
Abstract
Species are the fundamental category and the key to formulate conservation efforts. DNA and ecological niche modeling have become valuable tools for species delimitation. Wolf spiders include few web-living species, such as Aglaoctenus lagotis (Holmberg, 1876), a priority species for conservation in Uruguay. Behavioral and body coloration patterns of this species have allowed us to distinguish two groups (forms I and II). Here, we combine information from gene trees and multispecies coalescent analyses on mitochondrial (cox1, 12S, 16S + L1 + nad1) and nuclear (intron tif5A) DNA sequences, as well as from ecological niches comparisons, in order to clarify their taxonomic identity. We worked with localities in Uruguay and Argentina, including sympatric and allopatric areas. Gene trees were inferred with Maximum Likelihood, Bayesian, and statistical parsimony analyses. Molecular species delimitation analyses were conducted, and the species tree and divergence times were co-estimated. Characterization and comparison of the climatic requirements of both forms throughout annual and sexual periods were analyzed. Species delimitation and species tree analyses recovered three main lineages (Form I, Form IIa, and Form IIb). Form I is restricted to Uruguay and is closely related and sympatric with Form IIa. Form IIb is located in Argentina and in the Uruguayan west coast, generating a sympatric area of the three forms. Regarding to the sexual climatic niche, the three main lineages differ and do not overlap. Our results support the existence of more than one lineage within what is nowadays Aglaoctenus lagotis. Possible evolving processes explaining this scenario and the conservation consequences are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species are often claimed as the fundamental category in the biological world, the “units of evolution and biodiversity” (Pigliucci & Kaplan, 2006); that is why they are the first fundamental step of biological questions, including evolutionary processes and ecological systems, and are usually considered the key to formulate conservation efforts (Mace, 2004; see also Reydon, 2019). How to retain an objective criterion for species delimitation might require accepting that different disciplines in biology use different criteria (De Queiroz, 2007). Therefore, these days, it is more frequent to implement multidimensional studies that integrate more than one source of information (e.g., morphological, ecological, ethological, and genome-focused) (Network, 2012).
Incorporating information from DNA sequences for species delimitation and description has become a complementary, yet valuable tool for recognizing species entities, especially when traditional taxonomy has been unable to provide an accurate delimitation of species boundaries (Masta & Maddison, 2002; Montes de Oca et al., 2015; Padial et al., 2010; Sites & Marshall, 2004; Vogler & Monaghan, 2007). Also, studies that include more than a single gene have increased, especially in those species that seem to have diverged recently and which therefore have an increased risk of being incorrectly delimited as a result of incomplete lineage sorting or introgression (Hausdorf & Hennig, 2010; Satler et al., 2013). These kinds of studies (including more than a single gene) are now common in different groups (Pidancier et al., 2006) such as reptiles and amphibians (Ermakov et al., 2002; Hills, 2019), insects (Monaghan et al., 2005; Mullen et al., 2008), and spiders (Doménech et al., 2020; Kanesharatnam & Benjamin, 2019; Newton et al., 2020). Particularly in wolf spiders (i.e., family Lycosidae), these kinds of studies were performed in Pardosinae (Correa-Ramírez et al., 2010; Ivanov et al., 2018), Lycosinae (Bidegaray-Batista et al., 2017; Gonnet et al., 2021; Planas et al., 2013), and Sosippinae (Fontes, 2016; Macrini et al., 2015) subfamilies.
Characterizing the ecological niches, and particularly those considering temporal divergences among life forms, is a different kind of tool recently incorporated into the delimitation and characterization studies of species (Hendry & Day, 2005). Allochrony can significantly contribute to reproductive isolation, especially if populations have little match in breeding periods, and therefore little potential for gene flow (Taylor & Friesen, 2017). According to the seasonal asynchrony hypothesis, this asynchronous reproduction can lead to divergence between populations, when an increase in such asynchrony is positively related to the degree of differentiation and eventually reproductive isolation (Martin et al., 2009). Characterizing the annual climatic requirements of the species and then characterizing their seasonal requirements (corresponding to the months of sexual activity of each population, where females and males are present and matings occur) are accurate approaches to test if the periods are conserved between populations or have diverged in a temporal isolation. Seasonal divergence of breeding times appears as the most common mode of allochronic divergence, same which presents examples in plants, fungi, birds, amphibians, fish, corals, and several insects (Borzée et al., 2016; Rojas-Soto et al., 2021; Yamamoto & Sota, 2009). In spiders, there is valuable information but is not a commonly addressed topic nowadays (Gilman et al., 2018; Tretzel, 1955).
Wolf spiders are usually recognized as one of the most abundant and diverse families of spiders, with currently 127 genera and 2452 species (World Spider Catalog, 2022). Their members usually have wandering habits (Foelix, 2011); nevertheless, three of the eleven subfamilies currently reported (Piacentini & Ramirez, 2019) include species that live in webs in part or even their entire lives (Murphy et al., 2006). As a whole, these lycosids do not exceed 1% of the total species of the family (González et al., 2015a). Of the three subfamilies that build webs: Hippasinae is present in Southern Asia and Africa; Venoniinae occurs in Europe, part of Asia, North America, and Australasia; and Sosippinae is present in Europe, Eastern Asia, and the Americas (Piacentini & Ramirez, 2019). The natural history of the members of these subfamilies is scarcely known (Brach, 1976; Capocasale, 1982; Hodge & Marshall, 2018; Piacentini, 2011; Piacentini et al., 2017; Punzo & Haines, 2006; Santos & Brescovit, 2001; Wang et al., 2015; Yoo & Framenau, 2006). The exception is Aglaoctenus lagotis (Holmberg, 1876) for which there is information about behavior (e.g., Abregú et al., 2019; González, 2018; González & Toscano-Gadea, 2021; González et al., 2013, 2015b, 2019; Stefani & Del Claro, 2011; Stefani et al., 2011), taxonomy (Capocasale, 1982; Santos & Brescovit, 2001), and ecology (Sordi, 1996; Rubio et al., 2005; González et al., 2014; Stefani & Del Claro, 2014).
Aglaoctenus lagotis lives its whole life in a funnel web. This species has a wide distribution from Venezuela to Uruguay (Santos & Brescovit, 2001), country where it is considered a priority species for conservation (Ghione et al., 2017). It had been defined as highly variable, especially referring to female genitalia, body size, and coloration patterns (Santos & Brescovit, 2001). Furthermore, more recently, behavioral and ecological studies have pointed out that the species contains at least two differentiated groups: one mentioned as Southern Uruguay (González et al., 2013, 2014, 2015b) or Southern Uruguayan form (González et al., 2019), hereafter named as Form I, and the other mentioned as Central Argentina form (González et al., 2013, 2014, 2015b, 2019), hereafter named as Form II. Form I has been found in Uruguay and Form II in Argentina and Uruguay, being both sympatric in almost all the Uruguayan territory (González et al., 2015b). Both forms differ in body coloration patterns (González et al., 2015b), sexual behavior (González et al., 2013), genital traits (González, 2015; González et al., 2015b), general phenology, and in the microhabitats where they live (González et al., 2014). Moreover, reproductive isolation between individuals of both forms from distant localities has been already detected (González et al., 2013, 2015b).
The web wolf spider A. lagotis shows elements that suggest inconsistencies in its currently taxonomic identity (González et al., 2015b; Santos & Brescovit, 2001) and some authors have already pointed out this possibility, uncovering the existence of different lineages that could be different species (Macrini et al., 2015). In this context, integrating additional sources of evidence could be useful to clarify this question. We hereby combined information from mitochondrial and nuclear molecular markers to infer phylogenetic relationships. We also analyzed the climatic requirements of the species, during the annual period and during the sexual period, for both forms (Forms I and II) of the species. This novel approach allows us to clarify the limits between them and to go ahead in understanding the evolutionary diversification of this atypical (web living) wolf spider, a member of a little-known group and which additionally has a confusing taxonomic history.
Materials and methods
Identification of individuals
Individuals belonging to “Form I” have been characterized by an orange cephalothorax during the subadult stages which turns brown on reaching maturity. There are two dorsolateral white bands on the abdomen, from which several pairs of white lines, originating at an angle of 45°, converge in the mid-line to form forward-pointing “chevrons,” as well as two white circles proximal to the pedicel (González et al., 2015b) (Fig. 1A). Additionally, males’ bulbs have a spine-shaped median apophysis (González, 2015; González et al., 2015b). The sexual period (when both sexes are present and matings occur) happens during autumn, and the maternal period (when only females remain alive and care for their eggsacs and spiderlings) occurs during spring and summer (González et al., 2014). Finally, microhabitat preferences appear to be associated with grasslands (González et al., 2015b) (Fig. 1B).

A Body coloration patterns of juvenile, female and male of A. lagotis Form I. B Habitat of A. lagotis Form I, Lomas de Araminda, Canelones, Uruguay. C Habitat of both A. lagotis forms in the area of sympatry, showing the microhabitat each one occupies, Villa Serrana, Lavalleja, Uruguay. D Habitat of A. lagotis Form II, Lunarejo, Rivera, Uruguay. E Juvenile, female and male's body coloration patterns of A. lagotis Form II (photos: Carlos A. Toscano-Gadea)
Individuals belonging to “Form II” have been characterized by a brown cephalothorax during all their developmental stages. There are two dorsolateral white bands on the abdomen, from which several pairs of white lines, originating at an angle of 45°, are interrupted without meeting in the midline (González et al., 2015b) (Fig. 1E). Additionally, males’ bulb has a trapezius-shaped median apophysis (González, 2015; González et al., 2015b). The reproductive period (sexual plus maternal) takes place during spring and summer (González et al., 2014). Finally, microhabitat preferences appear to be associated with shrubs and trees in natural forests (Fig. 1D), but in Argentina, individuals are also present in grasslands (González et al., 2014, 2015b). In the areas where both forms are sympatric, Form I occupies the open grasslands and Form II mostly occupies the natural forest patches, on the herbaceous strata, on shrubs or trees (Fig. 1C).
We examined the individuals collected for this study and material of Aglaoctenus lagotis deposited in the Museo Argentino de Ciencias Naturales (MACN, Argentina) to identify as Form I or Form II. Additionally, we explored surveys for Aglaoctenus lagotis and Aglaoctenus sp. in Argentina, Uruguay, and Southern Brazil (Rio Grande do Sul) on the iNaturalist platform to also identify them as Form I or II, using the coloration pattern on the abdomen as a proxy.
Sampling and molecular data collection
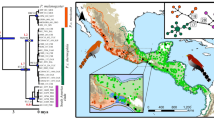
Aglaoctenus lagotis specimens were collected from 25 localities, 19 from Uruguay and six from Argentina (Table 1; Fig. 2). Individuals were stored in 95% ethanol at − 20 °C. Specimens of a close relative Aglaoctenus castaneus (Mello-Leitão, 1942) from Orellana (Ecuador) and Arequipa (Perú) were included in the analysis as outgroup. Most specimens were collected in the field by the authors and colleagues kindly provided some.

