
Abstract
Angiomatoid fibrous histiocytoma (AFH) is a rare soft tissue tumor, common in children and young adults, often misdiagnosed as either reactive or malignant. This study aims to highlight the clinico-radiological and pathological features of this uncommon entity. Eighteen cases of AFH diagnosed over a period of 20 years were analyzed and correlated with clinical data. The tumor had a wide age distribution with an M:F ratio of 3.5:1. Though swelling was the common clinical presentation, a subset of patients had constitutional symptoms like fever, weight loss, loss of appetite, and anemia. One patient was referred with deranged aPTT and hypergammaglobulinemia. The radiological features were also myriad ranging from infection in a discharging soft tissue swelling to lymphoma (in cases with nodal involvement) to sarcoma (angiosarcoma and telangiectatic osteosarcoma). Sites of occurrence were soft tissue of the upper limb, lower limb, head and neck, bone, and lung. Intra-operatively, these tumors run the risk of bleeding and may require pre-op embolization and support by blood and blood products. Wide local excision was the primary treatment offered. Macroscopically, the average size was 3.5 cm; the cut surface was nodular, cystic with hemorrhage, and gray–white to brownish–yellow in color. Microscopically, the tumors were circumscribed with a fibrous pseudo-capsule and showed mildly pleomorphic spindle cells insheets and fascicles, in a sclerotic to myxoid stroma. A peripheral cuff of lymphoplasmacytic cells was present in all cases. Atypical histological features observed were moderate nuclear pleomorphism, frequent mitosis, solid variant, and myxoid stroma. Immunohistochemically, they were most often positive for desmin, CD68, and EMA. Interestingly, about 55.5% cases had lymphadenopathy of which three showed metastatic tumor. Three of our cases harbored EWSR1 re-arrangement, proven by FISH. Follow up details were available for six patients and none showed recurrence. In conclusion, we emphasize the importance of improved recognition of this rare yet morphologically distinct neoplasm, with varied clinico-radiological presentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiomatoid fibrous histiocytoma (AFH) is a neoplasm of intermediate (rarely metastasizing) malignant potential, predominantly occurring in the superficial location of extremities in children and young adults. In its classic histologic form, comprising four key morphologic components: (1) nodules of mildly pleomorphic spindle to epithelioid cells, (2) blood-filled pseudo-angiomatous spaces, (3) thick fibrous pseudo-capsule, and (4) peripheral lymphoplasmacytic cuff, it is a fairly straightforward diagnosis, though it lacks specific immunohistochemical markers [1]. It is diagnostically challenging in small biopsies, when it presents at unusual sites, in older age groups, with variant histology, and in the absence of either one of its classic morphologic components. Though swelling is the most common symptom, its association with systemic symptoms like pyrexia, anemia, malaise, and weight loss confuses the clinical picture raising the possibility of infection to lymphoma, the latter due to its frequent occurrence in areas where lymph nodes are normally found. In addition, the absence of any specific radiologic features further contributes to the conundrum of this entity. The genetic abnormality detected in these tumors are a fusion of EWSR1 gene to either CREB1(in > 90%) or ATF1 [2, 3]. Cases with FUS-ATF1 have also been reported. In this case series, we aim to describe the varied clinical presentation, radiological features, histomorphology, immunohistochemical findings, and the differential diagnosis entertained before the diagnosis was clinched. The management and follow up of these patients, where available, have also been provided. Furthermore, three of our cases were confirmed by FISH for EWSR1 gene; the findings of which have been illustrated.
Material and Methods
The hospital information system (HIS) was used to retrieve 18 cases of AFH diagnosed over a period of 20 years (January 2002–December 2022). The diagnostic material was a combination of small biopsies, resection specimens, and patients referred here with slides/blocks from other hospitals.
Clinical information was obtained from the electronic medical records of the HIS. The following parameters were recorded: gender and age of the patients, site of the lesion, clinical presentation, radiology, type of treatment, and follow up where available.
