
Abstract
This study examines the influence of different food-grade n-3 PUFA-enriched simple emulsion (SE), double emulsion (DE) and gelled double emulsion (GDE) delivery systems on the extent of lipolysis, antioxidant capacity and the bioaccessibility of hydroxytyrosol (HTy). GDE emulsion offered better protection for HTy (89%) than the other systems (79% in SE and DE). The reducing capacity of the emulsions containing HTy were not altered during oral digestion. However, “in vitro” gastric and intestinal phases significantly reduced the antioxidant activity of all systems. The structural and physical state of GDE entailed a slowing-down of triacylglyceride hydrolysis (36.4%) in comparison with that of SE and DE (22.7 and 24.8% for SE and DE, respectively).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Emulsion technology is particularly suited for the design and fabrication of delivery systems of active components for use in food applications. In this regard a number of emulsion-based structured delivery systems have been developed for different purposes, including use as ingredients in the development of healthier and functional foods (Jiménez-Colmenero et al. 2015; McClements and Li 2010). In this connection, conventional oil-in-water emulsions (O/W) are currently the most widely used method because of their relative ease of preparation. These simple emulsions (SEs) have been extensively used as lipid carriers for compounds such as vitamins E and D and n-3 PUFAs in foods and beverages (McClements and Li 2010). Double (also denominated as multiple) or more specifically water-in-oil-in-water (W/O/W; DE) emulsions are more complex liquid dispersions in which oil globules, containing water droplets, are dispersed in a continuous aqueous phase. This more advanced technology offers a number of interesting opportunities for the food industry, including delivery of lipophilic compounds in aqueous systems, encapsulation of hydrophilic bioactive compounds and reduction of fat content (Garti 1997; Jiménez-Colmenero 2013). Depending on the intended food application, the instability and/or plastic properties of these systems are important issues. In this regard, novel proposals for liquid oil phase stabilization and structuring have recently been reported, among them structured emulsions such as gelled double emulsions (GDEs) with improved nutritional, technological and/or sensory characteristics (Li et al. 2014; Lobato-Calleros et al. 2008; Weiss et al. 2005). In this connection, cold-set gelation of emulsions by transglutaminase offers interesting possibilities for heat-labile bioactive compounds (Yang et al. 2013). This then offers the possibility of designing emulsion-based delivery systems that differ in structure, composition and physicochemical properties for use in a variety of food applications (Jiménez-Colmenero et al. 2015; Zetzl et al. 2012).
Scientific authorities and nutritional organizations recommend a higher consumption of unsaturated fatty acids, especially n-3 polyunsaturated fats, along with a reduction in the intake of dietary saturated fats (WHO/FAO Expert Consultation 2003). In this context, perilla oil, produced from perilla seeds (Perilla frutescens), is of special interest as it is one of the richest sources of α-linolenic acid (over 60 g/100 g oil). The health benefits of perilla oil are partially explained by its high α-linolenic acid content, but it seems to have other impacts on lipid metabolism via regulation of the expression of transcription factors (Jo et al. 2013). Therefore, the use of perilla oil offers interesting possibilities for the development of emulsion-based structured delivery systems. However, in view of the structural characteristics of emulsions (processing enlarges the oil/water interfacial area) and increased unsaturated fatty acid levels, these n-3 PUFA-enriched systems could be more prone to lipid oxidation. Among other strategies, addition of antioxidants is widely used as this has been found to be effective in reducing oxidation and/or retarding its onset (McClements and Decker 2000). Various synthetic antioxidants have proven very efficient, but given consumer demand for clean label foods and natural ingredients, different natural antioxidant sources have been studied (Ahn et al. 2002). In this connection, hydroxytyrosol (HTy) is a hydrophilic phenolic compound that has received considerable attention, not only because it is a very potent antioxidant but also because of anti-inflammatory and cancer-preventive effects attributed to it (De Leonardis et al. 2008; Fki et al. 2005; Gonzalez-Santiago et al. 2006). HTy, then, is an interesting bioactive compound that can also be used as an antioxidant to minimize oxidation in n-3 PUFA-rich foods (DeJong and Lanari 2009; Pazos et al. 2008).
Researchers have recently recognized the importance of food structure in the digestion and absorption of nutrients (Guo et al. 2014; Marze 2013). The design of food structures purposely to impact on lipid digestion has received increasing attention because of the need for solutions to tackle nutrition related concerns such as obesity and metabolic syndrome (Wooster et al. 2014). The rate and extent of lipid digestion can be affected by factors such as droplet size distribution, emulsifier type and lipid phase composition (McClements and Li 2010). In fact, it has been reported that slowing down gastric emptying increases satiation (Olivero-David et al. 2011). Then again, oxidation has been reported in the stomach (Olivero-David et al. 2011). Thus, an excessive slow-down of gastric emptying would negatively counterbalance those positive effects. Differences in structure, composition and physicochemical properties of emulsion-based delivery systems should therefore be considered as they can influence lipid digestion (e.g. lipase activity and accessibility) and the bioavailability of bioactive compounds (Mao and Miao 2015; Sánchez-Muniz et al. 2011). Previous papers studied the influence of food structures on the progression of lipid digestion by using a pH–stat (McClements and Li 2010; Wooster et al. 2014). However, as far as we know, no studies have been conducted on emulsion-delivery systems sequentially using a standardized static “in vitro” digestion method followed by high performance size exclusion chromatography (HPSEC) to assess triglyceride hydrolysis and quantify the hydrolytic compounds formed.
