
Abstract
Symbiotic diazotrophic bacteria collectively called “rhizobia” can associate with legumes and form specialized structures in the roots, called nodules, where the biological nitrogen fixation (BNF) process takes place. Common bean (Phaseolus vulgaris L.) is a major protein source in the diet of the population of many countries such as Brazil and can benefit from the BNF process, but there is still little knowledge about the diversity and effectiveness of indigenous microsymbionts. In this study, the diversity and the nitrogen fixation ability of bacterial isolates trapped by common bean nodules in 14 municipalities of Mato Grosso do Sul state, Central-Western region, comprising three Brazilian biomes were investigated. The DNA profiles (BOX-PCR) of 82 isolates indicated outstanding diversity, with 12 main clusters and 36 isolates occupying single positions, joined at a final level of similarity of less than 20%. The 16S rRNA phylogeny of 56 isolates representing the DNA profiles indicated ten genera, with 38 isolates identified as “classical rhizobia” and Agrobacterium, and the remaining 18 belonging to six other genera. The 38 isolates had their glnII gene sequenced and were evaluated for the capacity of nodulation and BNF with common bean, and only 12 formed effective nitrogen-fixing nodules, five positioned in the R. etli and six in the R. tropici clades, and one of Agrobacterium. These results highlight the promiscuity of common bean in capturing a variety of microbial species in their nodules, whose function has not been well elucidated yet. Only one-fifth of the isolates were effective in fixing nitrogen, which might explain the frequently reported low rates of contribution of the BNF with this legume, an intriguing paradigm in the evolution of the symbiosis.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Common bean (Phaseolus vulgaris L.) is one of the most important legumes cropped worldwide, and besides being of great social and economic importance, it represents the main source of protein in the diet of millions of people, mainly in developing countries (Myers and Kmiecik 2017; Shamseldin and Velázquez 2020). Common bean is also recognized for establishing promiscuous symbioses, being able to associate with a broad variety of rhizobial species; however, apparently several of them present low efficiency of biological nitrogen fixation (BNF) (Aserse et al. 2012; Dall’Agnol et al. 2013; Reinprecht et al. 2020; Shamseldin and Velázquez 2020).
Classification of rhizobia was initially based on phenotypic properties in vitro and on the capacity to nodulate host legumes, known as cross-inoculation groups (Peix et al. 2015; Velázquez et al. 2017). Based on this concept, all common bean symbionts were first classified as Rhizobium phaseoli (Frank 1889; Peix et al. 2015; Kaschuk and Hungria 2017). With the development of methodologies, including molecular techniques, the polyphasic taxonomy was confirmed as the most appropriate approach to obtain the correct taxonomic position of a strain or a group of strains (Colwell 1970), and nowadays integrates phenotypic, genotypic, and phylogenetic information (Thompson et al. 2013; Fournier et al. 2015; Hugenholtz et al. 2021). Based on this approach, several rhizobia of environmental and economic interest, such as Bradyrhizobium japonicum (Jordan 1982), Rhizobium tropici (Martínez-Romero et al. 1991), Bradyrhizobium elkanii (Kuykendall et al. 1992), Mesorhizobium ciceri (Jarvis et al. 1997), Rhizobium freirei (Dall’Agnol et al., 2013), among others, have been reclassified or described.
Studies aiming to characterize rhizobia are of great importance for understanding the origin, evolution, diversity, symbiotic behavior, among other properties that indicate the richness and biotechnological potential of this important group of bacteria. Mato Grosso do Sul (MS) state, located in the Central-Western region of Brazil is recognized as a hotspot of diversity of plants and animals, and certainly also of microorganisms. It encompasses three important biomes, Pantanal, Cerrado, and Mata Atlântica (Myers et al. 2000; Eisenlohr et al. 2015), but rhizobial diversity is still poorly known. Based on this information, this study evaluated the diversity and BNF capacity of symbionts trapped by nodules of common bean plants grown in soils from 14 municipalities of Mato Grosso do Sul, comprising these three biomes.
2 Materials and methods
2.1 Bacterial isolation
The 82 isolates were previously isolated by Fabio Martins Mercante (born 1963-died 2016), researcher at Embrapa Agropecuária Oeste (Dourados, Mato Grosso do Sul-MS, Brazil). Soil samples from 14 municipalities from MS (Fig. 1, Table 1), comprising three out of the six Brazilian biomes, Pantanal, Cerrados, and Mata Atlântica were collected and used as the substrate for growth of common bean (Phaseolus vulgaris L.) as the trap host. After growth up to the end of vegetative stage, nodules were collected, surface-disinfested, crushed in sterile saline solution, and streaked on culture medium for isolation of single colonies, as described before (Hungria et al. 2016). For long-term storage, bacterial isolates were cryopreserved at -80 °C and -150 °C in modified-yeast extract-mannitol (YM) culture medium (Hungria et al. 2016) with 30% glycerol (v/v), and lyophilized, as described (Delamuta et al. 2017).

Brazilian map showing the biomes and the municipalities from which the isolates from this study were obtained
For the analyses, the strains were grown in modified-yeast extract-mannitol-agar (YMA) medium (Hungria et al. 2016), and incubated at 28 °C for three to seven days, depending on the growth rate of each isolate. Regular bacterial maintenance was on modified-YMA medium at 4 °C.
All isolates are deposited at the “Diazotrophic and Plant Growth Promoting Bacteria Culture Collection of Embrapa Soja” (WFCC Collection # 1213, WDCM Collection # 1054), in Londrina, State of Paraná, Brazil.
2.2 Genotypic characterization
2.2.1 DNA extraction and BOX-PCR fingerprinting
The genomic DNAs of 82 isolates were extracted using the DNeasy Blood & Tissue kit (Qiagen), following the manufacturer's instructions, and DNA quality was verified by electrophoresis on agarose gels (1%) stained with ethidium bromide and visualized under UV light. BOX-PCR fingerprinting fragments were obtained by DNA amplification using the BOX-A1R primer (5'-CTACGGCAAGGCGACGCTGACG-3') (Versalovic et al. 1994) in a ProFlex PCR System Thermocycler (Applied Biosystems), following cycles as described before (Costa et al. 2018). The amplified fragments were separated by electrophoresis on 1.5% agarose gel, using 1 kb Plus Ladder (Invitrogen®) as molecular marker.
The fingerprinting profiles were used to build a dendrogram of similarity using the Bionumerics software (Applied Mathematics, Kortrijk, Belgium, v.7.6), applying the UPGMA (Unweighted Pair-Group Method with Arithmetic Mean) algorithm (Sneath and Sokal 1973), and the Jaccard coefficient (Jaccard 1912), with 2% tolerance.
2.2.2 Amplification and sequencing of 16S rRNA and glnII housekeeping genes
Based on the grouping according to the BOX-PCR fingerprinting dendrogram, 56 isolates were selected for sequencing of the 16S rRNA. The 16S rRNA genes were amplified using the pair of primers fD1 (5'-AGAGTTTGATCCTGGCTCAG-3') and rD1 (5'-CTTAAGGAGGTGATC CAGCC-3'), according to Weisburg et al. (1991) and sequenced with the primers 362f (5'-CTCCTACGGGAGGCAGCAGTGGGG-3') and 786r (5'CGAAAGCGTGGGGAGCAAACAGG-3'), according to Menna et al. (2006). The PCR products were purified with the PureLink kit (Invitrogen®), following the manufacturer's recommendations, and sequenced on an ABI 3500xL (Applied Biosystems), as described by Menna et al. (2006), with modifications proposed by Delamuta et al. (2017). Nucleotide sequences obtained were analyzed and corrected manually using the Bionumerics software (v.7.6), and compared with sequences of the Genbank database using the Blastn tool. This step allowed the identification of the genera of each isolate.