Map showing the sampling localities for genetic and ecological niche analyses of A. lagotis. Sampled locations are indicated by colored symbols according to the identification of the individuals (green circle: Form I, orange triangle: Form II). Symbols with black border denote the sampling localities used for the genetic analyses and are numbered as in Table 1
Genomic DNA was extracted from two legs of adult individuals following the protocol described in Medrano et al. (1990). Three mitochondrial fragments including the 5′ half of cytochrome c oxidase subunit I (cox1), the 3′ half of the 16S rRNA ribosomal subunit plus the complete tRNA-Leu plus 5′ half of the NADH dehydrogenase subunit I (16S + L1 + nad1), and a partial fragment of the small ribosomal unit (12S) were amplified and sequenced using the following primer pairs: (cox1) C1-J-1490 (Folmer et al., 1994) and C1-N-2776 (Hedin & Maddison, 2001), alternatively, as two overlapping fragments were used C1-J-1490 with C1-N-2198 (Folmer et al., 1994) and C1-J-1751 (Simon et al., 1994) with C1-N-2776; (16S + L1 + nad1) LR-N-13398 (Simon et al., 1994) and N1-J-12581 (Hedin & Maddison, 2001); and (12S) SR-J-14233 (Simon et al., 1994) and SR-N-14503 (Croom et al., 1991). The nuclear intron of the gene encoding translation initiation factor 5A (tif5A) was additionally sequenced for individuals of A. lagotis with the primer pairs TIF5AF and TIF5AR (Fontes, 2016). Polymerase chain reaction (PCR) was carried out following the protocol for Taq DNA Polymerase with Standard Taq Buffer (M0273, New England Biolabs Inc.). In a total volume of 25 µL, PCR amplification conditions for mitochondrial genes were as follows: initial denaturing step at 95 °C for 5 min; 5 × (95 °C for 30 s; 38 °C for 45 s for cox1 and 12S, and 41 °C for 45 s for 16S + L1 + nad1; 68 °C for 45 s) + 30 × (95 °C for 30 s; 42 °C for 45 s for cox1 and 12S, and 45 °C for 45 s for 16S + L1 + nad1; 68 °C for 45 s) and a final step at 68 °C for 5 min. PCR amplification conditions for the nuclear gene were as follows: initial denaturing step at 95 °C for 5 min and 35 × (95 °C for 30 s; 50 °C for 45 s; 68 °C for 45 s) and a final step at 68 °C for 5 min. PCR products were purified and sequenced by the Sequencing Service of Humanizing Genomics Macrogen, Seoul, Korea. DNA sequences were edited using the free trial version of the Geneious software.
Phylogenetic analyses and genetic diversity
The alignment of the partial fragment of cox1 sequences was trivial since no insertions/deletions (indels) were observed. The sequences of the 12S, 16S + L1 + nad1, and tif5A gene fragments were aligned using the online version of MAFFT v7 (Katoh et al., 2002) using the Q-ins-i algorithm. The alleles in heterozygous individuals for the tif5A intron were separated using the PHASE algorithm (Stephens & Donnelly, 2003; Stephens et al., 2001), as implemented in DnaSP v6. 12.03 (Rozas et al., 2017). Gaps of tif5A were scored as presence/absence characters using the method of Simmons and Ochoterena (2000) as implemented in SeqState (Müller, 2005). The resulting parsimony informative characters were included in the statistical parsimony analyses for the haplotype network reconstruction (see below). Nucleotide (πn) and haplotype (h) diversities were calculated using DnaSP v6.12.03 (Rozas et al., 2017), and genetic distances (p-distance) were estimated with MEGA X v10.0.1 (Kumar et al., 2018).
Concatenated mitochondrial gene trees
Maximum likelihood (ML) and Bayesian inference (BI) analyses were conducted on the concatenated mitochondrial sequences data set of A. lagotis and its congeneric relative A. castaneus. The best partition scheme and fitting model for each partition were selected with PartitionFinder2 (Lanfear et al., 2017). Maximum likelihood analyses were performed with RAxML v8 (Stamatakis, 2014) using independent GTR + G substitution models for each partition. The analyses were conducted selecting the rapid bootstrapping algorithm and the search for the best-scoring ML tree in one single run. The majority-rule tree criterion was used to automatically halt bootstrapping. PartitionFinder and ML analyses were run remotely at the CIPRES portal (Miller et al., 2010). Bayesian inference analyses were conducted using MrBayes v3.2.3 (Ronquist et al., 2012). Two independent runs of one million generations were carried out simultaneously, with six simultaneous MCMC chains, sampling trees every 100 generations. The program Tracer v1.7.1 (Rambaut et al., 2018) was used to ensure that the Markov chains had reached stationarity by examining the effective sample size (ESS) and to determine the number of generations of burn-in.
Haplotype networks
The haplotype network of the nuclear intron tif5A, the concatenated mitochondrial genes cox1 + 12S + 16S + L1 + nad1, and of a partial fragment of cox1 gene was estimated using statistical parsimony in TCS (Clement et al., 2002) and implemented in PopART v1.7 (Leigh & Bryant, 2015).
Species trees
Multispecies coalescent analyses were conducted including both mitochondrial and nuclear intron data sets using *BEAST option (Heled & Drummond, 2010) implemented in BEAST v2.6.3 (Bouckaert et al., 2019). Since this method requires a priori assignment of individuals to divergent populations (lineages or species), we defined as independent evolutionary lineages those recovered by the species delimitation analyses (see below). In *BEAST, the species tree, gene trees, and divergence times were co-estimated. The specifications of the analysis settings are described in the “Estimation of lineage divergence times” section.
Molecular species delimitation
Species delimitation was inferred using the Bayesian multispecies coalescent model implemented in the package STACEY (Jones, 2017) for BEAST2 v2.6.3 (Bouckaert et al., 2019). This method co-estimates gene trees and the species or minimal clusters trees (SMC-tree) along with the species delimitation. STACEY does not require a guide tree a priori or an assignment of individuals to species or lineages as minimal clusters; hence, the possible number of species can range from one to the total number of individuals (Jones, 2017).
The STACEY analyses were conducted including both the mitochondrial and nuclear intron data sets. Each individual was considered a priori as a minimal cluster. Parameters of the substitution models were specified in the priors for each gene fragment, and the mitochondrial clock models and mitochondrial trees were linked. A strict molecular clock model was specified for each partition. The bdcGrowthRate and popPriorScale priors were set with a lognormal distribution, and relativeDeathRateSpecies and collapseWeight priors were set with a uniform distribution (0–1). Two independent runs of 10 million generations, sampling every 1000 generations were carried out. The convergence and mixing of each MCMC chain were assessed with Tracer v1.7.1. Independent runs were combined with LogCombiner v2.6.3 (10% burn-in), and the TreeAnnotator v2.6.3 (both included in the BEAST 2 package) was used to summarize the information from the sampled trees. Results from STACEY were analyzed with the package speciesDA.jar setting the collapse height to 0.0001 and sim cutoff to 1. The priors for the analyses were selected following the STACEY package documentation (Jones, 2014).
Estimation of lineage divergence times
Lineage ages were estimated in *BEAST including both mitochondrial and nuclear genes of A. lagotis. The analyses were carried out using independent parameter models for each gene fragment but linking the mitochondrial trees. A Yule speciation process and a strict molecular clock model were specified as priors. We set a normal prior distribution for the substitution rates using the rate estimation described in Piacentini and Ramírez (2019) and Bidegaray-Batista and Arnedo (2011). Two independent runs of 10 million generations, sampling every 1000 generations, were carried out. The convergence and mixing between the runs were checked with Tracer v1.7.1. The results of the runs were combined with LogCombiner v2.6.3 (10% burn-in), and the information of the trees was summarized with TreeAnnotator v2.6.3.
Comparisons between climatic requirements
We used niche similarity test implemented in ecospat (Di Cola et al., 2017) in order to estimate and compare climatic requirements of the main lineages recovered in the molecular species delimitation analyses. To compare each form requires at least having more records than ecological variables considered. All the comparisons were done in two different ways: considering the annual climatic requirements of both forms and only considering those corresponding with their sexual activity. First, we estimated and compared climatic requirements considering annual climatic variables, which correspond to the climatic requirements that each form needs to subsist throughout the year. Second, to compare the climatic requirements of sexual periods, the estimations were based on monthly variables, considering for each form only those months of sexual activity. The sexual periods considered were March to May for the Form I and September to December for Form II (González et al., 2014).
Geographic information
The geographical sources of records of A. lagotis used to define the climatic requirements characterization included the following: (i) records obtained during field work of the authors and colleagues from Uruguay and Argentina, including the localities sampled for the molecular analyses; (ii) records from the arachnological collection of Facultad de Ciencias, Universidad de la República (Montevideo, Uruguay); and (iii) records published in Piacentini (2011) (Fig. 2; Table S1). These records were mapped using Quantum GIS software (Quantum GIS Development Team, 2020). The complete database encompasses 114 records: 59 presence records from Form I and 55 presence records from Form II (27 Form IIa and 28 Form IIb, see below the species delimitation results). Given the great importance of the geographic background in this kind of analyses (Barve et al., 2011), the niche for each climatic background was calculated as the 50-km buffer zone of those ecoregions containing at least one record of the species (sensu Olson et al., 2001).
Environmental variables
From the total set of seven monthly climatic variables (maximum, minimum and average temperature, precipitation, solar radiation, wind speed, water vapour pressure) available in Worldclim v2 (Fick & Hijmans, 2017), we selected four of them with low correlation (r Pearson < 0.7). Variables were downloaded at a spatial resolution of 2.5 arc-min. The selected variables were as follows: (1) maximum temperature (°C); (2) precipitation (mm); (3) solar radiation (kJ m−2 day−1); (4) wind speed (m s−1). With these layers, we generated two set of variables according to both approaches (annual and sexual requirements) by averaging monthly layers of the year as follows: (i) for the annual approach, we calculated the average layer of the layers corresponding to each month of the year; (ii) for the sexual approach, each average layer was calculated considering only those months of sexual activity for each form, in which females and males are present (copulations occurrence period; González et al., 2014).