The present paper hypothesizes that such emulsion-based delivery systems may play an important role in oil digestion and antioxidant activity by slowing down antioxidant losses and triglyceride hydrolysis. The aim of this study was to investigate the influence (composition/structure) of different food-grade n-3 PUFA-enriched delivery systems such as simple (SEs), double (DEs) and gelled double (GDEs) emulsions on the bioaccessibility of HTy, antioxidant capacity and lipolysis extent. These emulsions were prepared with perilla oil as a source of n-3 PUFAs and HTy as a natural antioxidant and bioactive compound.
Materials and methods
Materials
HTy (purity ≥99%) was purchased from Seprox Biotech (Madrid, Spain) and perilla oil was purchased from Grupo Nutracéutico Chiasa, SL (Meliana, Spain). The oil fatty acid composition, expressed as percentage, was as follows: palmitic acid 6.0 ± 0.29, stearic acid 1.7 ± 0.15, oleic acid 12.5 ± 0.39, cis-vaccenic acid 1.0 ± 0.03, linoleic acid 14.3 ± 0.12 and linolenic acid 64.6 ± 0.74. Upon arrival this oil was stored at 4 °C until preparation of the different emulsion systems. The hydroperoxide content was 0.22 ± 0.01 mmol cumene hydroperoxyde while thiobarbituric acid-reactive substances (TBARS) were under the limit of detection (Flaiz et al. 2016). Sodium caseinate was purchased from FrieslandCampina DMV (Excellion EM 7, Veghel, The Netherlands), polyglycerol polyricinoleate was from Lasenor Emul S.L. (Olesa de Montserrat, Spain), gelatine (type B, 200-220 bloom) was from Manuel Riesgo, S.A. (Madrid, Spain) and transglutaminase (Activa GS) was from Ajinomoto (Tokyo, Japan). Pepsin, pancreatin and bile extract, all porcine, chloroform, 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox), methanol, 2,4,6-tri-(2-pyridyl)-1,3,5-triazine (TPTZ), iron(III) chloride hexahydrate and tetrahydrofuran were purchased from Sigma-Aldrich (Madrid, Spain). All other reagents and solvents used were of a suitable grade for spectrophotometric or chromatographic analyses.
Preparation of simple (SE), double (DE) and gelled double (GDE) emulsions
The aqueous phase (WSE) of the SE consisted of 0.584 g NaCl, 0.04 g sodium azide, 0.441 g sodium caseinate and 44.1 mg HTy dissolved in 100 mL distilled water. The lipid phase (O) consisted of perilla oil (94 g/100 g) plus polyglycerol polyricinoleate (6 g/100 g). The simple (O/WSE) coarse emulsion was obtained by gentle addition of the lipid phase (32 g/100 g) to the WSE (68 g/100 g) in a Thermomix food processor (Vorwerk, Wuppertal, Germany) at setting 3 and 37 °C. Thereafter, this was passed twice through a two-stage high-pressure homogenizer (Panda Plus 2000, GEA NiroSoavi, Parma, Italy) at 15/3 MPa and the SE was collected in a beaker immersed in ice. Sample aliquots were transferred to screw-capped tubes and immediately stored at 4 °C.
A two-step procedure was used to prepare stable DEs, as reported elsewhere with minor modifications (Cofrades et al. 2013). The inner (W1) phase consisted of a 5.84 g/L aqueous solution of NaCl plus 375 mg/100 mL HTy. The outer (W2) phase was prepared by dispersing 5.84 g/L NaCl in distilled water at room temperature until fully dissolved. Thereafter, 0.04 g/100 mL sodium azide was added to the outer phase to prevent microbial growth and allow us to study their stability. The lipid phase (O) used was the same as in the SE.
Primary coarse emulsion (W1/O) was prepared by gentle addition of the inner (W1) aqueous phase (20 g/100 g) to the lipid phase (80 g/100 g) in the Thermomix food processor at 37 °C for 15 min, setting 6. This primary coarse emulsion was passed twice through a two-stage high pressure homogenizer at 55/7 MPa (Panda Plus 2000) and collected on ice. The resulting primary fine emulsion (W1/O) was immediately used for preparation of the DE by gradual (40 g/100 g) addition to the W2 (60 g/100 g) in the Thermomix food processor at 37 °C, setting 3. The resulting coarse W1/O/W2 emulsions were passed twice through a two-stage high pressure homogenizer (Panda Plus 2000) at 15/3 MPa to obtain the final DE, which was collected on ice. Sample aliquots were transferred to screw-capped tubes and immediately stored at 4 °C. Theoretically, the final DE contained 300 mg/kg HTy and 300.8 g/kg perilla oil. The pH of all SEs and DEs was in the range 6.8–7.0.