Based on the 16S rRNA results, 38 isolates were selected for further sequencing of the glnII housekeeping gene. The amplification of glnII was performed with the pair of primers TSglnIIf (5'-AAGCTCGAGTACATCTGGCTCGACGG-3') and TSglnIIr (5-'SGAGCCGTTCCAGTCGGTGTCG-3') and reaction cycles as described by Stepkowski et al. (2005). The purification, sequencing reactions, and correction of the sequences were performed as described for the 16S rRNA gene.
For the phylogenetic analysis, isolates from this study were analyzed along with the closest type strains, whose sequences were retrieved from the GenBank database, except for the 16S rRNA sequence of Bradyrhizobium shewense ERR11T, which was retrieved from the Joint Genome Portal (JGI). Multiple sequence alignment for each gene was obtained with MUSCLE (Edgar 2004) and the best evolutionary distance model was inferred by the lowest Bayesian information criterion scores (Schwarz 1978) for maximum likelihood (ML) (Felsenstein 1981) phylogenetic tree constructions in the MEGA software (Molecular Evolutionary Genetics Analysis, version 7.0) (Kumar et al., 2016). The statistical support of the phylogenetic trees was estimated by bootstrap analysis (Felsenstein 1985), with 1,000 replicates (Hedges 1992). The evolutionary models used to build the trees are described in the figure captions. For determination of nucleotide identity (NI) (Case et al. 2007) sequences were aligned in the Bioedit Sequence Alignment Editor software (v.7.2.5) (Hall 1999).
Sequences were deposited in the NCBI GenBank database and the access numbers are shown in the phylogenetic trees.
2.3 Morphophysiological characterization
The 38 isolates classified as “classical rhizobia” and Agrobacterium based on the 16S rRNA and glnII genes sequencing analyses were cultivated in Petri dishes on modified-YMA medium containing Congo red as indicator and pH adjusted to 6.8 to 7.0 (Hungria et al. 2016). Bacteria were also grown on modified-YMA medium with bromothymol blue used as indicator to assess the capacity of the strains to produce acid/alkaline reaction (Hungria et al. 2016). Growth was verified after 3 to 7 days of incubation at 28 °C, according to each bacteria. Tests were performed in triplicate. The following morphological properties were characterized: a) growth rate; b) colony diameter (mm); c) mucus production; d) colony shape; e) colony border; f) colony surface; g) colony elevation; h) consistency; i) optical details; j) acid or alkaline reaction; k) chromogenesis in both media, with bromothymol blue or Congo red indicators (Vincent 1970; Somasegaran and Hoben 1994; Hungria et al. 2016).
2.4 Nodulation capacity and nitrogen fixation efficiency
Evaluations were carried with Phaseolus vulgaris cultivar Esteio (black seeds). Seeds were surface-disinfested (Somasegaran and Hoben 1994; Hungria et al. 2016), pre-germinated on Germitest paper moistened with distilled water and kept at 25 °C for 72 h. After germination, one seedling was transplanted to each propylene bag containing a sheet of Germitest paper to serve as wick and 300 mL of sterilized N-free nutrient solution (Hungria et al. 2016). After transplanting, each seedling was inoculated with 1 mL of culture of each isolate, adjusted to approximately 108 cells mL−1. The experiment was performed with five replicates, under aseptic conditions in a glasshouse for 30 days.
The nodulation capacity was evaluated by the presence or absence of nodules. Nodules were then removed from roots and cut to verify the internal color. Nitrogen fixation efficiency was confirmed if the internal color of the nodule was red or pink, indicating functional leghemoglobin, and by the dark green color of the leaves.
3 Results
3.1 Genetic characterization
3.1.1 BOX-fingerprinting
DNA profiles were obtained in the BOX-PCR analyses for all 82 isolates from soils taken at 14 municipalities of the state of Mato Grosso do Sul, Brazil. Fragments within the range of 300 to 3,000 bp were considered in the analysis and a dendrogram of similarity was built (Fig. 2). Considering the similarity level of 70% as a cutoff, 48 distinct groups or single clades containing sole isolates were generated, indicating high genotypic diversity among the isolates. The dendrogram distributed 46 isolates in 12 groups and the other 36 occupied single positions joined at the final level of similarity of only 18.68%. Numbers presented in parentheses in the dendrogram (Fig. 2) refer to the municipalities from which the respective isolate was taken (Fig. 1, Table 1), showing high heterogeneity concerning the groups of isolates and their origin. The largest group (27) included 13 isolates belonging to the Cerrado and Mata Atlântica biomes, followed by group 3, with nine isolates from the Cerrado and Mata Atlântica biomes, and group 40, with four isolates belonging to the Cerrado, Mata Atlântica and Pantanal. The other groups had three (12 and 13) or two isolates (1, 7, 9, 17, 31, 32, 37) each, isolated from Mata Atlântica and Cerrado biomes.

Fingerprinting dendrogram of similarity based on the BOX-PCR profiles of the isolates of this study, using the UPGMA algorithm and the Jaccard coefficient with 2% tolerance (software Bionumerics 7.6). The numbers in parentheses represent the municipalities in the state of Mato Grosso do Sul, Brazil, from which each isolate was obtained, according to Fig. 1 and Table 1
It is worth mentioning that similarities of 100% were observed in only five groups, which included isolates CNPSo 3496, 3500, 3502, 3633, and 3634 (group 3); CNPSo 3494, 3499 and 3501 (group 3); CNPSo 3437 and 3457 (group 7); CNPSo 3493 and 3495 (group 13); CNPSo 4053, 4054 and 4055 (group 27); CNPSo 3971 and 3972 (group 27); and CNPSo 3460 and 3462 (group 40).
Based on the BOX-PCR dendrogram, 48 isolates were selected as representatives of the groups formed, in addition to other eight isolates showing great variability of the profiles, totalizing 56 isolates that were used for the following step of the polyphasic analysis.
3.2 Phylogenetic characterization
3.2.1 16S rRNA gene analysis
Sequences of 16S rRNA gene were obtained for the 56 isolates selected in the BOX-PCR analysis and were submitted to the Blastn tool, to identify their genera. Twenty-two out of 56 isolates were identified as representatives of three genera of the Alphaproteobacteria class known as “classical rhizobia”, including Rhizobium (20 isolates), Bradyrhizobium (1), and Mesorhizobium (1). The remaining 34 isolates were identified as members of the Agrobacterium (16), Herbaspirillum (11), Pseudomonas (3), Achromobacter (1), Brevibacillus (1), Burkholderia (1), and Enterobacter (1) genera, representing the Alpha-, Beta- and Gammaproteobacteria and Firmicutes. The distribution of the 56 isolates in ten genera confirmed high diversity among the isolates.
We proceeded with the characterization of the “classical rhizobia” and Agrobacterium, comprising 38 isolates. Four phylogenetic trees were built with the Agrobacterium, Rhizobium, Bradyrhizobium, and Mesorhizobium genera. The phylogenetic tree based on 16S rRNA gene sequences with the isolates belonging to the Agrobacterium genus (Fig. 3) formed a large group (G.I) comprising the 16 isolates of this study with the species A. salinitolerans YIC 5082 T and A. pusense NRCPB10T. Strains from G.I group share from 99.3 to 100% sequences similarity.