Niche similarity analyses
We analyzed the niche overlap of the main lineages based on the similarity tests presented by Broennimann et al. (2012) and available in the ecospat package, which assess niche difference against null model niches taken randomly within a given background area. We compared the niche overlap between Form Ia with Form IIa and IIb and between Form IIa and IIb. We did not include the comparison between Form Ia and Form Ib because of methodological limitations, as the comparisons require at least more records than the ecological variables considered in the study and we only have records of individuals of Form Ib for three localities (and worked with four ecological variables) where furthermore members of Form Ia are also present. The niche similarity test was performed in order to assess whether the niches are more similar than expected by chance through 100 random shifts of the niches within the available conditions for each lineage. First, we built a PCA based on the climatic variables within the environmental range defined for the study area and characterized the environmental space, within which we compared the lineages previously detected with the molecular species delimitation analyses. We projected the PCA scores of each lineage onto a grid of 100 × 100 cells, bounded by the minimum and maximum PCA scores in the study areas. The overlap between the niches was calculated using Schoener’s D and Hellinger’s I metrics, both ranging from 0 (no overlap between niches) to 1 (complete overlap; see Broennimann et al., 2012 for details). As stated, based on our goals, we calculated all these indexes considering the annual climatic requirements and the sexual ones. R-Scripts used are shown in Table S4.
Results
Morphological identification
We examined 85 specimens from MACN collection from the Argentinian provinces of Chaco, Córdoba, Corrientes, Entre Ríos, Jujuy, La Rioja, Santa Fé, and Tucumán and from the provinces of Canelones, Río Negro, San José, and Soriano in Uruguay. All the specimens of Form I were from Uruguay (N = 24). Specimens identified as Form II, at least following coloration patterns, were found in Uruguay (N = 6) and Argentina (N = 55) (Table S2). On the iNaturalist platform, we found 175 records, of which in 105, the dorsal views of the abdomen were clearly visible. Only two records showed the typical pattern of Form I and were from Maldonado and Canelones in Uruguay; the other 103 records showed the pattern associated with Form II (Table S3). From the material collected to carry out the molecular analyses, 39 were identified as belonging to Form I and 40 as belonging to Form II.
Sequence information
We sequenced gene fragments of 54 adult individuals of A. lagotis from 17 localities and two individuals of A. castaneus from two localities (Fig. 2; Table 1). Additionally, we included the information of 25 cox1 sequences of A. lagotis available in the BoldSystems database (SPDAR297-13 to SPDAR310-13, SPDAR312-13 to SPDAR314-13, SPDAR316-13 to SPDAR321-13, SPDAR340-14 and SPDAR925-15). We obtained 38 sequences from a short fragment of the mitochondrial cox1 gene (ON128450 to ON128487), 24 sequences from a long fragment of the mitochondrial cox1 gene (ON128488 to ON128511), 12S (ON149515 to ON149538) and 16S + L1 + nad1 (ON136032 to ON136055) mitochondrial genes, and 21 sequences of the nuclear intron tif5A (ON136056 to ON13607) (Table 1). The alignments of the cox1 long fragment, 12S and 16S + L1 + nad1 yielded 1218 characters, 264 and 685, respectively. The concatenation of the mitochondrial gene fragments of 24 individuals yielded a combined matrix of 2167 characters (including the outgroup). The alignment of the cox1 short fragment from 79 individuals yielded a matrix of 655 characters. We constructed two matrices to the aligned sequences of the nuclear intron tif5A: (i) one included 34 phased sequences and 389 characters and was used to infer the species trees, and (ii) the other included 42 phased sequences and 286 characters, including two informative gaps recorded as absence/presence, and was used to construct the haplotype network.
Phylogenetic inferences, species delimitation, and lineage age estimation
The best partition scheme and substitution model for each partition selected by PartitionFinder2 were as follows: K81UF + I for cox1 and TIM + I for 12S + 16S-L1-nad1. Bayesian inference and maximum likelihood analyses conducted on the concatenated mitochondrial gene tree converged on an identical topology (Fig. 3). Four well-supported clades were recognized in both BI and ML gene trees: the Form Ia clade included nine individuals previously identified as Form I from Uruguay (localities 1, 8–11, 13, 17, 18), the Form IIa clade included four individuals previously identified as Form II from Uruguay (localities 5, 9, 10), the Form Ib clade included three individuals previously identified as Form I from Uruguay (localities 6, 13, 10), and the Form IIb clade included six individuals previously identified as Form II, four from Argentina (localities 20, 23, 24, 25) and two from the localities 8 and 12 near the Uruguayan River in Uruguay. The clades Form Ia and Form IIa appear as sister groups with high support for both ML and BI analyses, and the clades Form Ib and Form IIb appear as sister groups in both analyses but with high support only for ML analyses (Fig. 3).

Maximum likelihood (ML) concatenated mitochondrial gene tree of A. lagotis. Bars on branches denote clade support from ML (left) and Bayesian Inference (right). Black bar indicates clade supported by ML bootstrap (> 70%) and Bayesian posterior probabilities (pp > 0.95), and gray bar indicates clade recovered but with support below the threshold values. The main mitochondrial lineages colors correspond to the identification of the individuals as explained in the material and methods section (i.e., Form I or Form I;, see Fig. 1) and match those of the localities in the map (Fig. 2). The two slash in the root indicate that the outgroup (A. castaneus) has been trimmed from the tree
The allele network from the nuclear intron tif5A showed 22 alleles connected by 1 to 6 mutational steps (Fig. 4). The A2, A8, and A12 alleles were the most frequent. Allele 12 was found in individuals identified as Form I and was connected by few mutational steps with the alleles found in the remaining Form I individuals (i.e., either from the Form Ia or Form Ib clades of the concatenated mitochondrial tree). A2 allele was found in almost all Form II individuals from Uruguay (i.e., corresponding to the Form IIa clade of the concatenated mitochondrial tree) and is connected by few mutational steps with alleles of the Form I individuals. A8 allele was found in Form II individuals from Argentina and is closely connected with the remaining alleles of the Form II from Argentina and from two localities near the Uruguayan River in Uruguay (i.e., corresponding to the Form IIb clade of the concatenated mitochondrial tree). The differences among Form I, Form IIb, and Form IIa included two informative gaps (recoded in the network as absence/presence). Those gaps that represent indels (insertions/deletions) in the alignment were exclusive to each lineage. One includes three base pairs (positions 294–296) that are absent in all individuals of Form IIa, and the other includes one base pair (position 286) that is absent in all the individuals of Form IIb. Those four base pairs are present in all the individuals of Form I.

Statistical parsimony network of tif5A alleles of A. lagotis. Colors of the pie plots correspond to the identifications of the individuals as in Figs. 1 and 2. The asterisks denote alleles that were present in the individuals that correspond to the Form Ib clade of the concatenated mitochondrial tree (see Fig. 3)
The concatenated mitochondrial network (cox1 + 12S + 16S + L1 + nad1) and cox1 network showed the main mitochondrial clades as described in the phylogenetic results of the concatenated mitochondrial gene tree (Figs. S1, S2). In both networks, fewer mutational steps connected the haplotypes of each main mitochondrial clade than between clades. However, in the concatenated mitochondrial network, the position of the Form Ib clade is connected with the Form Ia clade instead of with the Form IIb clade. The cox1 mitochondrial network included more individuals covering a wider distribution range and showed that all haplotypes from individuals of Argentina and some individuals from localities near the Uruguayan River in Uruguay (Hap_1, Hap_2, Hap_8, Hap_9, Hap_10, Hap_11 and Hap_12) belong to the Form IIb clade (Fig. S2). The remaining haplotypes were found in individuals from Uruguay and belong to the other clades.
The species delimitation analyses conducted with STACEY recovered that the number of species with the 95% highest posterior density (HPD) ranged from 3 to 7, with a median of 4 clusters (Fig. S3). The highest posterior probability (HPP) of clustering recognized 4 clusters (PP = 0.27), which include the following: (i) Form IIa clade, (ii) Form IIb clade, (iii) Form I clade (Ia + Ib), and (iv) the individual AlagNK52, included in the Form Ib clade of the mitochondrial analyses. The second HPP clustering recognized 3 clusters (PP = 0.12) that merge the individual AlagNK52 in a clade grouping all the individuals identified as Form I (i.e., Form Ia and Ib clades). A rerun of STACEY, in which individuals were associated with minimal clusters recovered in the first run, was performed. The same 4 clusters described above were recovered with a PP = 0.98. The clustering agrees with the results of ecological niche comparisons (see below), except for the individual AlagNK52 from Uruguay (locality 6; Fig. 2).
The species-tree analyses obtained with *BEAST resulted in a different topology to the ones reported for the concatenated mitochondrial analysis (Fig. 5). The species tree recovered a close relationship among the individuals of the Form I (Form Ia + Ib clades and AlagNK52), which were in turn the sister group of the Form IIa clade. The Form IIb clade was shown as sister to the other clades. The lineage age estimation obtained under the time-calibrated species tree revealed that the time of the most recent common ancestor (TMRCA) of the sampled A. lagotis individuals trace back to the Pleistocene (~ 2 Ma) and the TMRCA of the Forms I was dated at < 1 Ma (Fig. 5).

Species tree and chronogram inferred from mitochondrial genes (cox1, 12S, and 16S + L1 + nad1) and the nuclear intron tif5A with *BEAST under partition scheme by gene and strict clock. Numbers above branches denote Bayesian posterior probabilities clade support and below branches indicate node ages and in brackets their 95% HPD interval. Colors correspond to the identifications of the individuals as in Figs. 1 and 2. The names of clades denote main lineages identified in the concatenated mitochondrial gene tree
Genetic diversity
The average genetic distances (p-distance) among the four main mitochondrial clades for the concatenated cox1 + 12S + 16S + L1 + nad1 genes were from 1.5 to 3.6% (Table S5). The p-distance among the main clades for the nuclear intron tif5A (considering the Form I clades together; see Fig. 4) was from 1.1 to 3.3% (Table S5). The greater distances were between the Form IIb clade and the rest (i.e., Form IIa clade and Form I a + b clades) (Table S5). Intra clade genetic distances are also shown in Table S5. The nucleotide diversity was 0.020 for the concatenated mitochondrial genes and 0.019 for the nuclear intron tif5A. The haplotype diversity for the concatenated mitochondrial genes was 0.957, and the allelic diversity for tif5A was 0.928.
Climatic requirements comparisons
The comparisons between climatic niches performed by the similarity test analysis showed a high overlap between the annual set of variables of Form I and Form IIa clades (D = 0.55, I = 0.73), suggesting that niches are more similar than what expected under a null model (p = 0.01; Fig. 6A). At the same time, the similarity test analysis showed low overlap between niches of Form I and Form IIb clades (D = 0.08, I = 0.12) and between niches of Form IIa and Form IIb clades (D = 0.02, I = 0.03), further suggesting that niches are not more similar than what expected under a null model (p = 1.00 and p = 0.61, respectively) (Fig. 6B and C). Also, comparisons performed with the sexual climatic niches showed little or no niche overlap between all the three clades: Form I and Form IIa clades (D = 0.00, I = − 2.22E−16), Form I and Form IIb clades (D = 0.00, I = − 2.22E−16), and, more attenuated, Form IIa and Form IIb clades (D = 0.04, I = 0.08), suggesting that sexual niches are not more similar than that expected under a null model (p = 1.00, p = 1, p = 0.31, respectively) (Fig. 6A, B, and C). Likewise, each individual variable composing the niche showed a low general overlap when niches were compared annually, turning almost to an inexistent overlapping when the comparison took into account sexual periods of activity (sexual climatic niche; Fig. 6).

