST, Govt. of India, for the research infrastructure facilities provided to Centre of Biotechnology, Siksha O Anusandhan University.
Author information
Authors and Affiliations
Contributions
RKJ conceived and supervised the project. JNM collected samples, isolated RNA, performed cloning, sequencing and qPCR expression analyses. JNM and RKJ interpreted the data and prepared the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have declared that no competing interest exists.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Mohanty, J.N., Joshi, R.K. Molecular cloning, characterization and expression analysis of MADS-box genes associated with reproductive development in Momordica dioica Roxb.. 3 Biotech 8, 150 (2018). https://doi.org/10.1007/s13205-018-1176-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-018-1176-4