Maximum likelihood phylogeny based on the 16S rRNA alignment of the genus Agrobacterium (942 bp), using the Kimura 2-Parameter + G model. Accession numbers are indicated in parentheses. Isolates from this study are shown in bold. Bootstrap values > 70% are indicated at the nodes. Bradyrhizobium diazoefficiens USDA 110 T was used as outgroup. Bar indicates two substitutions per 100 nucleotide positions
In the phylogenetic tree with isolates of the Rhizobium genus (Fig. 4), two large groups were formed. Group G.I clustered nine isolates with 16 species of the clade R. etli/R. phaseoli/R. leguminosarum, called R. etli group, sharing NI from 98.4 to 100%. Group G.II clustered 11 isolates with 12 species belonging to the R. tropici group, with 94% bootstrap values, and sharing NI of 98 to 100%.

Maximum likelihood phylogeny based on the 16S rRNA alignment of the genus Rhizobium (944 bp), using Tamura 3-Parameter + G + I model. Accession numbers are indicated in parentheses. Isolates from this study are shown in bold. Bootstrap values > 70% are indicated at the nodes. Bradyrhizobium diazoefficiens USDA 110 T was used as outgroup. Bar indicates two substitutions per 100 nucleotide positions
The phylogenetic tree with the genus Bradyrhizobium (supplementary Fig. S1) clustered isolate CNPSo 3435 within the large clade of B. japonicum, showing the highest NI with the species B. japonicum USDA 6 T (99.7%). The tree with the genus Mesorhizobium (Fig. S2) grouped the isolate CNPSo 3975 with the species M. acaciae RITF741T, M. atlanticum CNPSo 3140 T and M. plurifarium LMG 11892 T, with 84% bootstrap values and sharing 100% NI.
3.2.2 glnII housekeeping gene analysis
To obtain a clearer taxonomic definition and better access to the diversity of the isolates, the phylogeny of the housekeeping gene glnII was analyzed. Again, four phylogenetic trees were constructed, with the genera Agrobacterium, Rhizobium, Bradyrhizobium, and Mesorhizobium.
In the phylogenetic tree of the Agrobacterium genus (Fig. 5), a large group (G.I) was formed with nine isolates from this study (CNPSo 4006, 4032, 3977, 3974, 3972, 3971, 4035, 4034, 3969), clustering again with the species A. pusense NRCPB10T and A. salinitolerans YIC 5082 T, sharing 97.6 to 100% of NI (Table 2), with 72% of statistical support. The other seven isolates (CNPSo 4058, 3529, 3973, 3436, 3966, 4001, 3498) occupied sole positions and their NIs were compared with the G.I, CNPSo 4058 shared 97.6 to 99.1% of NI with group G.I, the isolates CNPSo 3529, 3973 and 3436 shared 95.8 to 96.7% of NI; CNPSo 3966 shared 94.6 to 95.5% of NI, CNPSo 4001 shared 93.1 to 94.3% of NI and CNPSo 3498 shared 88.7 to 89.6% of NI with group G.I (Table 2).

Maximum likelihood phylogeny based on the glnII housekeeping gene alignment of the genus Agrobacterium (337 bp), using Tamura 3-Parameter + G model. Accession numbers are indicated in parentheses. Isolates from this study are shown in bold. Bootstrap values > 70% are indicated at the nodes. Bradyrhizobium diazoefficiens USDA 110 T was used as outgroup. Bar indicates five substitutions per 100 nucleotide positions
In the tree with the genus Rhizobium (Fig. 6), two large groups were formed. The large group G.I clustered nine isolates from this study with 15 species of the large clade R. etli. Two subgroups were formed, G.I.I clustered two isolates (CNPSo 3490 and 3982) sharing 96.5% of NI and showing 97.3 to 94.4% NI with the closest species R. esperanzae CNPSo 668 T. The second subgroup G I.II included seven isolates (CNPSo 3462, 3995, 3993, 3997, 4005, 4057, 4007) and the species R. phaseoli ATCC14482T, with 100% statistical support, sharing from 97.9 to 100% of NI (Table 3). The second large group (G.II) clustered 11 isolates from this study with seven species of the large clade R. tropici. Four subgroups were formed, beginning with G.II.I, with five isolates (CNPSo 3440, 3499, 3493, 3497, 3437) and the species R. leucaenae CFN 299 T with statistical support of 100% and sharing 99.7 to 100% of NI. G.II.II was positioned close to the G.II.I subgroup, sharing 94.4 to 95% of NI, and grouped the Rhizobium sp. CNPSo 3464 with the species R. paranaense PRF 35 T, with 82% of statistical support, and sharing 96.8% NI. Rhizobium sp. CNPSo 3968 occupied an isolated position, presenting 94.7% of NI with the species R. hainanense CCBAU 57015 T, and from 95 to 95.6% with the closest subgroup G.II.IV. Subgroup G.II.III grouped isolates CNPSo 4033 and CNPSo 4039 with statistical support of 75% and sharing 97.3% of NI among them, and from 96.2 to 97.3% NI with the closest subgroup G.II.IV. The last subgroup (G.II.IV) clustered isolates CNPSo 4062 and CNPSo 4063 with statistical support of 89%, and 98.8% NI (Table 3).

Maximum likelihood phylogeny based on glnII housekeeping gene alignment of the genus Rhizobium (346 bp), using Tamura-Nei + G + I model. Accession numbers are indicated in parentheses. Isolates from this study are shown in bold. Bootstrap values > 70% are indicated at the nodes. Bradyrhizobium diazoefficiens USDA 110 T was used as outgroup. Bar indicates two substitutions per 100 nucleotide positions
Regarding the Bradyrhizobium genus (Fig. S3) phylogeny, as in the phylogenetic tree of the 16S rRNA gene, isolate CNPSo 3435 was clustered with B. japonicum USDA 6 T in the glnII housekeeping gene phylogeny sharing 100% of NI and with 100% statistical support.
Finally, in the tree of the genus Mesorhizobum (Fig. S4) the strain CNPSo 3975 was clustered in the same branch as the species M. atlanticum CNPSo 3140 T, M. acaciae RITF741T, and M. shonense AC39aT, sharing NI of 96.1, 95.3 and 94.7%, respectively.
3.3 Morphophysiological characterization
Morphophysiological characterization in vitro was performed with the 38 strains classified as “classical rhizobia” and Agrobacterium isolates according to the sequencing analysis. The results are listed in supplementary Table S1. Among the 38 isolates, 22 showed fast (3 days), 14 intermediate (4 days), and two slow (6 and 7 days) growth. Regarding the acidic/alkaline reaction in modified-YM medium containing bromothymol blue as pH indicator, 34 isolates attributed to the Agrobacterium and Rhizobium genera showed neutral reaction. Isolates CNPSo 3975, 3995 and 4057, belonging to the Mesorhizobium and Rhizobium genera, showed acid reaction, and Bradyrhizobium sp. CNPSo 3435 alkaline reaction.
Regarding the chromogenesis of the colonies in modified-YM medium with Congo red as indicator, colonies of 19 isolates were red, 11 pink, and eight white. In the presence of bromothymol blue as indicator, 22 isolates resulted in colonies with yellow color, and 16 presented cream color.
In the evaluation of the properties of border, surface, shape and elevation of the colonies, all strains were similar, with smooth border and surface, with circular colonies and convex elevation. The diameter of the colonies ranged from 1.4 mm to 4.0 mm and the mucus production was considered moderate for 32 isolates, low for five isolates, and abundant for CNPSo 3977. For the optical details, 22 isolates were opaque and 16 translucent. The consistency of the growth mass was considered gummy for all strains tested, except for the isolates CNPSo 4005 and 4057, which showed viscous consistency.