GDEs were prepared by mixing the DE with gelatine (4% addition with respect to the initial weight of the DE) using an overhead stirrer (AGV-8 Bunsen, Madrid, Spain) and heating at 40 °C for 15 min until the gelatine was completely solubilized. The system was then placed on ice and cooled down to 35–37 °C. At this temperature, transglutaminase (2%) was added gradually while stirring for an extra 2 min after complete homogenization. Sample aliquots were transferred to 50 mL capacity bottles, screw capped and immediately stored at 4 °C. The addition of gelatine and transglutaminase to the continuous phase of the DE caused the system to gel resulting in a structured (hydrogelled) emulsion which was solid at room temperature and thermally stable due to the formation of covalent bonds. The pH of the gels was measured after diluting the ground gel 1:10 with distilled water while stirring thoroughly. The pHs of all gels were between 7.4 and 7.5.
These systems were characterized in more detail in a prior study (Flaiz et al. 2016).
Determination of hydroxytyrosol (HTy)
In SE and DE, 1 mL of sample was mixed with 100 μL of 85% orthophosphoric acid. In the case of GDE, 5 g of sample was homogenized for 30 s with 10 ml of 15% trichloroacetic acid dissolved in methanol. Samples were then centrifuged at 2000g for 10 min and the supernatant was collected for analysis.
HTy was analysed using an Agilent 1200 liquid chromatographic system equipped with an autosampler, quaternary pump and diode-array detector. A 250 mm × 4.6 mm i.d., 5-μm particle size Nucleosil 120 RP-18 column (Teknokroma) was used, preceded by a ODS precolumn. Elution was performed at a flow rate of 1.0 mL/min, using a mixture of 1% (v/v) formic acid in deionized water (solvent A), acetonitrile (solvent B) and methanol (solvent C) as mobile phase. The solvent gradient changed from 95% A—2.5% B—2.5% C to 70% A—15% B—15% C in 25 min, and to 0% A—50% B—50% C in 5 min, to 95% A—2.5% B—2.5% C in 5 min, followed by 5 min maintenance. Chromatograms were obtained at 280 nm. Injection volume was 20 μL. For quantification of HTy, a standard was prepared in a range of concentrations from 0.5 to 300 mg/kg, producing a linear response.
Simulated “in vitro” digestion
After one day of storage at 4 °C the different types of samples (SE, DE and GDE) had been subjected to the full (oral, gastric and intestinal phases) simulated “in vitro” digestion method following the recommendations and fluid composition described by Minekus et al. (2014). Briefly, oral phase: 5 mL (SE and DE) or 5 g (GDE freshly ground to particle sizes of 2 mm or less) of sample was mixed with 3.5 mL of simulated salivary fluid electrolyte solution, 0.025 mL of 0.3 M calcium chloride (CaCl2) and 0.975 mL water to reach a 1:1 ratio between food and digestion fluids and a total volume of 10 mL. The bolus was incubated at 37 °C for 2 min in a shaking incubator (311DS Labnet, Edison, NJ, USA) set at 80 rpm. Gastric phase: 7.5 mL of simulated gastric fluid electrolyte solution was added, followed by 5µL of 0.3 M CaCl2. The pH was adjusted to 3 using HCl (2 M). Thereafter, freshly prepared porcine pepsin was added to achieve 2000 U mL−1 in the final digestion mixture. If needed, pH was readjusted, and the necessary amount of water was added to obtain a final ratio of oral bolus to simulated gastric fluid plus other added recipients of 1:1 (v/v). The sample was incubated at 37 °C for 2 h with continuous agitation at 80 rpm. Intestinal phase: The gastric chyme (20 mL) was mixed with 11 mL of simulated intestinal fluid electrolyte solution and 0.04 mL CaCl2 (0.3 M), after adding bile extract (10 mM dissolved in 2.5 mL H2O plus 0.13 mL of 2 M NaOH). Thereafter, HCl (2 M) was added to bring pH up to 7. Freshly prepared pancreatin (lipase activity: 100 U/mL of the final mixture) and the necessary amount of water were added to obtain a final ratio of gastric bolus to simulated intestinal fluid plus other added recipients of 1:1 (v/v). A pH–stat (TitroMatic 1S, Crison, Alella, Spain) device was used to maintain pH 7 by adding NaOH (0.05 M), while this was incubated at 37 °C for 2 h with continuous agitation.
With respect to the analyses described below, it is important to note that sampling was conducted under continuous agitation in order to maximize their homogeneity.
Loss of HTy during “in vitro” digestion
The HTy content was determined in the sample before “in vitro” digestion, after the oral phase, the gastric phase, 1 h intestinal phase and at the end of the intestinal phase. The loss of HTy is expressed as the mass fraction percentage of the initial content. Bioaccessibility is defined as the fraction of a compound that is released from its matrix in the gastrointestinal tract and thus becomes available for intestinal absorption (Minekus et al. 2014).