Annual and sexual niches overlap comparisons between the three forms of A. lagotis: A comparisons between Form I and Form IIa (annual niche above, sexual niche below); B comparisons between Form I and Form IIb (annual niche above, sexual niche below); C comparisons between Form IIb and Form IIa (annual niche above, sexual niche below). The overlap is represented along two principal component analysis (PCA) calibrated axes, the solid and dashed contour lines illustrate 100% and 50%, respectively, of the available (background) environment. Color shading represents the density of the occurrences by cell. The similarity tests between the compared forms were calculated from 100 iterations
Discussion
More than one species within Aglaoctenus lagotis
In this study, the integration of molecular phylogenetic delimitation and macroecological modeling analyses support the hypothesis that there is more than one lineage inside what is currently defined as Aglaoctenus lagotis. These main lineages, referred here as Form I (in Uruguay), Form IIa (in Uruguay), and Form IIb (in Argentina and in the Uruguayan west coast bordering Argentina), may correspond to new species at present hidden within A. lagotis.
While the ML and BI conducted on the concatenated mitochondrial gene tree recognized four clades, with the Form I appearing divided in two groups (Form Ia and Ib) (see Fig. 3 and Fig. S2), the multilocus Bayesian analyses recovered three main lineages and show a close relationship among all individuals previously identified as Form I (see Fig. 5). This close relationship is also recovered in the allele network of the nuclear intron tif5A (see Fig. 4 and Fig. S1). Additionally, these three groups (Form I, Form IIa and Form IIb) show differences in their ecological niches. All of them differ in their sexual niches, which means that climatic variables analyzed are different for the forms during the mating period, although a greater similarity appears between Forms IIa and IIb. However, these two forms differ in their annual niche. At the same time, the sexual niches of Form I and Form IIa, which can be found in sympatry, significantly differ, but their annual niches are similar. Finally, Form I and Form IIb differ in both ecological types of niches (see Fig. 6). Overall, a non-overlapping of lineages at the sexual and/or annual ecological niche level is pointed out.
The fact that unlinked gene trees can be different and not necessarily congruent with the species trees (Fig. 5) is well described and can be explained by different process such as introgression, incomplete lineage sorting, horizontal gene transfer, gene duplication, sex-biased behaviors, and selective sweeps (Avise, 2009; Edwards, 2009; Ivanov et al., 2018; Maddison, 1997). Specifically, differences as the recovered in this study, between mitochondrial and nuclear markers (i.e., maternal vs. biparental inheritance, respectively), are very common (Avise, 2009; Ballard & Whitlock, 2004; Hare, 2001) and have been reported in several taxa, such as spiders (Bidegaray-Batista et al., 2016; Chang et al., 2007; Doménech et al., 2020; Ivanov et al., 2018; Peres et al., 2015), insects (Gómez-Zurita & Vogler, 2003; Mastrantonio et al., 2016), newts (Milá et al., 2010), and birds (Zhang et al., 2019), among others. In this study, the differences found might be due events of past introgression since some individuals (e.g., AlagNK52) were sampled in localities where Form I, Form IIb and Form IIa could coexist. However, we cannot rule out the incomplete lineage sorting, which is particularly expected in close related species with recent divergence times, as reported here for A. lagotis (the TMRCA is ~ 2 Ma). This kind of process is taken into account by the multispecies coalescent inference methods used in this study (i.e., Heled & Drummond, 2010; Jones, 2017).
Interestingly, individuals recognized phenotypically as Form II appear in two lineages (Form IIa and IIb) that are not sister groups in the species tree (see Fig. 5). Furthermore, we did not find an overlap between both lineages neither in their sexual nor in their annual climatic requirements. Based on the localities sampled in our study, these forms appear to have a parapatric distribution, and the Form IIa appears to be associated with shrubs and trees in natural forests, while Form IIb appears to be also present in grasslands (based on González et al., 2014, 2015b). Our results, together with the reports of Macrini et al. (2015) and Fontes (2016), raise the need for a greater number of studies on what today we call A. lagotis, which has a very wide distribution and probably contains more than one cryptic species, if we consider that the detection of these lineages have occurred only having studied part of the total distribution of “A. lagotis.” While as we mentioned above, differences between Form I and Form IIb had been pointed out in several previous studies, the existence of two cryptic lineages inside which was named as “Form II” was first detected in this study. Likely, differences were not detected in previous behavioral studies because the vast majority of the studies were carried out with individuals from what is now called Form IIb and Form I, but not between individuals from Form IIa and Form IIb. However, sexual studies with individuals from the locality 14 (Lavalleja, Uruguay), where Form IIa and I are sympatric, point out that the phenology of Form IIa is very similar, if not identical, to that reported for Argentinian individuals of Form IIb. And similarities also persist in relation to the sexual repertoire (González, pers. obs). Given our present results, these differences and similarities between both Form II deserve a more detailed study focusing on differences in other traits, as those already detected in molecular, sexual, and annual niches and microhabitat levels.
The differences reported between Form I and Form IIb are reaffirmed in the present study, but had also been detected previously based on other traits. González et al. (2013) showed differences in behavioral patterns of courtship and copulation durations. In addition, González et al. (2014) found differences in body coloration patterns (in adults and subadults), and the genitalia of males showed discrete differences in the median apophysis (González, 2015; González et al., 2015b). Additionally, sexual encounters between the Form I and Form IIb (in the laboratory and the field) strongly suggest a reproductive isolation between them (González et al., 2015b). Finally, the analysis of the microhabitats demonstrated that individuals of Form I occupy open areas as grasslands and the herbaceous stratus, while Form IIb can also occupy close areas and higher strata (González et al., 2014, 2015b). Another source of evidence that supports these differences is the distribution, clearly delimited in Form I, as was shown analyzing the material from the MACN collection and the images deposited in the iNaturalist digital platform. This material shows that the distribution of Form I is restricted to Uruguay. As we mentioned above, Form I and Form IIa are mostly sympatric (except for a southern area of Uruguay where we have only found the Form I until today), but always maintain different microhabitats. Additionally, their annual niches are similar but not the sexual niches. Since there was no overlap in the sexual niches or heterozygous individuals for the nuclear intron tif5A, it would appear that there is no gene flow between these two lineages (which could be consistent with the existing phenotypic differences between them). Therefore, mentioned characteristics of the somatic and genital morphology allow to clearly recognizing Form I and, together with the phenological, behavioral, and genetic characteristics mentioned, are enough to recognize this group as a different species. Other species of typical wandering lycosids of the Schizocosa genus (S. crassipes (Walckenaer, 1837), S. ocreata (Hentz, 1844), S. rovneri Uetz & Dondale, 1979, S. duplex Chamberlin, 1925, S. uetzi Stratton, 1997 and S. stridulans Stratton, 1984) have also shown differences in their sexual behavior and morphological structures, accompanying genetic differences (Hebets & Uetz, 1999; Miller et al., 1998; Stratton & Uetz, 1986). In web, lycosids of the Sosippus genus (S. floridanus Simon, 1898 and S. placidus Brady, 1972) from North and Central America have also been reported similar scenarios (Hodge & Marshall, 2018).
Recent origin and diversification
Our results show a recent origin of the sampled individuals of A. lagotis, with diversification times that trace back to the Early to Middle Pleistocene (~ 2 to 0.7 Ma). According to Turchetto-Zolet et al. (2013), several South American taxa underwent demographic and lineage splitting events that are related with Pleistocene climatic changes. These processes have also been reported in recent studies in spiders (Bartoleti et al., 2017, 2018; Campón et al., 2021; Peres et al., 2015; Postiglioni et al., 2019).
Specifically, during the Pleistocene, there were many changes in the sea level, where there were very large areas under water, with which many animals of Argentina and Uruguay would be isolated and reconnected again many times (Campón et al., 2021; Langone et al., 2016; Villamil et al., 2019). This scenario might explain the main split between Form IIb and the ancestral lineage of Forms I and Form IIa. Both Form I and Form IIa show restricted distributions, until now only reported for Uruguay. A plausible explanation could be that the large rivers function as barriers, as has been reported many times for other animals in the River Plate basin (Kopuchian et al., 2020; Marquez et al., 2006). Currently, the Uruguay River would be functioning in a similar way and would mainly affect the Form I, probably because it lives in grasslands and, conversely, coasts of the Plata River are generally shrubbier. The only form found on both sides of the River is Form IIb, but only found in the coastal zone of Uruguay, on the border with Argentina. In relation with the northern limit of Form I, it may have been distributed in southern Brazil, considering that until the late Holocene, that area was grassland, but now it is araucaria forests (Behling, 2002). These consequences of isolation and re-connection related with sea levels and climatic shifts have been already reported as causes of speciation process in web lycosids from North America (Hodge & Marshall, 2018).
Additionally, in Uruguay, the most southern area of the distribution, the climatic conditions, and the increase of grasslands from the Late Pleistocene (Behling, 2002; Ubilla et al., 2004) could have favored the success of Form I. As we mentioned above, this form is associated with open areas of grasslands and with a sexual period similar to other typical lycosids that inhabit similar areas (Costa & Simó, 2014). The hard conditions of being sedentary and going through the winter in open areas could be related to completely separating the sexual period from the egg sac and spiderlings period, something that does not occur in closed areas of natural forests with shrubs and trees. Moreover, as most differences encountered between the Forms (I and IIa) in relation to ecological niches exist between the sexual ones, these ones could be the efficient barrier to isolate them, even in a recent scenario of divergence as it appears in our analyses (~ 1 Ma; see Fig. 5). This allochrony in sexual periods, in other words, the impossibility of encountering between the different groups during matings, may have functioned as a quick reproductive barrier between these forms. The phenology assigned to Form IIa and Form IIb, with sexual period mainly during spring and immediately followed by the eggsac and spiderlings season, is the common pattern for spiders in Central and Northern Argentina and Southern and Central Brazil (Sordi, 1996) and is the most common for temperate zone spiders (Foelix, 2011). This trend is also observed in other arachnids such as most scorpions (Peretti, 1997; Polis, 1990; Toscano-Gadea, 2002) and harvestmen (Machado & Macías-Ordóñez, 2007; Toscano-Gadea & Simó, 2004). However, in Uruguay, the phenology reported in Form I, with an autumnal sexual period and a mainly period of egg sac and spiderlings during the spring, has been found in other spider species, such as the anyphaenid Sanogasta backhauseni (Simon, 1895), the caponid Caponina notabilis (Mello-Leitão, 1939), and the lycosid Schizocosa malitiosa (Tullgren, 1905) (Costa & Simó, 2014).