3.4 Nodulation and nitrogen fixation capacity
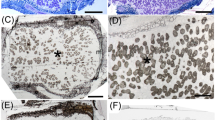
When the 38 isolates classified as “classical rhizobia” and Agrobacterium were evaluated for their capacity of nodulating and fixing nitrogen with Phaseolus vulgaris, only 13 (CNPSo 3437, 3440, 3462, 3464, 3490, 3493, 3497, 3498, 3499, 3982, 3993, 3997, 4005) were able to re-infect the host plant and form nodules. Verification of green color of the leaves and pink or red internal color of the nodules was performed as a first step to identify effective and ineffective strains (Fig. 7). All 13 isolates, except for Rhizobium sp. CNPSo 4005 formed nodules with internal color pink or red, indicating effective BNF.

A Green color of the leaves and internal pink color of the nodules indicating effectiveness of nitrogen fixation in common bean in comparison to (B) yellow leaves and ineffective nodules without the pink color that indicate an active leghemoglobin
4 Discussion
Particularly in the last decade, taxonomic studies of rhizobia symbionts of common bean have gained increasing notoriety worldwide and resulted in the description of several new species (e.g. Dall’Agnol et al. 2013, 2014; Ribeiro et al. 2013, 2015; Cordeiro et al. 2017; Huo et al. 2019; Helene et al. 2019). Although Brazil is not a genetic center of origin of common bean, the country embraces great diversity of rhizobia associated with this legume (e.g. Grange and Hungria 2004; Grange et al. 2007; Pinto et al. 2007; Stocco et al. 2008; Ribeiro et al. 2009, 2012; Dall’Agnol et al. 2013, 2014). The state of Mato Grosso do Sul is considered as a hotspot of biodiversity, encompassing three out of the six Brazilian biomes, Mata Atlântica, Cerrado, and Pantanal; however, although studies regarding rhizobial diversity point out high genetic richness (Pinto et al. 2007; Costa et al. 2018), they are still scarce. Additionally, indigenous rhizobial diversity in this Central-Western state of Brazil may represent an important source of strains with economic potential, as reported for common bean (Mercante et al. 2017).
The results obtained in this study highlight the outstanding rhizobial diversity in the Brazilian biomes. The DNA profiling by BOX-PCR of 82 isolates trapped in nodules of common bean grown in soils of 14 municipalities of MS generated 48 distinct groups or isolates occupying single positions, with a final level of similarity lower than 20%.
Following, we selected 56 isolates representative of the BOX-PCR profiles chosen to continue with the 16S rRNA phylogenetic analysis. It is worth mentioning that the 16S rRNA gene consists of about 1,500 bp, and for species definition the sequence of the whole gene is mandatory. However, in our study, the objective was to define the genera of the strains. Therefore, we used sequences of about 900 bp, because previous studies of diversity by our group have shown the viability of obtaining reliable classification at the genus level using shorter fragments, of about 1,000 bp (Costa et al. 2018; Chibeba et al. 2020; Klepa et al. 2021). In our study, one-third was composed by non-rhizobia, belonging to the genera Achromobacter (Yabuuchi and Yano 1981; Benata et al. 2008), Brevibacillus (Shida et al. 1996; Stajković et al. 2009), Burkholderia (Yabuuchi et al. 1992), Enterobacter (Hormaeche and Edwards 1960; Kan et al. 2007), Herbaspirillum (Baldani et al. 1986) and Pseudomonas (Yabuuchi et al. 1992). Noteworthy, some of these genera, such as Achromobacter, Burkholderia, Enterobacter and Pseudomonas may harbor severe human or plant pathogens. Following the Koch´s postulates, the non-rhizobia strains were not able to form nodules in common bean and this reinforces reports of endophytic bacteria coexisting with rhizobia in nodules, e.g. in Phaseolus lunatus (Chibeba et al., 2020), soybean (Glycine max (L.) Merr.) (Li et al. 2008; Zhao et al. 2017; Delamuta et al. 2020), common bean (Wang et al. 2016; Yan et al. 2017; Delamuta et al. 2020), and other legumes (Aserse et al. 2013; Tariq et al. 2014). Endophytes may contribute to plant growth by means of a variety of microbial processes (White et al. 2019; Verma et al. 2021). In our study the endophytic bacteria might contribute with nitrogen fixation, as it could be the case of the Herbaspirillum sp., but also with other microbial processes, such as the synthesis of phytohormones, as with Pseudomonas and Burkholderia, among others. The results confirm that there are far higher diversity of endophytes within the nodules as also shown in microbiome studies (Rocha et al. 2020), pointing out how far we are from understanding the role of this complex microbial community inhabiting legume nodules. Possible functions for these non-rhizobial nodule endophytes might include plant growth promotion by mechanisms such as synthesis of phytohormones, solubilization of phosphates, and tolerance to abiotic and biotic stresses (Schaedel et al. 2021).
Agrobacterium is another intriguing endophyte inhabiting nodules, reaching 28% of the isolates diversity based on the sequencing analyses. The genus includes several species commonly found in soil, generally pathogenic for several plants species (Singh and Prasad 2015; Barton et al. 2017). However, the genus includes as well non-pathogenic species, such as Agrobacterium fabacearum, also isolated from nodules, although, in general, they do not re-infect nodules in tests for nodulating capacity (Delamuta et al. 2020). Possible roles for endophytic Agrobacterium might include plant growth promotion, enhancement in nutrient acquisition, among other benefits (Chibeba et al. 2020; Delamuta et al. 2020; Dudeja et al. 2011).
The remaining 22 isolates whose 16S rRNA genes were sequenced were taxonomically positioned as “classical rhizobia”, comprising 20 Rhizobium, one Bradyrhizobium, and one Mesorhizobium. The genus Bradyrhizobium can associate symbiotically and endophytically with legumes such as Glycine max and Phaseolus lunatus (Durán et al. 2014; Chibeba et al. 2020), non-legumes as Oryza spp. and Parasponia spp. (Tan et al. 2001; Dupin et al. 2020), but is rarely reported as symbiont of common bean (Han et al. 2005; Cao et al., 2014; Wang et al. 2016). However, intriguingly, Da Conceição et al. (2018) reported that co-inoculation of common bean with Bradyrhizobium stimulated the symbiotic efficiency of Rhizobium. Therefore, a possible role for Bradyrhizobium in common bean nodules could rely on the improvement of the symbiotic performance of Rhizobium. The genus Mesorhizobium nodulates several legumes, such as chickpea (Cicer arietinum L.), another important legume for food security (Laranjo et al. 2014; Faridy et al. 2020), and although not usually reported, the genus has been isolated as an effective nitrogen-fixing symbiont of common bean (Helene et al. 2019).
The genus Rhizobium has a global distribution, being able to form symbiotic associations with a variety of legumes. They represent the main symbionts of P. vulgaris, and Brazilian soils are particularly rich in common bean Rhizobium diversity (e.g. Grange and Hungria 2004; Grange et al. 2007; Stocco et al. 2008; Ribeiro et al. 2009, 2012, 2013, 2015; Dall’Agnol et al. 2013, 2014; Gomes et al. 2015). In this study, about half of the Rhizobium isolates were positioned in the R. etli/R. leguminosarum/R. phaseoli clade, and the other half in the R. tropici clade. It has been suggested that the clade of R. tropici originated in the Andean region of South America, while species of the clade R. etli are from the Mesoamerican regions and Northern Argentina (Ribeiro et al. 2009, 2013; Gomes et al. 2015; Shamseldin and Velázquez 2020). In Brazil, bacteria belonging to both clades have been isolated from nodules of common bean all over the country, from the Northeast to the South (Grange and Hungria 2004; Grange et al. 2007; Pinto et al. 2007; Stocco et al. 2008).