Hematoxylin and eosin-stained slides/blocks of all 18 patients were retrieved from the archives. The gross findings that were recorded from the pathological reports included location, gross morphology including size, circumscription, nodularity, cystic change, intra-lesional hemorrhage, and cut surface. On microscopic examination, the features recorded were fibrous pseudo-capsule, nodularity, histological pattern, morphology of the cells—spindle/epithelioid, pleomorphism, mitotic activity (graded as frequent if > 5/10hpf) [4], pseudo-angiomatous spaces, stroma, and peripheral lympho-plasmacytic cuff including germinal center formation. The results of available immunohistochemical stains including desmin, EMA, CD68, CD99, vimentin, myogenin, Myo-D1, SOX10, MUC4 and CD34 were also recorded.
Molecular analysis for EWSR1 re-arrangement by FISH was available in three cases.
Results
The tumor had a wide age distribution (6 to 52 years) with M:F ratio of 3.5:1. The clinico-radiological and pathological features are enumerated in Table 1. Though swelling was the most common clinical presentation, a subset of patients (n = 4) had constitutional symptoms like fever, weight loss, loss of appetite, and anemia. The patient with lung mass presented with hemoptysis and shortness of breath. One patient was referred with deranged aPTT and was found to have hypergammaglobulinemia. Three patients presented with recurrent swelling and were operated on elsewhere with a diagnosis of hematoma in two of them. One patient presented with metastatic disease to lung and lymph nodes and was operated on multiple times elsewhere. Most of the cases occurred in the soft tissue of extremities (n = 8) followed by the head and neck (n = 3), and the trunk (n = 3). Of the non-somatic sites, three were in the bone and one was in the lung.
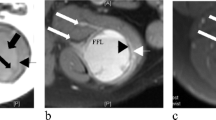
Macroscopically, the average size of the lesion was 3.9 cm (1.2 cm to 9.8 cm). Gross details were available for 15 cases. Almost all tumors were nodular, and cystic change was present in 10 of them, with evidence of hemorrhage in it (Fig. 1a). The cut surface was gray–white to brownish–yellow. The proximal humerus tumor involved the metaphysis and diaphysis, causing bone expansion with a soft brown cystic cut surface filled with hemorrhage (Fig. 1c). The lung tumor was endobronchial in a location with a firm gray–white cut surface and remarkably showed no cystic change.

a (gluteal region) and c (right humerus): Multinodular tumor with cystic change and hemorrhage. b MR images show a multi-lobulated lesion with blood-fluid levels (arrow) and thick septa in the subcutaneous plane of the left gluteal region. d MRI shows a large multi-lobulated cystic lesion with T2 hypointense solid areas and multiple blood-fluid levels in the proximal right humerus with focal transphyseal extension
Microscopically, the tumors were circumscribed (17/18) and nodular (15/18) with a fibrous pseudo-capsule (Fig. 2a, 16/18) and pseudo-angiomatous spaces (Fig. 2c, 15/18) and composed of mildly pleomorphic spindle-shaped cells in sheets and fascicles (Fig. 2d and e), set in a sclerotic to myxoid stroma. A peripheral cuff of lymphoplasmacytic cells was present in all cases (Fig. 2b) with some also displaying germinal centers (5/18). The atypical histological features observed were moderate nuclear pleomorphism (Fig. 2f, n = 7/18), frequent mitosis (n = 4/18), solid variant (3/18), and myxoid stroma (n = 3/18). Immunohistochemically, tumors were positive for desmin (n = 15/16), EMA (n = 9/11), CD68 (n = 8/11), CD99/MIC2 (n = 4/7), and vimentin (n = 5/6). Myogenin, MyoD1, SOX10, MUC4 and CD34 where available were negative. Figures 2g–i show desmin, EMA, and CD68 immunostains. SOX9 and ALK were available in two of our recent cases and found to be positive (Fig. 3). Of the 18 tumors, lymphadenopathy was found in 10 cases (55.5%). While 7 were reactive, 3 showed metastatic tumor, one of which was a patient who presented to us with nodal metastasis, with a history of multiple surgeries elsewhere. FISH was performed in three of eighteen cases. All the three demonstrated EWSR1 re-arrangement (100%) by FISH (Fig. 4).