At the same time, it is fair to say that with this study, we could not discard sympatric speciation as another process involved, especially explaining isolation between Form I and Form IIa. These closely related forms appear to differ in sexual behavior and body coloration pattern of their members (females and males) (González et al., 2015a, b; González pers. obs.). This differentiation could have evolved driven by sexual selection, as has been reported for other spiders (e.g., Barth & Schmitt, 1991; Masta & Maddison, 2002), although in this case, the analyses of the mitochondrial genes together with the nuclear one also showed genetic differentiation (Fig. 5; Fig. S3). In other wolf spiders supposed to be recently isolated by sexual selection, as Pardosa European species, the mitochondrial genes show very low differentiation (Astrin et al., 2016). Another possible scenario between these two forms is the occurrence of microallopatry, considering that both of them are biogeographically sympatric but occupy niches that are spatially exclusive (Fitzpatrick et al., 2008). Comparing potential ecological niches in the past of Form I and Form IIa will be very valuable to clarify which scenario underlies their differentiation.
Future perspectives
Although a formal description of the species is beyond the scope of this paper, future studies for its determination will be of great value. The holotypes of the proposed junior synonyms for A. lagotis should be reviewed in order to evaluate the presence of the diagnostic characters of the Form I and II. However, a problem related to this is the absence of type material of A. lagotis, added to the fact that several of the junior synonyms of A. lagotis have been described based on juveniles (Santos & Brescovit, 2001). Future studies resolving these problems and formally describing this new species will be valuable to advance in the taxonomic challenges of this spider family. Also, a phylogenomic approach from the whole geographic range of individuals that fit with the actual taxonomic concept of A. lagotis is a key aspect to species delimitation and to understand the underlying processes that shape their diversification. To clarify the scenario especially in the three forms’ sympatric area and to assess if there is introgression among the forms, a further study will be required, including a thorough sampling and genomic data such as single nucleotide polymorphisms (SNPs). Additionally, it is necessary to study the two Forms II discovered in this study, searching other traits (as sexual behavior and morphological structures) to understand how divergence process are acting to maintain both forms isolated, even in sympatry and with several morphological similarities. These mentioned studies become even more essential if we strive to clarify the already mentioned conservation issues of this species. Currently, the different lineages reported here (Form I and Form IIa) are considered as a unique species in the priority list of arachnids from Uruguay (Ghione et al., 2017). That is why solving these topics is so important, in order to take into account each form as independent evolving lineages to design appropriate conservation policies. Further studies of Form I and Form IIa, related with population structure, dispersal capacity, and species distribution models appear really promising in order to have a more complete idea about the evolutionary history and conservation situation of the species that inhabit in sympatry in their southernmost distribution.
Conclusions
Based on different sources of information, our findings support the existence of more than one lineage inside what is nowadays Aglaoctenus lagotis, defined here as Form I, Form IIa, and Form IIb. Form I appears restricted to Uruguay and closely related and sympatric with Form IIa, whereas Form IIb locates in Argentina and in the Uruguayan west coast, generating a sympatric area of the three forms. The three forms also show differences in the ecological niches. Our study sets a framework for further studies in this group in order to shed light on the evolutionary history and conservation of the species.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available in the GenBank repository (see corresponding GenBank numbers in the text in the “Results” section). Original sequence alignments used in the analyses were deposited at the Zenodo data repository: 10.5281/zenodo.6407406.
Code availability
Not applicable.
References
Abregú, D., Peretti, A. V., & González, M. (2019). Male performance and associated costs in successive sexual encounters in a polygynous web wolf spider. Acta Ethologica, 22, 175–186. https://doi.org/10.1007/s10211-019-00323-9
Astrin, J. J., Höfer, H., Spelda, J., Holstein, J., Bayer, S., Hendrich, L., Huber, B. A., Kielhorn, K. H., Krammer, H. J., Lemke, M., Monje, J. C., Morinière, J., Rulik, B., Petersen, M., Hannah Janssen, H., & Muster, C. (2016). Towards a DNA barcode reference database for spiders and harvestmen of Germany. PLoS One, 11, e0162624. https://doi.org/10.1371/journal.pone.0162624
Avise, J. C. (2009). Phylogeography: Retrospect and prospect. Journal of Biogeography, 36, 3–15. https://doi.org/10.1111/j.1365-2699.2008.02032.x
Ballard, J. W. O., & Whitlock, M. C. (2004). The incomplete natural history of mitochondria. Molecular Ecology, 13, 729–744. https://doi.org/10.1046/j.1365-294X.2003.02063.x
Barth, F. G., & Schmitt, A. (1991). Species recognition and species isolation in wandering spiders (Cupiennius spp.; Ctenidae). Behavioral Ecology and Sociobiology, 29(5), 333–339.
Bartoleti, L. F. d. M., Peres, E. A., Fontes, F. v. H. M., da Silva, M. J., & Solferini, V. N. (2018). Phylogeography of the widespread spider Nephila clavipes (Araneae: Araneidae) in South America indicates geologically and climatically driven lineage diversification. Journal of Biogeography, 45(6), 1246–1260. https://doi.org/10.1111/jbi.13217
Bartoleti, L. F. d. M., Peres, E. A., Sobral ÄêSouza, T., Fontes, F. v. H. M., Silva, M. r. J. d., & Solferini, V. N. (2017). Phylogeography of the dry vegetation endemic species Nephila sexpunctata (Araneae: Araneidae) suggests recent expansion of the Neotropical Dry Diagonal. Journal of Biogeography, 44(9), 2007–2020. https://doi.org/10.1111/jbi.12998
Barve, N., Barve, V., Jiménez-Valverde, A., Lira-Noriega, A., Maher, S. P., Peterson, A. T., Soberón, J., & Villalobos, F. (2011). The crucial role of the accessible area in ecological niche modelling and species distribution modeling. Ecological Modelling, 222(11), 1810–1819. https://doi.org/10.1016/j.ecolmodel.2011.02.011
Behling, H. (2002). South and southeast Brazilian grasslands during Late Quaternary times: A synthesis. Palaeogeography, Palaeoclimatology, Palaeoecology, 177, 19–27. https://doi.org/10.1016/S0031-0182(01)00349-2
Bidegaray-Batista, L., & Arnedo, M. A. (2011). Gone with the plate: The opening of the Western Mediterranean basin drove the diversification of ground-dweller spiders. BMC Evolutionary Biology, 11, 317. https://doi.org/10.1186/1471-2148-11-317
Bidegaray-Batista, L., Arnedo, M., Carlozzi, A., Jorge, C., Pliscoff, P., Postiglioni, R., Simó, M., & Aisenberg, A. (2017). Dispersal strategies, genetic diversity, and distribution of two wolf spiders (Araneae, Lycosidae): Potential bio-indicators of ecosystem health of coastal dune habitats of South America. In: Viera C, Gonzaga M. (eds) Behaviour and Ecology of Spiders. Springer, Cham. https://doi.org/10.1007/978-3-319-65717-2_5
Bidegaray-Batista, L., Sánchez-Gracia, A., Santulli, G., Maiorano, L., Guisan, A., Vogler, A. P., Arnedo, A., & M.A. (2016). Imprints of multiple glacial refugia in the Pyrenees revealed by phylogeography and palaeodistribution modelling of an endemic spider. Molecular Ecology, 25(9), 2046–2064. https://doi.org/10.1111/mec.13585
Borzée, A., Kim, J. Y., Cunha, M. A. M., Lee, D., Sin, E., Oh, S., Yi, Y., & Jang, Y. (2016). Temporal and spatial differentiation in microhabitat use: Implications for reproductive isolation and ecological niche specification. Integrative Zoology, 11, 375–387. https://doi.org/10.1111/1749-4877.12200
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., Heled, J., Jones, G., Kühnert, D., De Maio, N., Matschiner, M., Mendes, F. K., Müller, N. F., Ogilvie, H. A., du Plessis, L., Popinga, A., Rambaut, A., Rasmussen, D., Siveroni, I., Suchard, M. A., Wu, C. H., Xie, D., Zhang, C., Stadler, T., & Drummond, A. J. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Computational Biology, 15, e1006650. https://doi.org/10.1371/journal.pcbi.1006650
Brach, V. (1976). Subsocial behavior in the funnel-web spider Sosippus floridanus (Araneae: Lycosidae). The Florida Entomologist, 59(3), 225–229.
Brady, A. R. (1972). Geographic variation and speciation in the Sosippus floridanus species group (Araneae: Lycosidae). Psyche, Cambridge, 79(1–2), 27–48.
Broennimann, O., Fitzpatrick, M. C., Pearman, P. B., Petitpierre, B., Pellissier, L., Yoccoz, N. G., Thuiller, W., Fortin, M. J., Randin, C., Zimmermann, N. E., Graham, C. H., & Guisan, A. (2012). Measuring ecological niche overlap from occurrence and spatial environmental data. Global Ecology and Biogeography, 21, 481–497. https://doi.org/10.1111/j.1466-8238.2011.00698.x
Campón, F. F., Solferini, V. N., Carrara, R., Marvaldi, A., & Confalonieri, V. (2021). Phenotypic plasticity and the colonization of new habitats: A study of a colonial spider in the Chaco region and the Cerrado. Evolutionary Ecology, 35, 235–251. https://doi.org/10.1007/s10682-021-10105-0
Capocasale, R. M. (1982). Las especies del género Porrimosa Roewer, 1959 (Araneae, Hippasinae). Journal of Arachnology, 10, 145–156.
Chamberlin, R. V. (1925). Diagnoses of new American Arachnida. Bulletin of the Museum of Comparative Zoology, 67, 209–248.
Chang, J., Song, D., & Zhou, K. (2007). Incongruous nuclear and mitochondrial phylogeographic patterns in two sympatric lineages of the wolf spider Pardosa astrigera (Araneae: Lycosidae) from China. Molecular Phylogenetics and Evolution, 42(1), 104–121.
Clement, M., Snell, Q., Walker, P., Posada, D., & Crandall, K. (2002). TCS: Estimating gene genealogies 2: 0184–0184.
Correa-Ramírez, M. M., Jiménez, M. L., & García-De León, F. J. (2010). Testing species boundaries in Pardosa sierra (Araneae: Lycosidae) using female morphology and COI mtDNA. The Journal of Arachnology, 38, 538–554. https://doi.org/10.1636/Sh09-15.1
Costa, F. G., & Simó, M. (2014). Fenología de las arañas epígeas de una zona costera del sur de Uruguay: Un estudio bianual con trampas de caída. Boletín De La Sociedad Zoológica De Uruguay, 23(1), 1–15.