As the 16S rRNA gene is highly conserved, the phylogenetic definition can be improved with the analysis of housekeeping genes, and the glnII (glutamine synthetase II) has proven to considerably help in taxonomic definition of rhizobia (e.g. Roma Neto et al. 2010). In our study, the main taxonomic position of rhizobia and Agrobacterium was confirmed and improved with the analysis of the glnII gene, except for the isolate CNPSo 3498 of Agrobacterium sp., indicating a putative event of horizontal transfer (HGT) of this gene.
Regarding nodulation and nitrogen fixation capacity, only 13 out of 38 “classical rhizobia” and Agrobacterium were able to re-infect the host plant. According to the analysis of the 16S rRNA and glnII genes, six were positioned in the R. etli clade, another six in the R. tropici clade, and one was classified as Agrobacterium sp. All isolates, except for CNPSo 4005, positioned in the R. etli clade, established effective symbiosis, characterized by nodules with pink or red internal color and plant shoots with leaves of dark green color, indicating adequate biological N supply.
There are reports showing that in Brazilian soils, predominantly acid, strains of the R. tropici clade are more competitive, characterized by higher tolerance of abiotic stresses, genetically more stable, and with higher capacity of BNF; consequently, commercial strains recommended for this crop in Brazil carry only species belonging to this clade (Hungria et al. 2000, 2003; Gomes et al. 2015; Mercante et al. 2017). Unfortunately, in our study R. tropici represented only a small percentage of the isolates, what can explain in part the low efficiency of BNF in common bean in the field.
Our study confirms that the Brazilian biomes represent a rich repository of microbial species with importance for plant growth promotion and nutrition. It also confirms the promiscuous nature of the symbiosis with common bean, which establishes interactions with a variety of effective and non-effective nitrogen-fixing rhizobia. Intriguingly, apparently the non-effective strains overtake in number the effective ones. Due to the broad range of bacterial species identified, characterized by different mechanisms that could explain plant infection, it is feasible to conclude that the symbiotic promiscuity in common bean is controlled by the host plant and not by the bacteria. Understanding the genetic mechanisms in common bean that allow this promiscuity might represent the most promising way to increase the contribution of BNF in this crop. In addition, it might also pave ways to manipulate other plant species, facilitating plant–microbe interactions.
5 Conclusions
The results obtained in this study highlight outstanding genetic diversity in bacteria isolated from common bean nodules cultivated in soils of 14 municipalities in Mato Grosso do Sul, in the Central-Western region of Brazil. Estimates were that 32% of the isolates were non-rhizobia endophytes, which might play further roles in the symbiosis, such as plant growth promotion, and tolerance to abiotic and biotic stresses. The remaining isolates were classified as Agrobacterium and “classical rhizobia", but only 31% were able to form effective nitrogen-fixing nodules when re-inoculated in common bean. The effective strains were positioned in the R. etli and R. tropici clades and one as Agrobacterium. These results emphasize the high promiscuity of common bean, which allows nodule colonization by a variety of bacterial species, most of them not efficient in nitrogen fixation, limiting the contribution of the BNF to the crop.
References
Aserse AA, Räsänen LA, Assefa F, Hailemariam A, Lindström, K (2012) Phylogeny and genetic diversity of native rhizobia nodulating common bean (Phaseolus vulgaris L.) in Ethiopia. Systematic and Applied Microbiology 35(2):120–131. https://doi.org/10.1016/j.syapm.2011.11.005.
Aserse AA, Räsänen LA, Assefa F, Hailemariam A, Lindström K (2013) Diversity of sporadic symbionts and nonsymbiotic endophytic bacteria isolated from nodules of woody, shrub, and food legumes in Ethiopia. Appl Microbiol Biotechnol 97(23):10117–10134. https://doi.org/10.1007/s00253-013-5248-4
Baldani JI, Baldani VLD, Seldin L, Döbereiner J (1986) Characterization of Herbaspirillum seropedicae gen. nov., sp. nov., a root-associated nitrogen-fixing bacterium. International Journal of Systematic and Evolutionary Microbiology 36(1):86–93. https://doi.org/10.1099/00207713-36-1-86.
Barton IS, Fuqua C, Platt TG (2017) Ecological and evolutionary dynamics of a model facultative pathogen: Agrobacterium and crown gall disease of plants. Environ Microbiol 20(1):16–29. https://doi.org/10.1111/1462-2920.13976
Benata H, Mohammed O, Noureddine B, Abdelbasset B, Abdelmoumen H, Muresu R, Squartini A, El Idrissi MM (2008) Diversity of bacteria that nodulate Prosopis juliflora in the eastern area of Morocco. Syst Appl Microbiol 31(5):378–386. https://doi.org/10.1016/j.syapm.2008.08.002
Cao Y, Wang ET, Zhao L, Chen WM, Wei GH (2014) Diversity and distribution of rhizobia nodulated with Phaseolus vulgaris in two ecoregions of China. Soil Biol Biochem 78:128–137. https://doi.org/10.1016/j.soilbio.2014.07.026
Case RJ, Boucher Y, Dahllof I, Holmstrom C, Doolittle WF, Kjelleberg S (2007) Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol 73(1):278–288. https://doi.org/10.1128/AEM.01177-06
Chibeba AM, Pereira CS, Antunes JEL, Ribeiro RA, De Almeida Lopes AC, Gomes RLF, Hungria M, Araujo ASF (2020) Polyphasic characterization of nitrogen-fixing and co-resident bacteria in nodules of Phaseolus lunatus inoculated with soils from Piauí State. Northeast Brazil Symbiosis 80(3):279–292. https://doi.org/10.1007/s13199-020-00672-1
Colwell RR (1970) Polyphasic taxonomy of the genus Vibrio: numerical taxonomy of Vibrio cholerae, Vibrio parahaemolyticus, and related Vibrio species. J Bacteriol 104(1):410–433. https://doi.org/10.1128/jb.104.1.410-433.1970
Cordeiro AB, Ribeiro RA, Helene LCF, Hungria M (2017) Rhizobium esperanzae sp. nov., a N2-fixing root symbiont of Phaseolus vulgaris from Mexican soils. International Journal of Systematic and Evolutionary Microbiology 67(10):3937–3945. https://doi.org/10.1099/ijsem.0.002225.
Costa MR, Chibeba AM, Mercante FM, Hungria M (2018) Polyphasic characterization of rhizobia microsymbionts of common bean [Phaseolus vulgaris (L.)] isolated in Mato Grosso do Sul, a hotspot of Brazilian biodiversity. Symbiosis 76(2):163–176. https://doi.org/10.1007/s13199-018-0543-6.
Da Conceição JE, De Almeida LR, Do Amaral Bastos R, Da Silva Aragão OO, Araújo AP (2018) Co-inoculation of Bradyrhizobium stimulates the symbiosis efficiency of Rhizobium with common bean. Plant Soil 425(1):201–215. https://doi.org/10.1007/s11104-017-3541-1
Dall’Agnol RF, Ribeiro RA, Ormeno-Orrillo E, Rogel MA, Delamuta JRM, Andrade DS, Martínez-Romero E, Hungria M (2013) Rhizobium freirei sp. nov., a symbiont of Phaseolus vulgaris that is very effective at fixing nitrogen. International Journal of Systematic and Evolutionary Microbiology 63(11):4167–4173. https://doi.org/10.1099/ijs.0.052928-0.