a (H&E, 40 ×) and b (H&E, 100 ×): Circumscribed tumor with fibrous pseudo-capsule and peripheral lympho-plasmacytic cuff. c (H&E, 40 ×): Classic type with pseudo-angiomatous space. d (H&E, 100 ×): Sheets of histiocytoid cells. e (H&E, 400 ×): Epithelioid to histiocytoid tumor cells. f (H&E, 400 ×): Tumor cells with moderate nuclear atypia. g (40 ×): Desmin. h (200 ×): EMA. i (400 ×): CD68

a (H&E, 40 ×): AFH of right axilla with positivity for SOX9 (b 200 ×) and ALK (c 200 ×). d (H &E, 40 ×): AFH of left knee with positivity for SOX9 (e 200 ×) and ALK (f 200 ×)

a (H&E, 40 ×): Solid variant of AFH with peripheral lympho-plasmacytic cuff, right supraclavicular region. b (H&E, 200 ×): Fascicles of spindle cells in a myxoid stroma. c (200 ×): Diffuse EMA positivity. d EWSR1 FISH demonstrating split signals in the tumor cells
Wide local excision was the primary treatment offered with one patient receiving adjuvant chemotherapy and another receiving radiotherapy. Follow up details were available for six patients (1 year to 6 years). All of them had clinically enlarged regional lymph nodes (6/6). On histological examination, two were metastatic disease (2/6; 33.3%) while the other four were reactive. None of these patients had metastasis to the lung. All of the six patients were doing well with no evidence of recurrence, at the time of the last follow up.
Discussion
AFH is a rare soft tissue neoplasm, of uncertain differentiation, accounting for 0.3% of all soft tissue tumors. It was first described by Enzinger in 1979, as a distinctive tumor of adolescence and early adult life and called angiomatoid “malignant” fibrous histiocytoma [5]. Though the peak incidence is in the first two decades of life, the tumor has a wide age distribution, reflected in our case series as well. The mean age in our case series was 17.5 years, with the majority falling in the first two decades of life. We found a male predominance (~ 76%), comparable with literature [1, 4, 6,7,8]. The site of occurrence was predominantly in soft tissue of extremities with four cases noted in non-somatic sites: three in bone and one in lung. Though these tumors typically occur in the superficial soft tissues of the extremities, uncommonly they can occur outside somatic soft tissue, like the lung, mediastinum, vulva, retroperitoneum, and ovary [9].
All the patients presented with swelling as the main symptom, with a few also displaying constitutional symptoms like fever (n = 4), anemia (n = 2), loss of appetite (n = 1), and weight loss (n = 2). AFH is one of the translocation-positive rare soft tissue neoplasm of intermediate biology that has been linked to paraneoplastic syndromes with elevated levels of IL-6, suggested as being responsible for the aforementioned symptoms [10]. Polyclonal gammapathy has also been consistently reported in some of the patients [5, 6]. Rarer systemic symptoms include membranous nephropathy, migraine-like headache with aura, platelet storage pool deficiency, and bleeding disorder [10]. One of our patients presented with deranged aPTT, probably attributed to hypergammaglobulinemia, which resolved with the removal of the tumor. Three patients presented with recurrent swelling, operated elsewhere, and presented to us with a diagnosis of hematoma, the latter being a common misconception as these tumors can be predominantly blood-filled and cystic [1].