Antioxidant capacity during “in vitro” digestion
The reducing capacity of samples during “in vitro” digestion was determined using the FRAP assay (Pulido et al. 2000) setup, by automated analysis in a plate reader. The reducing power of samples was measured by the increase in absorbance at 595 nm of the complex tripyridyltriazine/Fe(II) in the presence of reducing agents. 10 μL of either trolox or test sample or water as blank were added to a 96-well microplate followed by addition of 30 μL of distilled water and 200 μL of 0.3 M acetate buffer, pH 3.6. The microplate reader (Bio-Tek, Winooski, VT, USA) was programmed to record every 90 s for 30 min at 595 nm after addition of 60 μL of FRAP reagent, which was freshly prepared by mixing 2.5 mL of a 10 mM TPTZ solution in 40 mM HCl plus 2.5 mL of 20 mM FeCl3.H2O and 7.5 mL of 0.3 M acetate buffer, pH 3.6. The temperature was maintained at 37 °C. Each value is the average of four determinations. Trolox was used as a standard and results were expressed as μM of Trolox equivalents.
Extent of lipolysis during “in vitro” digestion
To gain a better understanding of the effect of the delivery system on the lipolysis extent, two types of control samples were included in this experiment: the oil and the gel (GC) controls. The latter control (GC) consisted of a mixture of the oil itself and the gelatine gel containing the same ingredients of the GDE except the lipid phase. This was used to check if gelatine and other compounds used in the composition of the double emulsion have per se any effect on the oil lipolysis in comparison to GDE.
The lipid composition was determined in the oil, SE, DE, GC and GDE samples before “in vitro” digestion, after 1 h intestinal phase and at the end of the digestion (after 2 h intestinal phase). In all cases, the same amount of fat was used for “in vitro” digestions for better comparison.
Fat extraction
Briefly, samples were mixed with chloroform/methanol (1:1, v/v) then washed again with chloroform. The organic phase was finally purified using a chloroform/methanol/0.58% NaCl solution mix (vol:vol, 3/48/47) and dehydrated by filtration through anhydrous sodium sulphate. The solvent was evaporated to dryness in a water bath at 40–50 °C under nitrogen atmosphere.
High-performance size-exclusion liquid chromatography (HPSEC)
HPSEC was performed to elucidate lipid composition as described by Dobarganes et al. (2000). Briefly, the previously isolated samples (10–15 mg mL−1 tetrahydrofuran) were applied in a high-performance liquid chromatograph (HPLC) (Agilent 1100 series, Madrid, Spain) with a 20 μL sample loop. A refractive index detector (Agilent Technologies 1260 infinity, Madrid, Spain) and two serially-connected 300 mm × 7.5 mm i.d. (5 μm particle size), 0.01 and 0.05 μm, PL gel columns (Agilent, Bellefonte, PA, USA) were operated at 40 °C. HPLC grade tetrahydrofuran was used as the mobile phase at a flow of 1 mL/min. Triacylglycerides (TAG), diacylglycerides (DAG), monoacylglycerides (MAG) and free fatty acids (FFA) were quantified at the initial time and in the intestinal phase (t = 1 or 2 h). Digestibility of samples was also calculated following the formula:
Statistical analysis
All experiments were carried out in triplicate. Data were analysed using the SPSS 21.0 statistical package. A two-way analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) post hoc test were used to ascertain differences in HTy content and FRAP values during the different stages of the simulated “in vitro” digestion and different delivery systems. Pearson’s correlation was conducted to assess the relationship between HTy content and FRAP values. As for the lipid hydrolysis values, the repeated measures test followed by the LSD post hoc test was used for statistical comparisons between samples in the course of “in vitro” digestion. A paired Student t test was used for comparisons between digestion times (0–1–2 h). Spearman correlations were performed to assess relationships between the degree of oil hydrolysis and the losses of antioxidant capacity by the systems. A p ≤ 0.05 was deemed significant in all calculations.
Results and discussion
HTy content and antioxidant activity changes during “in vitro” digestion
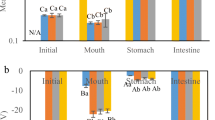
The HTy contents of SE, DE and GDE immediately after preparation (299, 298 and 292 mg/kg respectively) coincided closely with the amount of HTy added (300 mg/kg). Additionally, there was some loss of HTy during storage (1 day) of these emulsion systems prior to exposure to “in vitro” digestion (Table 1). This loss was greater in GDE and can be attributed to an increasing interfacial region percentage (McClements and Decker 2000) produced by the higher degree of compartmentalization and preparation conditions. Despite that, the oxidation levels found in these systems before digestion (1.4 ± 0.5, 1.3 ± 0.6 and 7.3 ± 0.7 nmol malondialdehyde/g in SE, DE and GDE respectively) were very low (Flaiz et al. 2016).
There were no changes in HTy content after oral digestion in any of the systems. In previous work, the amount of HTy in the same emulsions was found to decrease over the course of storage at 4 °C (as low as 163 mg/kg in GDE after 22 days), whereas the oxidation status remained unchanged up to 30 days (Flaiz et al. 2016). Therefore, as HTy loss occurred before the onset of oxidation it is reasonable to infer that the oxidation status of these systems remained constant during the oral step as this only takes 2 min. However, gastric and intestinal phases contributed to the partial degradation of HTy in the course of digestion, which may have a bearing on the increased oxidation found in the stomach in short-term digestibility studies (Olivero-David et al. 2011). Despite the foregoing findings and the well-known protective effect of HTy, further studies are necessary to determine if this antioxidant is also capable of preventing oxidation during digestion as in such systems when kept in cold storage.