Dall’Agnol RF, Ribeiro RA, Delamuta JRM, Ormeño-Orrillo E, Rogel MA, Andrade DS, Martínez-Romero E, Hungria M (2014) Rhizobium paranaense sp. nov., an effective N2-fixing symbiont of common bean (Phaseolus vulgaris L.) with broad geographical distribution in Brazil. International Journal of Systematic and Evolutionary Microbiology 64(Pt_9):3222–3229. https://doi.org/10.1099/ijs.0.064543-0.
Dall’Agnol RF, Bournaud C, De Faria SM, Béna G, Moulin L, Hungria M 2017. Genetic diversity of symbiotic Paraburkholderia species isolated from nodules of Mimosa pudica (L.) and Phaseolus vulgaris (L.) grown in soils of the Brazilian Atlantic Forest (Mata Atlântica). FEMS Microbiology Ecology 93(4): fix027. https://doi.org/10.1093/femsec/fix027.
Delamuta JRM, Menna P, Ribeiro RA, Hungria M (2017) Phylogenies of symbiotic genes of Bradyrhizobium symbionts of legumes of economic and environmental importance in Brazil support the definition of the new symbiovars pachyrhizi and sojae. Syst Appl Microbiol 40(5):254–265. https://doi.org/10.1016/j.syapm.2017.04.005
Delamuta JRM, Scherer AJ, Ribeiro RA, Hungria M (2020) Genetic diversity of Agrobacterium species isolated from nodules of common bean and soybean in Brazil, Mexico, Ecuador and Mozambique, and description of the new species Agrobacterium fabacearum sp. nov. Int J Syst Evol Microbiol 70(7):4233–4244. https://doi.org/10.1099/ijsem.0.004278
Dudeja SS, Giri R, Saini R, Suneja-Madan P, Kothe E (2011) Interaction of endophytic microbes with legumes. J Basic Microbiol 52(3):248–260. https://doi.org/10.1002/jobm.201100063
Dupin SE, Geurts R, Kiers ET (2020) The non-legume Parasponia andersonii mediates the fitness of nitrogen-fixing rhizobial symbionts under high nitrogen conditions. Front Plant Sci 10:1779. https://doi.org/10.3389/fpls.2019.01779
Durán D, Rey L, Mayo J, Zúñiga-Dávila D, Imperial J, Ruiz-Argüeso T, Martínez-Romero E Ormeño-Orrillo E (2014) Bradyrhizobium paxllaeri sp. nov., and Bradyrhizobium icense sp. nov., nitrogen-fixing rhizobial symbionts of Lima bean (Phaseolus lunatus L.) in Peru. International Journal of Systematic and Evolutionary Microbiology. 64(6):2072–2078. https://doi.org/10.1099/ijs.0.060426-0.
Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Eisenlohr PV, De Oliveira-Filho AT, Prado J (2015) The Brazilian Atlantic Forest: new findings, challenges and prospects in a shrinking hotspot. Biodivers Conserv 24(9):2129–2133. https://doi.org/10.1007/s10531-015-0995-4
Faridy JCM, Stephanie CGM, Gabriela MMO, Cristian JM (2020) Biological activities of chickpea in human health (Cicer arietinum L.). A review. Plant Foods for Human Nutrition 75(2):142–153. https://doi.org/10.1007/s11130-020-00814-2.
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17(6):368–376. https://doi.org/10.1007/BF01734359
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39(4):783–791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x
Fournier PE, Lagier JC, Dubourg G, Raoult D (2015) From culturomics to taxonomogenomics: a need to change the taxonomy of prokaryotes in clinical microbiology. Anaerobe 36:73–78. https://doi.org/10.1016/j.anaerobe.2015.10.011
Frank B (1889) Über die Pilzsymbiose der Leguminosen. Berichte Der Deutschen Botanischen Gesellschaft 7:332–346. https://doi.org/10.1111/j.1438-8677.1889.tb05711.x
Gomes DF, Ormeño-Orrillo E, Hungria M (2015) Biodiversity, symbiotic efficiency and genomics of Rhizobium tropici and related species. In: de Bruijn FJ (ed). Biological Nitrogen Fixation. Hoboken, New Jersey: John Wiley & Sons, Inc., pp 747–756. https://doi.org/10.1002/9781119053095.ch74.
Grange L, Hungria M (2004) Genetic diversity of indigenous common bean (Phaseolus vulgaris) rhizobia in two Brazilian ecosystems. Soil Biol Biochem 36(9):1389–1398. https://doi.org/10.1016/j.soilbio.2004.03.005
Grange L, Hungria M, Graham PH, Martínez-Romero E (2007) New insights sinto the origins and evolution of rhizobia that nodulate common bean (Phaseolus vulgaris) in Brazil. Soil Biol Biochem 39(4):867–876. https://doi.org/10.1016/j.soilbio.2006.10.008
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Han SZ, Wang ET, Chen WX (2005) Diverse bacteria isolated from root nodules of Phaseolus vulgaris and species within the genera Campylotropis and Cassia grown in China. Syst Appl Microbiol 28(3):265–276. https://doi.org/10.1016/j.syapm.2004.12.005
Hedges SB (1992) The number of replications needed for accurate estimation of the bootstrap P value in phylogenetic studies. Mol Biol Evol 9(2):366–369. https://doi.org/10.1093/oxfordjournals.molbev.a040725
Helene LCF, Dall’Agnol RF, Delamuta JRM, Hungria M (2019) Mesorhizobium atlanticum sp. nov., a new nitrogen-fixing species from soils of the Brazilian Atlantic Forest biome. International Journal of Systematic and Evolutionary Microbiology 69(6):1800–1806. https://doi.org/10.1099/ijsem.0.003397.
Hormaeche E, Edwards PR (1960) A proposed genus Enterobacter. International Bulletin of Bacteriological Nomenclature and Taxonomy 10(2):71–74. https://doi.org/10.1099/0096266X-10-2-71
Hugenholtz P, Chuvochina M, Oren A, Parks DH, Soo RM (2021) Prokaryotic taxonomy and nomenclature in the age of big sequence data. ISME J 15:1879–1892. https://doi.org/10.1038/s41396-021-00941-x
Hungria M, Andrade DS, Chueire LMO, Probanza A, Guttierrez-Mañero FJ, Megías M (2000) Isolation and characterization of new efficient and competitive bean (Phaseolus vulgaris L.) rhizobia from Brazil. Soil Biol Biochem 32:1515–1528. https://doi.org/10.1016/S0038-0717(00)00063-8
Hungria M, Campo RJ, Mendes IC (2003) Benefits of inoculation of the common bean (Phaseolus vulgaris) crop with efficient and competitive Rhizobium tropici strains. Biol Fertil Soils 39(1):88–93. https://doi.org/10.1007/s00374-003-0682-6
Hungria M, O'Hara G, Zilli JE, Araujo RS, Deaker R, Howieson JG (2016)Isolation and growth of rhizobia. In: Howieson JG, Dilworth MJ (eds). Working with Rhizobia. Australian Center for International Agricultural Research (ACIAR), Canberra, Australia, pp 39–60.
Huo Y, Tong W, Wang J, Wang F, Bai W, Wang E, Shi P, Chen W, Wei G (2019) Rhizobium chutanense sp. nov., isolated from root nodules of Phaseolus vulgaris in China. International Journal of Systematic and Evolutionary Microbiology 69(7):2049–2056. https://doi.org/10.1099/ijsem.0.003430.