The imaging findings of AFH are non-specific. Magnetic resonance (MR) imaging is superior to radiographs and computed tomography that show a heterogeneous soft tissue mass, and possibly hint at cystic and enhancing components. Even in MR, the findings are non-specific; however soft tissue mass intra-lesional blood-filled cystic spaces with fluid–fluid levels, enhancing fibrous pseudo-capsule and hemosiderin deposition in the extremity of a child or adolescent should prompt the consideration of AFH in the differential [11]. Radiologically, it is often mistaken for a benign condition such as a hematoma or hemangioma [12]. The radiological details were available in 13 cases in our series. The tumors in soft tissue showed non-specific thickening/mass-forming lesions with cystic change (Fig. 1b). Radiologically, the differential diagnosis was myriad ranging from infection in a discharging soft tissue swelling to lymphoma to vascular tumors including angiosarcoma. Intra-osseous AFH shows expansile, lytic, blood-filled multiple large cystic spaces with fluid-filled levels and a pseudo-capsule. It can also be associated with an extensive extraosseous component [13]. While the fluid–fluid levels raise the differential of aneurysmal bone cyst (ABC), it can also be potentially misdiagnosed as malignancy (angiosarcoma and telangiectatic osteosarcoma). Of the three AFH of bone in our series, two were radiologically aggressive, and one of them was radiologically considered to be telangiectatic osteosarcoma/angiosarcoma (Fig. 1d).
Grossly, these tumors are small, firm, multinodular to multi-cystic masses, with a wide size range (0.7–12 cm) and a median size of 2 cm. The cut surface can also be solid with a yellowish–tan or white fleshy appearance [5, 6, 14]. The average size of the lesion in our series was 3.9 cm (1.2 to 9.8 cm). Gross details were available for 15 cases of which 66.6% showed cystic spaces filled with hemorrhage. The cut surface was gray–white to brownish–yellow, the latter reflecting the presence of hemosiderin deposits within the tumor.
Angiomatoid fibrous histiocytomas are circumscribed and lobulated tumors that show a wide morphologic spectrum. They are often surrounded by fibrous pseudo-capsules and the only constant finding is sheets and short fascicles of ovoid, epithelioid, or spindle cells with bland, vesicular nuclei that have a fibroblastic or histiocytoid appearance with moderate amounts of eosinophilic cytoplasm. Mitosis is usually infrequent, although atypical forms may be present. The tumors may show marked pleomorphism, which may be diffuse but nuclear atypia and increased mitosis are not associated with a worse clinical outcome. The characteristic blood-filled pseudo-angiomatous spaces are due to intra-lesional hemorrhage, lined by flattened neoplastic cells and not endothelial cells. This leads to hemosiderin deposition which can be a prominent feature. Upto 80% show peripheral dense lymphoplasmacytic cuff, with or without germinal center formation.Approximately one-third show completely solid histology without pseudo-angiomatous spaces. Some cases lack any inflammatory component. The stroma may be myxoid to sclerotic or desmoplastic-like. Rare findings include tumoral giant cells and reactive osteoclast-like giant cells within pseudo-angiomatous spaces. The other unusual morphologic features are nuclear grooving, clear cells, rhabdomyoblast-like cells, small blue round cells resembling undifferentiated round cell sarcoma, and pulmonary edema-like and reticular patterns of cells in myxoid stroma. Schwannoma-like features, including nuclear palisading and stroma containing prominently hyalinized vessels, have also been described [15]. However, there are no reliable histopathological parameters that predict clinical behavior [1, 14]. Our series showed that the tumors were circumscribed (94.4%), nodular (83.3%), with fibrous pseudo-capsule (88.8%), pseudo-angiomatous spaces(83.3%), with spindle-shaped (100%) to epithelioid (23%) cells, with mild nuclear pleomorphism, in sheets (100%) and fascicles (23%), set in a sclerotic (77%) to myxoid (23%) stroma. A peripheral cuff of lymphoplasmacytic cells was present in all cases except one (Fig. 2b, 94%) with some also displaying germinal centers (5/18; 27.7%). The atypical histological features observed were moderate nuclear pleomorphism (38.8%), frequent mitosis (22.2%), solid variant (16.6%), and myxoid stroma (16.6%). Excluding the patient who presented with nodal metastasis, concomitant lymphadenopathy was found in nine patients (52%), two of which showed metastatic tumor in the lymph nodes.