Croom, H. B., Gillespie, R. G., & Palumbi, S. R. (1991). Mitochondrial DNA sequences coding for a portion of the RNA of the small ribosomal subunits of Tetragnatha mandibulata and Tetragnatha hawaiensis (Araneae, Tetragnathidae). Journal of Arachnology, 19, 210–214. https://www.jstor.org/stable/3705892
De Queiroz, K. (2007). Species concepts and species delimitation. Systematic Biology, 56(6), 879–886. https://doi.org/10.1080/10635150701701083
Di Cola, V., Broennimann, O., Petitpierre, B., Breiner, F. T. D., Amen, M., Randin, C., Engler, R., Pottier, J., Pio, D., Dubuis, A., Pellissier, L., Mateo, R. G., Hordijk, W., Salamin, N., & Guisan, A. (2017). ecospat: An R package to support spatial analyses and modeling of species niches and distributions. Ecography, 40, 001–014. https://doi.org/10.1111/ecog.02671
Doménech, M., Crespo, L. C., Enguídanos, A., & Arnedo, M. A. (2020). Mitochondrial discordance in closely related Theridion spiders (Araneae, Theridiidae), with description of a new species of the T. melanurum group. Zoosystematics and Evolution, 96(1), 159–173. https://doi.org/10.3897/zse.96.49946
Edwards, S. V. (2009). Is a new and general theory of molecular systematics emerging? Evolution, 63, 1–19. https://doi.org/10.1111/j.1558-5646.2008.00549.x
Ermakov, O. A., Surin, V. L., Titov, S. V., Tagiev, A. F., Luk’yanenko, A. V., & Formozov, N. A. (2002). A molecular genetic study of hybridization in four species of ground squirrels (Spermophilus: Rodentia, Sciuridae). Russian Journal of Genetics, 38, 796–809. https://doi.org/10.1023/A:1016395722664
Fick, S. E., & Hijmans, R. J. (2017). Worldclim 2: New 1-km spatial resolution climate surfaces for global land areas. International Journal of Climatology, 37(12), 4302–4315.
Fitzpatrick, B. M., Fordyce, J. A., & Gavrilets, S. (2008). What, if anything, is sympatric speciation? Journal of Evolutionary Biology, 21(6), 1452–1459.
Foelix, R. F. (2011). Biology of spiders (3rd ed., p. 419). Oxford University Press.
Folmer, O., Black, M., Hoeh, W., Lutz, R., & Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294–299.
Fontes, F. V. H. M. (2016). Análise filogeográfica de duas espécies do gênero Aglaoctenus (Araneae, Lycosidae). Phylogeographical analysis of two Aglaoctenus species (Araneae, Lycosidae), Universidade Estadual de Campinas. Instituto de Biologia.
Ghione, S., Coelho, L., Costa, F. G., García, L. F., González, M., Jorge, C., Laborda, A., Montes de Oca, L., Pérez-Miles, F., Postiglioni, R., Simó, M., Toscano-Gadea, C. A., Viera, C., & Aisenberg, A. (2017). Arácnidos prioritarios para la conservación en Uruguay. Boletín de la Sociedad Zoológica del Uruguay (2ª época), 26(1), 1–8. https://doi.org/10.26462/26.1.1
Gilman, R. T., Fowler-Finn, K., & Hebets, E. A. (2018). A probable case of incipient speciation in Schizocosa wolf spiders driven by allochrony, habitat use, and female mate choice. The American Naturalist, 192(3), 332–346. https://doi.org/10.1086/698302
Gómez-Zurita, J., & Vogler, A. (2003). Incongruent nuclear and mitochondrial phylogeographic patterns in the Timarcha goettingensis species complex (Coleoptera, Chrysomelidae). Journal of Evolutionary Biology, 16, 833–843. https://doi.org/10.1046/j.1420-9101.2003.00599.x
Gonnet, V., Bidegaray-Batista, L., Aisenberg, A., Laborda, Á., Hagopián, D., Izquierdo, M. A., Piacentini, L. N., & Simó, M. (2021). A wolf spider from South American grasslands: Phylogenetic placement and redescription of Paratrochosina amica (Mello-Leitão 1941). Zoologischer Anzeiger, 295, 1–11. https://doi.org/10.1016/j.jcz.2021.08.009
González, M. (2015) Aspectos reproductivos de Aglaoctenus lagotis: estudio interpoblacional de una araña lobo sedentaria de gran variabilidad fenotípica. PhD thesis, Facultad de Ciencias Exactas, Físicas y Naturales, Universidad Nacional de Córdoba, Argentina, p 254.
González, M. (2018). Are multiple copulations harmful? Damage to male pedipalps in the funnel-web wolf spider Aglaoctenus lagotis (Araneae: Lycosidae). The Journal of Arachnology, 46, 162–164. https://doi.org/10.1636/JoA-S-17-017.1
González, M., Costa, F. G., & Peretti, A. V. (2014). Strong phenological differences between two populations of a Neotropical funnel-web wolf spider. Journal of Natural History, 48(35–36), 2183–2197. https://doi.org/10.1080/00222933.2014.908974
González, M., Costa, F. G., & Peretti, A. V. (2019). Different levels of polyandry in two populations of the funnel-web wolf spider Aglaoctenus lagotis from South America. Journal of Ethology, 37, 325–333. https://doi.org/10.1007/s10164-019-00606-5
González, M., Peretti, A. V., & Costa, F. G. (2015b). Reproductive isolation between two populations of Aglaoctenus lagotis, a funnel-web wolf spider. Biological Journal of the Linnean Society, 114, 646–658. https://doi.org/10.1111/bij.12448
González, M., Peretti, A.V., & Costa, F.G. (2015a). Efecto del sustrato sobre el cortejo de dos arañas lobo, una de tela y otra errante. Boletín de la Sociedad Zoológica del Uruguay (2ª época), 24(2), 57–72.
González, M., Peretti, A. V., Viera, C., & Costa, F. G. (2013). Differences in sexual behavior of two distant populations of the funnel-web wolf spider Aglaoctenus lagotis. Journal of Ethology, 31, 175–184. https://doi.org/10.1007/s10164-013-0365-1
González, M., & Toscano-Gadea, C.A. (2021). Can’t even trust the family? The web of the unusual web-building wolf spider Aglaoctenus lagotis (Araneae: Lycosidae) invaded by typical wandering wolf spiders. Arachnology, 18(7), 710–714. https://doi.org/10.13156/arac.2020.18.7.710
Hare, M. P. (2001). Prospects for nuclear gene phylogeography. Trends in Ecology and Evolution, 16, 700–706. https://doi.org/10.1016/S0169-5347(01)02326-6
Hausdorf, B., & Hennig, C. (2010). Species delimitation using dominant and codominant multilocus markers. Systematic Biology, 59(5), 491–503. https://doi.org/10.1093/sysbio/syq039
Hebets, E. A., & Uetz, G. W. (1999). Female responses to isolated signals from multimodal male courtship displays in the wolf spider genus Schizocosa (Araneae: Lycosidae). Animal Behaviour, 57(4), 865–872. https://doi.org/10.1006/anbe.1998.1048
Hedin, M. C., & Maddison, W. P. (2001). A combined molecular approach to phylogeny of the jumping spider subfamily Dendryphantinae (Araneae: Salticidae). Molecular Phylogenetics and Evolution, 18, 386–403. https://doi.org/10.1006/mpev.2000.0883
Heled, J., & Drummond, A. J. (2010). Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution, 27, 570–580. https://doi.org/10.1093/molbev/msp274
Hendry, A. P., & Day, T. (2005). Population structure attributable to reproductive time: Isolation by time and adaptation by time. Molecular Ecology, 14(4), 901–916. https://doi.org/10.1111/j.1365-294X.2005.02480.x
Hentz, N. M. (1844). Descriptions and figures of the araneides of the United States. Boston Journal of Natural History, 4, 386–396.
Hills, D. M. (2019). Species Delimitation in Herpetology. Journal of Herpetology, 53(1), 3–12. https://doi.org/10.1670/18-123
Hodge, M. A., & Marshall, S. D. (2018). Habitat associations of the web-building wolf spiders Sosippus floridanus and Sosippus placidus (Lycosidae: Sosippinae): A widespread generalist versus an endemic specialist. Journal of Arachnology, 46(3), 428–431. https://doi.org/10.1636/JoA-S-17-056.1
Holmberg, E. L. (1876). Arácnidos argentinos. Anales De Agricultura De La República Argentina, 4, 1–30.
Ivanov, V., Lee, K. M., & Mutanen, M. (2018). Mitonuclear discordance in wolf spiders: Genomic evidence for species integrity and introgression. Molecular Ecology, 27, 1681–1695. https://doi.org/10.1111/mec.14564
Jones, G. (2014). STACEY package documentation: Species delimitation and species tree estimation with BEAST2. http://www.indriid.com/software.html
Jones, G. (2017). Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. Journal of Mathematical Biology, 74, 447–467. https://doi.org/10.1007/s00285-016-1034-0
Kanesharatnam, K., & Benjamin, S. P. (2019). Multilocus genetic and morphological phylogenetic analysis reveals a radiation of shiny South Asian jumping spiders (Araneae, Salticidae). ZooKeys, 839, 1–81. https://doi.org/10.5061/dryad.b04s6t7
Katoh, K., Misawa, K., & Kuma, K. I. (2002). MAFFT: A novel method for rapid multiple sequence alignment on fast Fourrier transform. Nucleic Acids Research, 30, 3059–3066. https://doi.org/10.1093/nar/gkf436
Kopuchian, C., Campagna, L., Lijtmaer, D. A., Cabanne, G. S., García, N. C., Lavinia, P. D., Tubaro, P. L., Lovette, I., & Di Giacomo, A. S. (2020). A test of the riverine barrier hypothesis in the largest subtropical river basin in the Neotropics. Molecular Ecology, 29(12), 2137–2149. https://doi.org/10.1111/mec.15384
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Molecular Biology and Evolution, 35(6), 1547–1549. https://doi.org/10.1093/molbev/msy096
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., & Calcott, B. (2017). PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Molecular Biology and Evolution, 34, 772–773. https://doi.org/10.1093/molbev/msw260
Langone, J., Camargo, A., & de Sá, R. O. (2016). High genetic diversity but low population structure in the frog Pseudopaludicola falcipes (Hensel, 1867) (Amphibia, Anura) from the Pampas of South America. Molecular Phylogenetics and Evolution, 95, 137–151. https://doi.org/10.1016/j.ympev.2015.11.012
Leigh, J. W., & Bryant, D. (2015). PopART: Full-feature software for haplotype network construction. Methods in Ecology and Evolution, 6, 1110–1116. https://doi.org/10.1111/2041-210X.12410
Mace, G. M. (2004). The role of taxonomy in species conservation. Philosophical Transactions of the Royal Society B, 359, 711–719. https://doi.org/10.1098/rstb.2003.1454
Machado, G., & Macías-Ordóñez, R. (2007). Reproduction in: Harvestmen: The biology of Opiliones (Pinto da Rocha, R., Machado, G. & Giribet, G. eds.): 414–454. Cambridge (MA): Harvard University Press.