It is worth noting that nutrient bioaccessibility may be affected by droplet size (Marze 2013). However, the emulsification conditions in this experiment were similar, and hence the emulsion systems presented similar distributions with no differences in their volume-weighted mean diameter (d43) (Flaiz et al. 2016). In this regard, they are therefore comparable and the existing differences can be attributed to other factors. The total effect of “in vitro” gastric and intestinal phases on HTy stability was similar in SE and DE, reducing HTy content after gastric digestion by 11 and 14% in SE and DE respectively, and after 2 h of intestinal digestion by 10 and 7% in SE and DE respectively, giving a total HTy loss of 21% in both emulsions. However, GDE emulsion, with the lowest initial content but with a similar final HTy concentration (Table 1), proved to offer better protection for HTy than the other systems (SE and DE), as there was limited HTy loss from the beginning till the end of the gastric phase (4%) and less degradation after 2 h of intestinal digestion (7%). Therefore, the embedding of DE oil droplets in the protein gel may afford better protection against HTy than in DE and SE. It may therefore be assumed that the HTy is more bioaccessible when this bioactive compound is incorporated in the inner phase of a GDE. It seems reasonable to assume that pepsin and pancreatin require more time to fully destroy the gel network formed than in the other systems. Consequently, the exposure of the GDE encapsulated HTy in gastric and intestinal fluids is delayed, which explains the lower degradation rate. During digestion, proteins such as gelatine produce different peptides and amino acids with antioxidant properties (Gómez-Guillén et al. 2011) and thus additionally help increase HTy stability with respect to SE and DE.
These results partly agree with an evaluation of the “in vitro” digestive stability of HTy alone by Pereira-Caro et al. (2012), who observed high stability after gastric and intestinal digestions, resulting in a total apparent loss of 20.3%. The fact that this is in line with the total HTy loss of 21% when incorporated in both DE and SE emulsions but significantly higher than the HTy loss of 12% observed in GDE system confirms the positive effect of the gel on HTy stability. It is worth noting that no 3,4-dihydroxyphenyl acetic acid (DOPAC) derived from HTy oxidation was detected in the present study, contrary to Pereira-Caro et al. (2012), where part of the HTy recovered was converted to DOPAC (22%). Therefore, the absence of DOPAC suggests that there is a potential benefit in using these systems (and perhaps in other complex food matrices) to deliver bioactive HTy when compared with direct intake.
Regarding the antioxidant activity of the emulsions containing HTy, oral digestion did not modify the reducing capacity of samples (Table 2). Conversely, “in vitro” gastric and intestinal phases significantly reduced the antioxidant activity of the emulsions (SE, DE and GDE), consistent with the reported losses of HTy during these digestion steps. Therefore, the antioxidant activity of digested emulsions is mainly affected by the HTy content, which explains its high correlation (Pearson’s r = 0.82, p < 0.001, n = 45). This suggests that the benefits of the “in vivo” antioxidant derived from HTy intake can be modulated by preserving this bioactive compound during digestion. In this connection, the use of GDE may offer some advantages, as this compound was found to be more stable than the SE or the DE, which registered similar losses upon digestion (Table 1).
Extent of lipolysis during “in vitro” digestion
The impact of the three systems (SE, DE and GDE) on the extent of lipolysis was evaluated by HPSEC analysis of the composition of digestion products. The evaluation included the disappearance of TAG and the formation of hydrolytic compounds (DAG, and the sum of MAG and FFA). This analytical method is helpful in understanding “in vitro” and “in vivo” lipid digestion (Sánchez-Muniz et al. 2011), as it helps to quantify the degree of hydrolysis and the amount of different hydrolytic compounds released (Arroyo et al. 1996; Dobarganes et al. 2000).
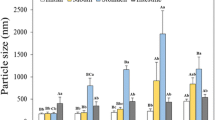
Table 3 shows the acyl composition of different samples, initially (oil) and after 1 or 2 h of intestinal digestion. During the first hour, all samples showed pronounced changes in their composition, leading to a considerable decrease in TAG concentration and an increase in the presence of hydrolytic compounds. However, the TAG concentration was significantly higher in GDE than in DE. DAG concentration was significantly lower in GDE than in SE, DE and GC, suggesting a different degree of GDE digestion from the other systems. The inclusion of GC in this study allows us to tell if the new products used to prepare the GDE affected the digestion. The potential effects of structural changes and interactions with the oil are excluded in GC samples because the oil was added directly to the digestion solutions, separately from the gel matrix. Regarding the extent of lipid hydrolysis, the digestion of GC sample did not differ from that of the oil, demonstrating the lack of effect of gel ingredients (gelatine and transglutaminase) on digestion enzymes. The changes in fat composition over the course of digestion are in line with those reported by Zhang et al. (2015). The hydrolytic effect on TAG was lower during the second hour, probably due to a negative feed-back control. “In vivo”, FFA and MAG are formed and absorbed sequentially, whereas in the “in vitro” approach the digestion products are accumulated, reducing the activity of the lipase enzyme (Lindblad 1988).