Immunohistochemically, these tumors show myoid differentiation [16] with approximately 50% of cases, immunoreactive for desmin, which may be focal or diffuse. Expression of smooth muscle actin, calponin, and rarely h-caldesmon has also been described. Epithelial membrane antigen, CD99, and CD68 are variably expressed, ranging from approximately 40 to 50% of lesions. They are consistently negative for skeletal muscle markers such as myogenin and MyoD1 and vascular endothelial markers such as CD31, CD34, and factor VIII-related antigens. CD35, S100 protein, cytokeratins, and lysozyme are also negative in these tumors [1, 14]. In our series, expression of markers was as follows: desmin (n = 15/16; 93.7%), EMA (n = 9/11; 81.9%), CD68 (n = 8/11; 72.7%), CD99/MIC2 (n = 4/7; 57.1%), and vimentin (n = 5/6; 83.3%). None of the tumors were positive for myogenin, MyoD1, SOX10, MUC4, and CD34. The utility of SOX9 in diagnosing AFH and overexpression of ALK (anaplastic lymphoma kinase) have been described only recently, and two of our cases were positive for SOX9 and ALK. SOX9 is a transcription factor essential for chondrogenesis and is predominantly used to delineate tumors of chondroid differentiation. Recently, the role of EWS in regulation of SOX9 expression has been reported. A recent study by Berklite et al. found SOX9 to be specific for AFH, though they found weak expression of SOX9 in a subset of aneurysmal dermatofibroma. However, it is absent in other myofibroblastic lesions, except calcifying aponeurotic fibroma [17]. ALK expression has been described in AFH [4, 18, 19], particularly with clones D5F3 and 5A4 antibodies, which are found to be highly sensitive. The underlying mechanism of ALK expression in AFH is unclear, and none of the ALK immunopositive AFH showed ALK re-arrangement or amplification [19].
Small biopsy of this entity can be particularly challenging. A 14-year-old boy in our series, with a 3-year history of right arm swelling and features of paraproteinemia, was radiologically diagnosed as ABC/telangiectatic osteosarcoma of the humerus. His first small biopsy showed features of chronic osteomyelitis. The repeat biopsy had a histiocytoid population of cells with a very focal and scant lymphoid infiltrate. This proved to be a difficult diagnostic dilemma, in view of his clinico-radiological profile. However, the morphology, in conjunction with his immunoprofile and also his rare presentation with hypergammaglobulinemia, helped arrive at the possibility of AFH. The resection showed classical features of AFH, in the rare intra-osseous location.
A 52-year-old gentleman with a right axillary mass had been variably diagnosed as hematoma/necrotic material/abscess/angiosarcoma on radiology. He had undergone multiple failed attempts elsewhere, to completely excise the mass, due to profuse intra-operative bleeding. He was pre-operatively embolized at our center, and debulking was done with multiple post-operative blood transfusions. Histopathology with immunohistochemistry was diagnostic of AFH, and few months later, he underwent a complete excision of the mass with pre-op embolization and transfusions.
Follow up details were available for 6/18 patients (1 year to 6 years), and all of them were doing well with no evidence of recurrence. Only one of our patients (5%) presented with metastatic disease to the lung, while three had metastasis to the lymph nodes (16.6%). AFH is mostly known to follow an indolent course, with local recurrence in nearly 15% of cases and metastasis in < 2–5% of cases, predominantly to locoregional lymph nodes and rarely to the lungs, liver, and brain. Local recurrence is related to infiltrative margins, invasion into deep fascia or muscle, and head and neck location. Extrasomatic AFHs appear to show higher recurrence rates compared with those of somatic soft tissue. Wide local excision with follow-up is the definitive treatment, although adjuvant radiation therapy or chemotherapy may be indicated for metastatic or unresectable disease [14].