Macrini, C. M. T., Peres, E. A., & Solferini, V. N. (2015). Cryptic diversity of Agloactenus lagotis (Araneae, Lycosidae) in the Brazilian Atlantic rainforest: Evidence from microsatellite and mitochondrial DNA sequence data. Journal of Applied Biology and Biotechnology, 3, 009–014. https://doi.org/10.7324/JABB.2015.3602
Maddison, W. P. (1997). Gene trees in species trees. Systematic Biology, 46, 523–536. https://doi.org/10.1093/sysbio/46.3.523
Marquez, A., Maldonado, J. E., González, S., Beccaceci, M. D., Garcia, J. E., & Duarte, J. M. B. (2006). Phylogeography and Pleistocene demographic history of the endangered marsh deer (Blastocerus dichotomus) from the Río de la Plata Basin. Conservation Genetics, 7(4), 563–575. https://doi.org/10.1007/s10592-005-9067-8
Martin, P. R., Bonier, F., Moore, I. T., & Tewksbury, J. J. (2009). Latitudinal variation in the asynchrony of seasons: Implications for higher rates of population differentiation and speciation in the tropics. Ideas in Ecology and Evolution, 2, 9–17. https://doi.org/10.4033/iee.2009.2.3.n
Masta, S. E., & Maddison, W. P. (2002). Sexual selection driving diversification in jumping spiders. Proceedings of the National Academy of Sciences USA, 99(7), 4442–4447. https://doi.org/10.1073/pnas.072493099
Mastrantonio, V., Porretta, D., Urbanelli, S., Crasta, G., & Nascetti, G. (2016). Dynamics of mtDNA introgression during species range expansion: Insights from an experimental longitudinal study. Scientific Reports, 6, 1–8. https://doi.org/10.1038/srep30355
Medrano, J., Aasen, E., & Sharrow, L. (1990). DNA extraction from nucleated red blood cells. BioTechniques, 8, 43.
Mello-Leitão, C. F. (1939). Les arachnides et la zoogéographie de l’Argentine. Physis, Revista De La Sociedad Argentina De Ciencias Naturales, 17, 601–630.
Mello-Leitão, C. F. (1942). Cinco aranhas novas do Perú. Revista Brasileira De Biologia, 2, 429–434.
Milá, B., Carranza, S., Guillaume, O., & Clobert, J. (2010). Marked genetic structuring and extreme dispersal limitation in the Pyrenean brook newt Calotriton asper (Amphibia: Salamandridae) revealed by genome-wide AFLP but not mtDNA. Molecular Ecology, 19, 108–120. https://doi.org/10.1111/j.1365-294X.2009.04441.x
Miller, M. A., Pfeiffer, W., & Schwartz, T. (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees, 1–8. https://doi.org/10.1145/2016741.2016785
Miller, G. L., Stratton, G. E., Miller, P. R., & Hebets, E. (1998). Geographical variation in male courtship behavior and sexual isolation in wolf spiders of the genus Schizocosa. Animal Behaviour, 56, 937–951. https://doi.org/10.1006/anbe.1998.0851
Monaghan, M. T., Balke, M., Gregory, T. R., & Vogler, A. P. (2005). DNA-based species delineation in tropical beetles using mitochondrial and nuclear markers. Philosophical Transactions of the Royal Society London B Biological Sciences, 360, 1925–1933. https://doi.org/10.1098/rstb.2005.1724
Montes de Oca, L., & D`Elía, G., & Pérez-Miles, F. (2015). An integrative approach for species delimitation in the spider genus Grammostola (Theraphosidae, Mygalomorphae). Zoologica Scripta, 45(3), 322–333. https://doi.org/10.1111/zsc.12152
Mullen, S. P., Dopman, E. B., & Harrison, R. G. (2008). Hybrid zone origins, species boundaries, and the evolution of wing-pattern diversity in a polytypic species complex of North American admiral butterflies (Nymphalidae: Limenitis). Evolution, 62(6), 1400–1417. https://doi.org/10.1111/j.1558-5646.2008.00366.x
Müller, K. (2005). SeqState: Primer design and sequence statistics for phylogenetic DNA datasets. Applied Bioinformatics, 4, 65–69. https://doi.org/10.2165/00822942-200504010-00008
Murphy, N. P., Framenau, V. W., Donellan, S. C., Harvey, M. S., Park, Y. C., & Austin, A. D. (2006). Phylogenetic reconstruction of the wolf spiders (Araneae: Lycosidae) using sequences from the 12S rRNA, 28S rRNA, and NADH1 genes: Implications for classification, biogeography, and the evolution of web building behavior. Molecular Phylogenetics and Evolution, 38, 583–602. https://doi.org/10.1016/j.ympev.2005.09.004
Network, T. M. C. S. (2012). What do we need to know about speciation?. Trends in Ecology & Evolution, 27(1), 27–39. https://doi.org/10.1016/j.tree.2011.09.002
Newton, L. G., Starrett, J., Hendrixson, B. E., Derkarabetian, S., & Bond, J. E. (2020). Integrative species delimitation reveals cryptic diversity in the southern Appalachian Antrodiaetus unicolor (Araneae: Antrodiaetidae) species complex. Molecular Ecology, 29(12), 2269–2287. https://doi.org/10.1111/mec.15483
Olson, D. M., Dinerstein, E., Wikramanayake, E. D., Burgess, N. D., Powell, G. V. N., Underwood, E. C., D’amico, J. A., Itoua, I., Strand, H. E., Morrison, J. C., Loucks, C. J., Allnutt, T. F., Ricketts, T. H., Kura, Y., Lamoreux, J. F., Wettengel, W. W., Heda, P., & Kassem, K. R. (2001). Terrestrial ecoregions of the world: A new map of life on Earth: A new global map of terrestrial ecoregions provides an innovative tool for conserving biodiversity. BioScience, 51(11), 933–938. https://doi.org/10.1641/0006-3568(2001)051[0933:TEOTWA]2.0.CO;2
Padial, J., & M., Miralles, A., De la Riva, I., & Vences, M. (2010). The integrative future of taxonomy. Frontiers in Zoology, 7, 1–14. https://doi.org/10.1186/1742-9994-7-16
Peres, E. A., Sobral-Souza, T., Perez, M. F., Bonatelli, I. A., Silva, D. P., & Silva, M.r.J., & Solferini, V.N. (2015). Pleistocene Niche Stability and Lineage Diversification in the Subtropical Spider Araneus omnicolor (Araneidae). PLoS ONE, 10, e0121543. https://doi.org/10.1371/journal.pone.0121543
Peretti, A. V. (1997). Alternativas de gestación y producción de crías en escorpiones argentinos (Arachnida, Scorpiones). Iheringia, Série Zoologia, 82, 25–32.
Piacentini, L. (2011). Three new species and new records in the wolf spider subfamily Sosippinae from Argentina (Araneae: Lycosidae). Zootaxa, 3018, 27–49. https://doi.org/10.11646/zootaxa.3018.1.4
Piacentini, L. N., & Ramírez, M. J. (2019). Hunting the wolf: A molecular phylogeny of the wolf spiders (Araneae, Lycosidae). Molecular Phylogenetics and Evolution, 136, 227–240. https://doi.org/10.1016/j.ympev.2019.04.004
Piacentini, L. N., Scioscia, C. L., Carbajal, M. N., Ott, R., Brescovit, A. D., & Ramírez, M. J. (2017). A revision of the wolf spider genus Diapontia Keyserling, and the relationships of the subfamily Sosippinae (Araneae: Lycosidae). Arthropod Systematics and Phylogeny, 75(3), 387–415.
Pidancier, N., Jordan, S., Luikart, G., & Taberlet, P. (2006). Evolutionary history of the genus Capra (Mammalia, Artiodactyla): Discordance between mitochondrial DNA and Y-chromosome phylogenies. Molecular Phylogenetics and Evolution, 40, 739–749. https://doi.org/10.1016/j.ympev.2006.04.002
Pigliucci, M., & Kaplan, J. (2006). Species as family resemblance concepts: The (dis-)solution of the species problem? In Making sense of evolution: The Conceptual foundations of evolutionary biology. The University of Chicago Press, EEUU, 207–226. https://doi.org/10.1002/bies.10284
Planas, E., Fernandez-Montraveta, C., & Ribera, C. (2013). Molecular systematics of the wolf spider genus Lycosa (Araneae: Lycosidae) in the Western Mediterranean Basin. Molecular Phylogenetics and Evolution, 67(2), 414–428. https://doi.org/10.1016/j.ympev.2013.02.006
Polis, G. A. (1990). The biology of scorpions (p. 587). Stanford University Press.
Postiglioni, R., Bidegaray-Batista, L., Simó, M., & Arnedo, M. A. (2019). Move to stay: Genetic structure and demographic history of a wolf spider inhabiting coastal sand dunes of southern South America. Systematics and Biodiversity, 17(7), 635–649. https://doi.org/10.1080/14772000.2019.1689197
Punzo, F., & Haines, L. (2006). Body size, duration of embryonic development, growth rate, mother-offspring interaction, and diet in Sosippus floridanus Simon (Araneae: Lycosidae). Bulletin-British Arachnological Society, 13(9), 365–371.
Quantum GIS Development Team. (2020). QGIS Geographic Information System. Open Source Geospatial Foundation Project. https://www.qgis.org
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., & Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67, 901–904. https://doi.org/10.1093/sysbio/syy032
Reydon, T. A. C. (2019). Are species good units for biodiversity studies and conservation efforts? In D. Vecchi (Ed.), Casetta E, Marques da Silva J (pp. 167–193). From assessing to conserving biodiversity. Springer International Publishing.
Rojas-Soto, O. R., Baldo, D., Lescano, J., Encarnación-Luévano, A., Leynaud, G., & Nori, J. (2021). Seasonal dissociation in fossorial activity between the Llanos’ frog populations as a survival strategy in arid subtropical environments. Journal of Herpetology, 55, 442–451. https://doi.org/10.1670/20-096
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., Larget, B., Liu, L., Suchard, M. A., & Huelsenbeck, J. P. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542.