The digestion of GDE sample was significantly reduced with respect to SE and DE. In fact, GDE was the only sample that failed to reach 70% digestibility after 2 h (Table 3). The present results show that after 2 h digestion of GDE samples, DAG and MAG + FFA levels were similar to those in the other samples after just 1 h digestion, clearly supporting the hypothesis that gelation in the GDE system significantly delayed the lipase action observed in the other samples. Wang et al. (2013) also proved that the gelation of simple emulsions can delay lipid digestion. A detailed comparison of the hydrolysis occurring over 1–2 h in the GDE and the other systems shows that DAG formation was highest and that of MAG + FFA lowest, suggesting that the last step in the TAG → DAG → MAG +FFA sequence was delayed (Fig. 1).

Changes in fat composition of oil, simple (SE) and double (DE) emulsions, gelled double emulsion (GDE) and gel blank (GDEB) during the second hours of “in vitro” digestion. ANOVA, followed by the LSD post hoc test was used for statistical comparisons between the samples. Different letters (a, b) designate significant differences (p ≤ 0.05)
Previous studies have suggested that the bioavailability of lipid components depends on their physicochemical properties as well as on the structural organization of the lipids in the matrix (Porter et al. 2007). The slowing-down observed in “in vitro” digestion of GDE sample is therefore presumably due to its gelation structure. Although the proteins were partially digested in the stomach and intestinal phases, oil droplets, being less accessible to bile and lipase activity, probably remained in the gel matrix, causing a delay in digestion.
Non-significant differences were found between SE and DE systems and the tested oil after 1 or 2 h hydrolysis, suggesting that the systems used were unable to modify the oil digestion. This suggests that the non-gelled double systems were severely disrupted during “in vitro” digestion, thus affording digestive enzymes free access to the system oil.
At present we have no clear evidence as to the exact phase or moment when disruption of the emulsion occurs; however, the gastric phase and the 1st hour intestinal period seem likely candidates. In fact protein digestion occurs at both levels; thus, casein should be partially or totally disaggregated by the gastric and intestinal enzymes, permitting the oil to flow to the digestion system and/or the enzymes to penetrate the “system-droplets”. In the case of GDE, gelation seems to delay access of enzymes to the inner oil-phase. This hypothesis needs to be corroborated by electron microscopy in future studies. A potential negative effect of the double gel-compounds on digestions (e.g. inhibition of enzyme action, oil emulsion disturbance) cannot be entertained as the GC system displayed a similar degree of digestion to the other systems and the oil itself. What the present results confirm, then, is that although GDE samples were formed from DE, the changes in structure and physical state significantly slowed down lipid digestion. This effect should be taken into account when designing a novel food that incorporates emulsion-filled gels.
This study suggests that the structure of a lipid system could limit the degree of digestion. The digestion model used simulated the entire process, involving the mouth, stomach and small intestine fluids, and thus is arguably a good approach. Further “in vivo” feeding studies need to be conducted to check present results.
As noted earlier, it has been reported that significant lipid oxidation occurs during digestion (Olivero-David et al. 2011). To test such an effect in our experimental conditions, the losses of HTy and antioxidant capacity during the 1st and 2nd hours were correlated with the digestibility of oil. FRAP and HTY losses after 1 and 2 h correlated significantly (Spearman’s r = 0.733, p = 0.025, n-9; r = 0.867, p = 0.002, n = 9 respectively). In addition, a correlation was established between FRAP and HTy losses and digestibility in which the greater the digestibility, the greater were antioxidant losses. Thus, after 1 h losses correlated with oil digestibility (FRAP: Spearman’s r = 0.833, p = 0.005, n = 9; HTy: r = 0.700, p = 0.036, n = 9). And after 2 h the correlations were stronger (FRAP: Spearman’s r = 0.900, p = 0.001, n = 9; HTy: r = 0.767, p = 0.016, n = 9), supporting previous findings by our group (Olivero-David et al. 2011).
Conclusion
Among the studied delivery systems, GDE was found to protect HTy during digestion more efficiently than DE and SE. Conversely, DE gelation may be disadvantageous in a system intended to deliver n-3 PUFAs, as the entrapment of emulsion droplets in the gel matrix impedes oil digestibility. However, the decrease in the extent of lipolysis may be an additional benefit when trying to produce low-calorie foods by reducing fatty acid uptake. Therefore, the particular structuring of GDE offers interesting opportunities for the protection and delivery of certain labile compounds as well as for the development of soft-solid ingredients that can be used in certain food applications.
Work now continues on the physical and oxidative stability of these n-3 PUFA enriched (perilla oil) and HTy emulsion-based delivery systems (SEs, DEs and GDEs).