Jaccard P (1912) The distribution of the flora in the alpine zone. New Phytol 11(2):37–50. https://doi.org/10.1111/j.1469-8137.1912.tb05611.x
Jarvis BDW, van Berkum P, Chen WX, Nour SM, Fernandez MP, Cleyet-Marel JC, Gillis M (1997) Transfer of Rhizobium loti, Rhizobium huakuii, Rhizobium ciceri, Rhizobium mediterraneum, and Rhizobium tianshanense to Mesorhizobium gen. nov. International Journal of Systematic and Evolutionary Microbiology 47(3):895–898. https://doi.org/10.1099/00207713-47-3-895.
Jordan DC (1982) Transfer of Rhizobium japonicum Buchanan 1980 to Bradyrhizobium gen. nov., a genus of slow-growing, root nodule bacteria from leguminous plants. International Journal of Systematic and Evolutionary Microbiology 32(1):136–139. https://doi.org/10.1099/00207713-32-1-136.
Kan FL, Chen ZY, Wang ET, Tian CF, Sui XH, Chen WX (2007) Characterization of symbiotic and endophytic bacteria isolated from root nodules of herbaceous legumes grown in Qinghai-Tibet plateau and in other zones of China. Arch Microbiol 188(2):103–115. https://doi.org/10.1007/s00203-007-0211-3
Kaschuk G, Hungria M (2017) Diversity and importance of diazotrophic bacteria to agricultural sustain ability in the tropics. In: Azevedo JL, Quecine MC (eds). Diversity and Benefits of Microorganisms From the Tropics. Part III. Springer International Publishing, pp 269–292. https://doi.org/10.1007/978-3-319-55804-2_12.
Klepa MS, Janoni V, Paulitsch F, Silva AR, Carmo MRB, Delamuta JRM, Hungria M (2021) Batista JSS (2021) Molecular diversity of rhizobia-nodulating native Mimosa of Brazilian protected areas. Arch Microbiol 203:5533–5545. https://doi.org/10.1007/s00203-021-02537-7
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33(7):1870–1879. https://doi.org/10.1093/molbev/msw054.
Kuykendall LD, Saxena B, Devine TE, Udell SE (1992) Genetic diversity in Bradyrhizobium japonicum Jordan 1982 and a proposal for Bradyrhizobium elkanii sp. nov. Canadian Journal of Microbiology 38(6):501–505. https://doi.org/10.1139/m92-082.
Laranjo M, Alexandre A, Oliveira S (2014) Legume growth-promoting rhizobia: an overview on the Mesorhizobium genus. Microbiol Res 169(1):2–17. https://doi.org/10.1016/j.micres.2013.09.012
Li JH, Wang ET, Chen WF, Chen WX (2008) Genetic diversity and potential for promotion of plant growth detected in nodule endophytic bacteria of soybean grown in Heilongjiang province of China. Soil Biol Biochem 40(1):238–246. https://doi.org/10.1016/j.soilbio.2007.08.014
Martínez-Romero E, Segovia L, Mercante FM, Franco AA, Graham P, Pardo MA (1991) Rhizobium tropici, a novel species nodulating Phaseolus vulgaris L. beans and Leucaena sp. trees. International Journal of Systematic and Evolutionary Microbiology 41(3):417–426. https://doi.org/10.1099/00207713-41-3-417.
Menna P, Hungria M (2011) Phylogeny of nodulation and nitrogen-fixation genes in Bradyrhizobium: supporting evidence for the theory of monophyletic origin, and spread and maintenance by both horizontal and vertical transfer. Int J Syst Evol Microbiol 61(12):3052–3067. https://doi.org/10.1099/ijs.0.028803-0
Menna P, Hungria M, Barcellos FG, Bangel EV, Hess PN, Martínez-Romero E (2006) Molecular phylogeny based on the 16S rRNA gene of elite rhizobial strains used in Brazilian commercial inoculants. Syst Appl Microbiol 29(4):315–332. https://doi.org/10.1016/j.syapm.2005.12.002
Mercante FM, Otsubo AA, Brito OR (2017) New native rhizobia strains for inoculation of common bean in the Brazilian savanna. Revista Brasileira de Ciência do Solo 41:e0150120. https://doi.org/10.1590/18069657rbcs20150120
Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GA, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403(6772):853–858. https://doi.org/10.1038/35002501
Myers JR, Kmiecik K (2017) Common bean: Economic importance and relevance to biological science research. In: Pérez de la Vega M, Santalla M, Marsolais F (eds). The Common Bean Genome. Springer, Cham. pp 1–20. https://doi.org/10.1007/978-3-319-63526-2_1.
Peix A, Ramírez-Bahena MH, Velázquez E, Bedmar EJ (2015) Bacterial associations with legumes. Crit Rev Plant Sci 34(1–3):17–42. https://doi.org/10.1080/07352689.2014.897899
Pinto FGS, Hungria M, Mercante FM (2007) Polyphasic characterization of Brazilian Rhizobium tropici strains effective in fixing N2 with common bean (Phaseolus vulgaris L.). Soil Biol Biochem 39:1851–1864. https://doi.org/10.1007/s00253-011-3708-2
Reinprecht Y, Schram L, Marsolais F, Smith TH, Hill B, Pauls KP (2020) Effects of nitrogen application on nitrogen fixation in common bean production. Front Plant Sci 11:1172. https://doi.org/10.3389/fpls.2020.01172
Ribeiro RA, Barcellos FG, Thompson FL, Hungria M (2009) Multilocus sequence analysis of Brazilian Rhizobium strains microsymbionts of common beans (Phaseolus vulgaris) reveals unexpected taxonomic diversity. Res Microbiol 160:297–306. https://doi.org/10.1016/j.resmic.2009.03.009
Ribeiro RA, Rogel MA, López-López A, Ormeño-Orrillo E, Barcellos FG, Martínez J, Thompson FL, Martìnez-Romero E, Hungria M (2012) Reclassification of Rhizobium tropici type A strains as Rhizobium leucaenae sp. nov. Int J Syst Evol Microbiol 62:1180–1185. https://doi.org/10.1099/ijs.0.032912-0
Ribeiro RA, Ormeño-Orrillo E, Dall’Agnol RF, Graham PH, Martínez-Romero E, Hungria M, (2013) Novel Rhizobium lineages isolated from root nodules of common bean (Phaseolus vulgaris L.) in Andean and Mesoamerican areas. Res Microbiol 164:740–748. https://doi.org/10.1016/j.resmic.2013.05.002
Ribeiro RA, Martins TB, Ormeño-Orrillo E, Delamuta JRM, Rogel MA, Martínez-Romero E, Hungria M (2015) Rhizobium ecuadorense sp. nov., an indigenous N2-fixing symbiont of the Ecuadorian common bean (Phaseolus vulgaris L.) genetic pool. International Journal of Systematic and Evolutionary Microbiology 65(9):3162–3169. https://doi.org/10.1099/ijsem.0.000392.
Rocha SMB, Mendes LW, De Souza Oliveira LM, Melo VMM, Antunes JEL, Araujo FF, Hungria M, Araujo ASF (2020) Nodule microbiome from cowpea and lima bean grown in composted tannery sludge-treated soil. Appl Soil Ecol 151:103542. https://doi.org/10.1016/j.apsoil.2020.103542
Roma Neto IVR, Ribeiro RA, Hungria M (2010) Genetic diversity of elite rhizobial strains of subtropical and tropical legumes based on the 16S rRNA and glnII genes. World J Microbiol Biotechnol 26(7):1291–1302. https://doi.org/10.1007/s11274-009-0300-3
Schaedel M, Hidrobo G, Grossman J (2021) From microns to meters: Exploring advances in legume microbiome diversity for agroecosystem benefits. Frontiers in Sustainable Food Systems 5:668195. https://doi.org/10.3389/fsufs.2021.668195
Schwarz G (1978) Estimating the dimension of a model. The Annals of Statistics 6(2):461–464. https://doi.org.jstor.org/stable/2958889.