The large majority of AFH is characterized by re-arrangement of EWSR1(demonstrated in all three of our cases with FISH), with one member of the CREB/ATF family of transcription factors, the EWSR1-CREB1 fusion gene being the most frequent molecular abnormality [2, 3]. EWSR1-ATF1 is more frequently associated with AFH of extra somatic soft tissues [1]. The type of gene fusion does not appear to correlate with clinical, histologic, or immunohistochemical differences. It is notable that EWSR1-CREB1 fusion is seen in two other neoplasms, clear cell sarcoma-like tumor of the gastrointestinal tract (CCSLGT) and primary pulmonary myxoid sarcoma (PPMS), while EWSR1-ATF1 fusion is found in clear cell sarcoma of soft tissue [14].
The differential diagnosis of AFH is wide, and it can often be misdiagnosed as reactive/infective conditions, benign neoplasms to malignancy. This could be due to the fact that the only consistent finding in AFH is sheets of ovoid to spindled cells, while the other characteristic features such as the fibrous capsule, pseudo-angiomatous spaces, and the lymphoplasmacytic infiltrate may be absent or not sampled in small biopsies. The reactive/infective conditions that it can mimic include organized hematoma in long-standing cases osteomyelitis to granulomatous inflammation. The benign neoplasms that come into the differential diagnosis due to its superficial location in the dermis and subcutis are aneurysmal benign fibrous histiocytoma and spindle cell hemangioma. The dense lymphoplasmacytic inflammation and expression of ALK give rise to the differential diagnosis of inflammatory myofibroblastic tumor (IMT). Malignant differentials like Kaposi sarcoma, angiosarcoma, and telangiectatic osteosarcoma also have been entertained. In the lymph node, the benign mimicker is palisaded myofibroblastoma and the malignant one is follicular dendritic cell sarcoma. The other sarcomas of the pediatric age group that need to be excluded include Ewing sarcoma and rhabdomyosarcoma, due to their immunopositivity for CD99 and desmin, respectively. A prominent lymphoid cuff with germinal centers could lead to diagnostic confusion as metastatic tumor deposits within lymph nodes. However, the absence of surrounding true nodal architecture, including subcapsular and medullary sinuses, and evenly distributed germinal centers could aid in the diagnosis. In addition, a history of primary cancer or the finding of a primary site after a clinico-radiological correlation would clear the confusion. The last of the tumors that need to be considered, a proportion of which also share EWSR1 re-arrangement, is soft tissue myoepithelial neoplasms. The latter, though positive for EMA like AFH, also shows co-expression of S100 and cytokeratins, unlike AFH [14]. Careful histological examination, rational interpretation of immunohistochemistry, and in difficult cases, demonstration of EWSR1 re-arrangement/fusion transcripts could differentiate the above entities from AFH.
Conclusion
AFH is a morphologically distinct neoplasm, with a wide spectrum of clinical and radiological presentations. It is imperative for the pathologist to consider it in the differential diagnosis of spindle cell tumors of children and adolescents who present with cystic lesions with fluid–fluid levels, hemorrhage and constitutional symptoms. The recommended immunohistochemistry, though not completely specific, is a panel of desmin, EMA, and CD68. This case series reiterates the need to be aware of the unique diagnostic histopathology of this entity, in spite of the varied clinico-radiological features, to avoid mismanagement.