References
Ahn J, Grun IU, Fernando LN (2002) Antioxidant properties of natural plant extracts containing polyphenolic compounds in cooked ground beef. J Food Sci 67:1364–1369. doi:10.1111/j.1365-2621.2002.tb10290.x
Arroyo R, Sánchez-Muniz FJ, Cuesta C, Burguillo FJ, Sánchez-Montero JM (1996) Hydrolysis of used frying palm olein and sunflower oil catalyzed by porcine pancreatic lipase. Lipids 31:1133–1139. doi:10.1007/bf02524287
Cofrades S, Antoniou I, Solas MT, Herrero AM, Jiménez-Colmenero F (2013) Preparation and impact of multiple (water-in-oil-in-water) emulsions in meat systems. Food Chem 141:338–346. doi:10.1016/j.foodchem.2013.02.097
De Leonardis A, Aretini A, Alfano G, Macciola V, Ranalli G (2008) Isolation of a hydroxytyrosol-rich extract from olive leaves (Olea Europaea L.) and evaluation of its antioxidant properties and bioactivity. Eur Food Res Technol 226:653–659. doi:10.1007/s00217-007-0574-3
DeJong S, Lanari MC (2009) Extracts of olive polyphenols improve lipid stability in cooked beef and pork: contribution of individual phenolics to the antioxidant activity of the extract. Food Chem 116:892–897. doi:10.1016/j.foodchem.2009.03.053
Dobarganes MC, Velasco J, Dieffenbacher A (2000) Determination of polar compounds, polymerized and oxidized triacylglycerols, and diacylglycerols in oils and fats: results of collaborative studies and the standardized method (Technical report). Pure Appl Chem 72:1563. doi:10.1351/pac200072081563
Fki I, Allouche N, Sayadi S (2005) The use of polyphenolic extract, purified hydroxytyrosol and 3,4-dihydroxyphenyl acetic acid from olive mill wastewater for the stabilization of refined oils: a potential alternative to synthetic antioxidants. Food Chem 93:197–204. doi:10.1016/j.foodchem.2004.09.014
Flaiz L, Freire M, Cofrades S, Mateos R, Weiss J, Jimenez-Colmenero F, Bou R (2016) Comparison of simple, double and gelled double emulsions as hydroxytyrosol and n-3 fatty acid delivery systems. Food Chem 213:49–57. doi:10.1016/j.foodchem.2016.06.005
Garti N (1997) Double emulsions—scope, limitations and new achievements. Colloid Surf A 123–124:233–246. doi:10.1016/S0927-7757(96)03809-5
Gómez-Guillén MC, Giménez B, Lopez-Caballero ME, Montero MP (2011) Functional and bioactive properties of collagen and gelatin from alternative sources: a review. Food Hydrocoll 25:1813–1827. doi:10.1016/j.foodhyd.2011.02.007
Gonzalez-Santiago M et al (2006) One-month administration of hydroxytyrosol, a phenolic antioxidant present in olive oil, to hyperlipemic rabbits improves blood lipid profile, antioxidant status and reduces atherosclerosis development. Atherosclerosis 188:35–42. doi:10.1016/j.atherosclerosis.2005.10.022
Guo Q, Ye A, Lad M, Dalgleish D, Singh H (2014) Effect of gel structure on the gastric digestion of whey protein emulsion gels. Soft Matter 10:1214–1223. doi:10.1039/c3sm52758a
Jiménez-Colmenero F (2013) Potential applications of multiple emulsions in the development of healthy and functional foods. Food Res Int 52:64–74. doi:10.1016/j.foodres.2013.02.040
Jiménez-Colmenero F, Salcedo-Sandoval L, Bou R, Cofrades S, Herrero AM, Ruiz-Capillas C (2015) Novel applications of oil-structuring methods as a strategy to improve the fat content of meat products. Trends Food Sci Technol 44:177–188
Jo H, Kim M, Lee J, Kim H, Song YO (2013) Anti-atherogenic properties of emulsified perilla oil (EPO) in apo E KO mice and plasma lipid lowering effects of rice porridge containing EPO in healthy young adults. Food Sci Biotechnol 22:79–85. doi:10.1007/s10068-013-0011-2
Li J, Cheng Y, Tatsumi E, Saito M, Yin L (2014) The use of W/O/W controlled-release coagulants to improve the quality of bittern-solidified tofu. Food Hydrocoll 35:627–635. doi:10.1016/j.foodhyd.2013.08.002
Lindblad BS (1988) Perinatal nutrition. Academic Press Inc., Orlando
Lobato-Calleros C, Sosa-Pérez A, Rodríguez-Tafoya J, Sandoval-Castilla O, Pérez-Alonso C, Vernon-Carter EJ (2008) Structural and textural characteristics of reduced-fat cheese-like products made from W1/O/W2 emulsions and skim milk. LWT-Food Sci Technol 41:1847–1856. doi:10.1016/j.lwt.2008.01.006
Mao L, Miao S (2015) Structuring food emulsions to improve nutrient delivery during digestion. Food Eng Rev 7:439–451. doi:10.1007/s12393-015-9108-0
Marze S (2013) Bioaccessibility of nutrients and micronutrients from dispersed food systems: impact of the multiscale bulk and interfacial structures. Crit Rev Food Sci Nutr 53:76–108. doi:10.1080/10408398.2010.525331