Shamseldin A, Velázquez E (2020) The promiscuity of Phaseolus vulgaris L. (common bean) for nodulation with rhizobia: a review. World Journal of Microbiology and Biotechnology 36(5):1–12. https://doi.org/10.1007/s11274-020-02839-w.
Shida O, Takagi H, Kadowaki K, Komagata K (1996) Proposal for two new genera, Brevibacillus gen. nov. and Aneurinibacillus gen. nov. International Journal of Systematic and Evolutionary Microbiology 46(4):939–946. https://doi.org/10.1099/00207713-46-4-939.
Singh RK, Prasad M (2015) Advances in Agrobacterium tumefaciens-mediated genetic transformation of graminaceous crops. Protoplasma 253(3):691–707. https://doi.org/10.1007/s00709-015-0905-3
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principles and practice of numerical classification. W.H Freeman and Company San Francisco, USA, p 573
Somasegaran P, Hoben HJ (1994) Handbook for Rhizobia: Methods in Legume-Rhizobium Technology. Springer Science & Business Media, 450 p. https://doi.org/10.1007/978-1-4613-8375-8.
Stajković O, De Meyer S, Miličić B, Willems A, Delić D (2009) Isolation and characterization of endophytic non-rhizobial bacteria from root nodules of alfalfa (Medicago sativa L.). Botanica Serbica 33(1):107–114.
Stepkowski T, Moulin L, Krzyžańska A, Mcinnes A, Law IJ, Howieson J (2005) European origin of Bradyrhizobium populations infecting lupins and serradella in soils of western Australia and South Africa. Appl Environ Microbiol 71(11):7041–7052. https://doi.org/10.1128/AEM.71.11.7041-7052.2005
Stocco P; Santos JCP; Vargas VP, Hungria M (2008) Avaliação da biodiversidade de rizóbios simbiontes do feijoeiro (Phaseolus vulgaris L.) em Santa Catarina. (Assessment of biodiversity in rhizobia symbionts of common bean (Phaseolus vulgaris L.) in Santa Catarina, Brazil). Revista Brasileira de Ciência do Solo 32(8):1107–1120. https://doi.org/10.1590/S0100-06832008000300019.
Tan Z, Hurek T, Vinuesa P, Müller P, Ladha JK, Reinhold-Hurek B (2001) Specific detection of Bradyrhizobium and Rhizobium strains colonizing rice (Oryza sativa) roots by 16S–23S ribosomal DNA intergenic spacer-targeted PCR. Appl Environ Microbiol 67(8):3655–3664. https://doi.org/10.1128/AEM.67.8.3655-3664.2001
Tariq M, Hameed S, Yasmeen T, Zahid M, Zafar M (2014) Molecular characterization and identification of plant growth promoting endophytic bacteria isolated from the root nodules of pea (Pisum sativum L.). World Journal of Microbiology and Biotechnology 30(2):719–725. https://doi.org/10.1007/s11274-013-1488-9.
Thompson C, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL (2013) Microbial genomic taxonomy. BMC Genomics 14:913. https://doi.org/10.1186/1471-2164-14-913
Velázquez E, García-Fraile P, Ramírez-Bahena MH, Rivas R, Martínez-Molina E (2017) Current status of the taxonomy of bacteria able to establish nitrogen-fixing legume symbiosis. In Zaidi A, Khan M, Musarrat J (eds) Microbes for Legume Improvement (pp. 1–43). Springer, Cham., pp 1–43. https://doi.org/10.1007/978-3-319-59174-2_1.
Verma SK, Sahu PK, Kumar K, Pal G, Gond SK, Kharwar RN, White JF (2021) Endophyte roles in nutrient acquisition, root system architecture development and oxidative stress tolerance. J Appl Microbiol 131(5):2161–2177. https://doi.org/10.1111/jam.15111
Versalovic J, Schneider M, de Bruijn FJ, Lupsky JR (1994) Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods in Molecular and Cellular Biology 5(1):25–40
Vincent JM (1970) A Manual for the Practical Study of Root-Nodule Bacteria. International Biological Program Handbook No. 15. Blackwell Science Publications, Oxford.
Wang ET, Tian CF, Chen WF, Young JPW, Chen WX (2019) Ecology and Evolution of Rhizobia. Springer, Singapore. https://doi.org/10.1007/978-981-32-9555-1
Wang L, Cao Y, Wang ET, Qiao YJ, Jiao S, Liu ZS, Wei GH (2016) Biodiversity and biogeography of rhizobia associated with common bean (Phaseolus vulgaris L.) in Shaanxi Province. Systematic and Applied Microbiology 39(3):211–219. https://doi.org/10.1016/j.syapm.2016.02.001.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697703. https://doi.org/10.1128/jb.173.2.697-703.1991
White JF, Kingsley KL, Zhang Q, Verma R, Obi N, Matthew SD, Elmore MT, Verma SK, Gond SK, Kowalski KP (2019) Review: Endophytic microbes and their potential applications in crop management. Pest Manag Sci 75(10):2558–2565. https://doi.org/10.1002/ps.5527
Yabuuchi E., Yano I (1981) Achromobacter gen. nov. and Achromobacter xylosoxidans (ex Yabuuchi and Ohyama 1971) nom. rev. International Journal of Systematic and Evolutionary Microbiology 31(4):477–478. https://doi.org/10.1099/00207713-31-4-477.
Yabuuchi E, Kosako Y, Oyaizu H, Yano I, Hotta H, Hashimoto Y, Ezaki T, Arakawa M (1992) Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiology and Immunology 36(12):1251–1275. https://doi.org/10.1111/j.1348-0421.1992.tb02129.x
Yan J, Yan H, Liu LX, Chen WF, Zhang XX, Verástegui-Valdés MM, Wang ET, Han XZ (2017) Rhizobium hidalgonense sp. nov., a nodule endophytic bacterium of Phaseolus vulgaris in acid soil. Arch Microbiol 199(1):97–104. https://doi.org/10.1007/s00203-016-1281-x.
Zhao L, Xu Y, Lai X (2018) Antagonistic endophytic bacteria associated with nodules of soybean (Glycine max L.) and plant growth-promoting properties. Braz J Microbiol 49(2) 269–278. https://doi.org/10.1016/j.bjm.2017.06.007.
Acknowledgements
We thank Dr. Jakeline R. M. Delamuta on the 16S rRNA and glnII housekeeping genes analyses; Ligia M. O. Chueire for her help during the execution of the methodologies; Embrapa Soja for supplying structure and materials. Capes (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and Fundação Araucária for granting the M.Sc. fellowship to F.T. Moura. M.A. Nogueira and M. Hungria are also CNPq research fellows (National Council for Scientific and Technological Development).
Funding
Partially financed by INCT—Plant Growth Promoting Microorganisms for Agricultural Sustainability and Environmental Responsibility (CNPq 465133/2014–4, Fundação Araucária-STI 043/2019, CAPES).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing or conflicting interests
The authors declare that they have no competing or conflicting interests, or ethical conflicts.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moura, F.T., Ribeiro, R.A., Helene, L.C.F. et al. So many rhizobial partners, so little nitrogen fixed: The intriguing symbiotic promiscuity of common bean (Phaseolus vulgaris L.). Symbiosis 86, 169–185 (2022). https://doi.org/10.1007/s13199-022-00831-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13199-022-00831-6