References
Fletcher CDM, Unni KK, Merton F (2020) World Health Organization classification of tumours: pathology and genetics of tumours of soft tissue and bone, 5th edn. IARC Press, Lyon, pp 271–273
Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, Fletcher CD et al (2007) EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosom Cancer 46(12):1051–1060
Rossi S, Szuhai K, Ijszenga M, Tanke HJ, Zanatta L, Sciot R et al (2007) EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res Off J Am Assoc Cancer Res 13(24):7322–7328
Maqbool H, Bashir S, Hassan U, Hussain M, Mushtaq S, Ishtiaq S (2022) Angiomatoid fibrous histiocytoma: a tumor with uncertain behavior and various clinicopathological presentations. Cureus 14(9):e28985
Enzinger FM (1979) Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 44(6):2147–2157
Costa MJ, Weiss SW (1990) Angiomatoid malignant fibrous histiocytoma. A follow-up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol 14(12):1126–32
Rekhi B, Adamane S, Ghodke K, Desai S, Jambhekar NA (2016) Angiomatoid fibrous histiocytoma: clinicopathological spectrum of five cases, including EWSR1-CREB1 positive result in a single case. Indian J Pathol Microbiol 59(2):148–152
Saito K, Kobayashi E, Yoshida A, Araki Y, Kubota D, Tanzawa Y et al (2017) Angiomatoid fibrous histiocytoma: a series of seven cases including genetically confirmed aggressive cases and a literature review. BMC Musculoskelet Disord 18(1):31
Chen G, Folpe AL, Colby TV, Sittampalam K, Patey M, Chen MG, et al (2011) Angiomatoid fibrous histiocytoma: unusual sites and unusual morphology. Mod Pathol Off J U S Can Acad Pathol Inc 24(12):1560–70
Agaimy A (2019) Paraneoplastic disorders associated with miscellaneous neoplasms with focus on selected soft tissue and undifferentiated/rhabdoid malignancies. Semin Diagn Pathol 36(4):269–278
Yikilmaz A, Ngan BY, Navarro OM (2015) Imaging of childhood angiomatoid fibrous histiocytoma with pathological correlation. Pediatr Radiol 45(12):1796–1802
Bauer A, Jackson B, Marner E, Gilbertson-Dahdal D (2012) Angiomatoid fibrous histiocytoma: a case report and review of the literature. J Radiol Case Rep 6(11):8–15
Petrey WB, LeGallo RD, Fox MG, Gaskin CM (2011) Imaging characteristics of angiomatoid fibrous histiocytoma of bone. Skeletal Radiol 40(2):233–237
Thway K, Fisher C (2015) Angiomatoid fibrous histiocytoma: the current status of pathology and genetics. Arch Pathol Lab Med 139(5):674–682
Kao YC, Lan J, Tai HC, Li CF, Liu KW, Tsai JW et al (2014) Angiomatoid fibrous histiocytoma: clinicopathological and molecular characterisation with emphasis on variant histomorphology. J Clin Pathol 67(3):210–215
Fletcher CD (1991) Angiomatoid “malignant fibrous histiocytoma”: an immunohistochemical study indicative of myoid differentiation. Hum Pathol 22(6):563–568
Berklite L, John I, Ranganathan S, Parafioriti A, Alaggio R (2020) SOX9 Immunohistochemistry in the distinction of angiomatoid fibrous histiocytoma from histologic mimics: diagnostic utility and pitfalls. Appl Immunohistochem Mol Morphol AIMM 28(8):635–640
De Noon S, Fleming A, Singh M (2020) Angiomatoid fibrous histiocytoma with ALK expression in an unusual location and age group. Am J Dermatopathol 42(9):689–693
Cheah AL, Zou Y, Lanigan C, Billings SD, Rubin BP, Hornick JL et al (2019) ALK expression in angiomatoid fibrous histiocytoma: a potential diagnostic pitfall. Am J Surg Pathol 43(1):93–101
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Badrinath T. is currently working at Composite Hospital, Trivandrum.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ponmar, M., T., B., A., R. et al. Case Series of Angiomatoid Fibrous Histiocytoma (AFH)—A Clinico-Radiological and Pathological Conundrum. Indian J Surg Oncol (2024). https://doi.org/10.1007/s13193-024-02016-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13193-024-02016-7