McClements DJ, Decker EA (2000) Lipid oxidation in oil-in-water emulsions: impact of molecular environment on chemical reactions in heterogeneous food systems. J Food Sci 65:1270–1282. doi:10.1111/j.1365-2621.2000.tb10596.x
McClements DJ, Li Y (2010) Structured emulsion-based delivery systems: controlling the digestion and release of lipophilic food components. Adv Colloid Interf Sci 159:213–228. doi:10.1016/j.cis.2010.06.010
Minekus M et al (2014) A standardised static in vitro digestion method suitable for food—an international consensus. Food Funct 5:1113–1124
Olivero-David R et al (2011) Effect of thermally oxidized oil and fasting status on the short-term digestibility of ketolinoleic acids and total oxidized fatty acids in rats. J Agric Food Chem 59:4684–4691. doi:10.1021/jf1048063
Pazos M, Alonso A, Sanchez I, Medina I (2008) Hydroxytyrosol prevents oxidative deterioration in foodstuffs rich in fish lipids. J Agric Food Chem 56:3334–3340
Pereira-Caro G, Sarriá B, Madrona A, Espartero JL, Escuderos ME, Bravo L, Mateos R (2012) Digestive stability of hydroxytyrosol, hydroxytyrosyl acetate and alkyl hydroxytyrosyl ethers. Int J Food Sci Nutr 63:703–707. doi:10.3109/09637486.2011.652943
Porter CJH, Trevaskis NL, Charman WN (2007) Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–248
Pulido R, Bravo L, Saura-Calixto F (2000) Antioxidant activity of dietary polyphenols as determined by a modified ferric reducing/antioxidant power assay. J Agric Food Chem 48:3396–3402
Sánchez-Muniz FJ, Benedí J, Bastida S, Olivero-David R, Gonzalez-Muñoz MJ (2011) Enzymes and thermally oxidized oils and fats. In: Boskou D, Elmadfa I (eds) Frying of foods: oxidation, nutrient and non-nutrient antioxidants, biologically active compounds and high temperatures. CRC Press, Taylor and Francis group LLC, Boca Raton, pp 105–155
Wang Z, Neves MA, Kobayashi I, Uemura K, Nakajima M (2013) Preparation, characterization, and in vitro gastrointestinal digestibility of oil-in-water emulsion-agar gels. Biosci Biotechnol Biochem 77:467–474. doi:10.1271/bbb.120659
Weiss J, Scherze I, Muschiolik G (2005) Polysaccharide gel with multiple emulsion. Food Hydrocoll 19:605–615. doi:10.1016/j.foohyd.2004.10.023
WHO/FAO Expert Consultation (2003) Diet, nutrition and the prevention of chronic diseases. World Health Organization. http://www.who.int/dietphysicalactivity/publications/trs916/download/en/. Accessed 15 Feb 2017
Wooster TJ, Day L, Xu M, Golding M, Oiseth S, Keogh J, Clifton P (2014) Impact of different biopolymer networks on the digestion of gastric structured emulsions. Food Hydrocoll 36:102–114. doi:10.1016/j.foodhyd.2013.09.009
Yang M, Liu F, Tang C-H (2013) Properties and microstructure of transglutaminase-set soy protein-stabilized emulsion gels. Food Res Int 52:409–418. doi:10.1016/j.foodres.2011.11.012
Zetzl AK, Marangoni AG, Barbut S (2012) Mechanical properties of ethylcellulose oleogels and their potential for saturated fat reduction in frankfurters. Food Funct 3:327–337. doi:10.1039/C2FO10202A
Zhang R, Zhang Z, Zhang H, Decker EA, McClements DJ (2015) Influence of emulsifier type on gastrointestinal fate of oil-in-water emulsions containing anionic dietary fiber (pectin). Food Hydrocoll 45:175–185. doi:10.1016/j.foodhyd.2014.11.020
Acknowledgements
R. B. thanks the Spanish Ministry of Economy and Competitiveness for his contract through the Ramón y Cajal Program and A. G. for the BES-2012-054752 (FPI) pre-doctoral fellowship. This research was supported under Project AGL2011-29644-C02-01, AGL2014-53207-C2-1-R and AGL2014-53207-C2-2-R of the Plan Nacional de Investigación Cientifica, Desarrollo e Innovación Tecnologica (I + D+I), Ministerio de Economia y Competitividad, and Intramural project CSIC: 201470E056.
Author information
Authors and Affiliations
Corresponding author
Additional information
Susana Cofrades and Ricard Bou contributed equally to this work.
Rights and permissions
About this article

Cite this article
Cofrades, S., Bou, R., Flaiz, L. et al. Bioaccessibility of hydroxytyrosol and n-3 fatty acids as affected by the delivery system: simple, double and gelled double emulsions. J Food Sci Technol 54, 1785–1793 (2017). https://doi.org/10.1007/s13197-017-2604-x
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-017-2604-x