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., & Sánchez-Gracia, A. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Molecular Biology and Evolution, 34, 3299–3302. https://doi.org/10.1093/molbev/msx248
Rubio, G., Meza Torres, E., & Ávalos, G. (2005). Aspectos ecológicos de Aglaoctenus lagotis (Araneae: Lycosidae) con énfasis en la preferencia de hábitat. Corrientes Argentina. Revista Colombiana De Entomología, 31, 229–232.
Santos, A. J., & Brescovit, A. D. (2001). A revision of the South American spider genus Aglaoctenus Tullgren, 1905 (Araneae, Lycosidae, Sosippinae). Andrias, 15, 75–90.
Satler, J. D., Cartens, B. C., & Hedin, M. (2013). Multilocus species delimitation in a complex of morphologically conserved trapdoor spiders (Mygalomorphae, Antrodiaetidae, Aliatypus). Systematic Biology, 62(6), 805–823. https://doi.org/10.1093/sysbio/syt041
Simmons, M. P., & Ochoterena, H. (2000). Gaps as characters in sequence-based phylogenetic analyses. Systematic Biology, 49, 369–381. https://doi.org/10.1093/sysbio/49.2.369
Simon, E. (1895). Arachnides recueillis a la Terre-de-feu par M. Carlos Backhausen. Anales del Museo Nacional de Buenos Aires, 4(ser. 2, t. 1), 167–172.
Simon, E. (1898). Descriptions d’arachnides nouveaux des familles des Agelenidae, Pisauridae, Lycosidae et Oxyopidae. Annales De La Société Entomologique De Belgique, 42, 5–34.
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H., & Flooket, P. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America, 87, 651–701. https://doi.org/10.1093/aesa/87.6.651
Sites, J. W., & Marshall, J. C. (2004). Operational criteria for delimiting species. Annual Review of Ecology, Evolution, and Systematics, 35, 199–227. https://doi.org/10.1146/annurev.ecolsys.35.112202.130128
Sordi, S. (1996). Ecologia de populaçoes da aranha Porrimosa lagotis (Lycosidae) nas reservas Mata de Santa Genebra, Campinas (SP) e Serra do Japi, Jundai (SP). PhD Thesis, Universidade Estadual de Campinas, Sao Paulo, Brasil.
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stefani, V., & Del-Claro, K. (2011). Oviposition and post-embryonic development of Aglaoctenus lagotis (Araneae: Lycosidae). Zoologia, 28, 565–570. https://doi.org/10.1590/S1984-46702011000500003
Stefani, V., & Del-Claro, K. (2014). The effects of forest fragmentation on the population ecology and natural history of a funnel-web spider. Journal of Natural History, 49(3–4), 211–231. https://doi.org/10.1080/00222933.2014.909068
Stefani, V., Del-Claro, K., Silva, L. A., Guimaraes, B., & Tizo-Pedroso, E. (2011). Mating behavior and maternal care in the tropical savanna funnel-web spider Aglaoctenus lagotis Holmberg (Araneae: Lycosidae). Journal of Natural History, 45, 1119–1129. https://doi.org/10.1080/00222933.2011.552802
Stephens, M., & Donnelly, P. (2003). A comparison of Bayesian methods for haplotype reconstruction from population genotype data. American Journal of Human Genetics, 73, 1162–1169. https://doi.org/10.1086/379378
Stephens, M., Smith, N. J., & Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics, 68, 978–989. https://doi.org/10.1086/319501
Stratton, G. E. (1984). Morphological and behavioral correlates in Schizocosa: Behavior as a taxonomic tool in wolf spiders. American Zoologist, 24, 52A.
Stratton, G. E. (1997). A new species of Schizocosa from the southeastern USA (Araneae, Lycosidae). Journal of Arachnology, 25, 84–92.
Stratton, G. E., & Uetz, G. W. (1986). The inheritance of courtship behavior and its role as a reproductive isolating mechanism in two species of Schizocosa wolf spiders (Araneae; Lycosidae). Evolution, 40(1), 129–141. https://doi.org/10.1111/j.1558-5646.1986.tb05724.x
Taylor, R. S., & Friesen, V. L. (2017). The role of allochrony in speciation. Molecular Ecology, 26(13), 3330–3342. https://doi.org/10.1111/mec.14126
Toscano-Gadea, C. A. (2002). Fenología y distribución de la escorpiofauna del Cerro de Montevideo, Uruguay: Un estudio de dos años con trampas de caída. Revista Ibérica De Aracnología, 5, 77–82.
Toscano-Gadea, C. A., & Simó, M. (2004). La fauna de opiliones de un área costera del Río de la Plata (Uruguay). Revista Ibérica De Aracnología, 10, 157–162.
Tretzel, E. (1955). Intragenerische isolation und interspezifische konkurrenz bei spinnen. Zeitschrift Für Morphologie Und Ökologie Der Tiere, 44, 43–162.
Tullgren, A. (1905). Aranedia from the Swedish expedition through the Gran Chaco and the Cordilleras. Arkiv För Zoologi, 2(19), 1–81.
Turchetto-Zolet, A. C., Pinheiro, F., Salgueiro, F., & Palma-Silva, C. (2013). Phylogeographical patterns shed light on evolutionary process in South America. Molecular Ecology, 22(5), 1193–1213. https://doi.org/10.1111/mec.12164
Ubilla, M., Perea, D., Aguilar, C. G., & Lorenzo, N. (2004). Late Pleistocene vertebrates from northern Uruguay: Tools for biostratigraphic, climatic and environmental reconstruction. Quaternary International, 114(1), 129–142. https://doi.org/10.1016/S1040-6182(03)00048-X
Uetz, G. W., & Dondale, C. D. (1979). A new wolf spider in the genus Schizocosa (Araneae: Lycosidae) from Illinois. Journal of Arachnology, 7, 86–88.
Villamil, J., Avila, L. J., Morando, M., Sites, J. W., Jr., Leaché, A. D., Maneyro, R., & Camargo, A. (2019). Coalescent-based species delimitation in the sand lizards of the Liolaemus wiegmannii complex (Squamata: Liolaemidae). Molecular Phylogenetics and Evolution, 138, 89–101. https://doi.org/10.1016/j.ympev.2019.05.024
Vogler, A. P., & Monaghan, M. T. (2007). Recent advances in DNA taxonomy. Journal of Zoological Systematics and Evolutionary Research, 45, 1–10. https://doi.org/10.1111/j.1439-0469.2006.00384.x
Walckenaer, C. A. (1837). Histoire naturelle des insectes. Aptères. Tome premier. Roret, Paris, 682 pp., pl. 1–15.
Wang, L. Y., Li, Z. X., Zhou, K. X., & Zhang, Z. S. (2015). Redescription of three Hippasa species from China (Araneae: Lycosidae), with a proposed species group-division and diagnosis. Zootaxa, 3974(2), 231–244. https://doi.org/10.11646/zootaxa.3974.2.7
World Spider Catalog. (2022). World Spider Catalog. Version 23.5. Natural History Museum Bern, online at http://wsc.nmbe.ch, Accessed July 2022. 10.24436/2.
Yamamoto, S., & Sota, T. (2009). Incipient allochronic speciation by climatic disruption of the reproductive period. Proceedings of the Royal Society b: Biological Sciences, 276(1668), 2711–2719. https://doi.org/10.1098/rspb.2009.0349
Yoo, J. S., & Framenau, V. W. (2006). Systematics and biogeography of the sheet-web building wolf spider genus Venonia (Araneae: Lycosidae). Invertebrate Systematics, 20, 675–712. https://doi.org/10.1071/IS06013
Zhang, D., Tang, L., Cheng, Y., Hao, Y., Xiong, Y., Song, G., Qu, Y., Rheindt, F. E., Alström, P., & Jia, C. (2019). “Ghost introgression” as a cause of deep mitochondrial divergence in a bird species complex. Molecular Biology and Evolution, 36, 2375–2386. https://doi.org/10.1093/molbev/msz170
Acknowledgements
We would like to thank A. Aisenberg, A. Albín, M. Casacuberta, A. Laborda, C.A. Toscano-Gadea, and M. Trillo for their valuable help during the process of collecting material. We are also very grateful to Miguel Simó and Alvaro Laborda for helping us in the material revision and for loaning the specimens of Aglaoctenus deposited in the arachnological collection of Sección Entomología, Facultad de Ciencias, Montevideo, Uruguay. Diana Silva also loaned specimens deposited in the Pontifical Catholic University of Perú. Eduardo Aisenberg has kindly reviewed linguistic terms. We also thank the two anonymous reviewers for their critical reading and valuable comments on the manuscript.
Funding
The project was undertaken with financial supports of the HLMFAR-American Arachnological Society, grant to M. González, and of the Programa de Desarrollo de las Ciencias Básicas (PEDECIBA) and Sistema Nacional de Investigadores, Agencia Nacional de Investigación e Innovación (ANII, Uruguay) to L. Bidegaray-Batista and M. González as well as from the Dr. Carlos Carbajal Campi Fund, FAICE GENBIO. N. Kacevas also received financial support from the ANII postgraduate grant (POS_NAC_2018_1_151155), J. Nori from the Foncyt (PICT 2017 2666) and Secyt, and L. Piacentini from CONICET iBol Argentina 2012 (PICT 2015–0283).
Author information
Authors and Affiliations
Contributions
All authors whose names appear on the submission (1) made substantial contributions to the conception or design of the work or to the acquisition, analysis, or interpretation of data, (2) drafted the work or revised it critically for important intellectual content, (3) approved the version to be published, and (4) agree to be made accountable for all the work aspects hereby ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval
No approval of research ethics committees was required in order to accomplish the goals of this study given that experimental work was conducted with an unregulated invertebrate species.
Consent to participate and for publication
All authors agreed with the content, gave explicit consent to submit the present manuscript, and have obtained consent from the responsible authorities at the institutes where the work has been carried out.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
13127_2022_586_MOESM1_ESM.docx
Supplementary file1 Table S1. Geographical records of A. lagotis and their respective localities used for the ecological niche comparisons. These comparisons were developed between the main lineages recovered in the molecular species delimitation analyses (Form I, Form IIa and Form IIb). (DOCX 14 KB)
13127_2022_586_MOESM2_ESM.docx
Supplementary file2 Table S2. Examined specimens of A. lagotis deposited in the Museo Argentino de Ciencias Naturales (MACN, Argentina) and identified as Form I or Form II. (DOCX 27 KB)
13127_2022_586_MOESM3_ESM.docx
Supplementary file3 Table S3. Examined photograph material of A. lagotis from the iNaturalist platform and identified as Form I or Form II, based on the coloration pattern on the abdomen as a proxy. (DOCX 28 KB)
13127_2022_586_MOESM4_ESM.txt
Supplementary file4 Table S4. R-Scripts used in the ecospat-analyses performed during the niche similarity analyses. (TXT 16 KB)
13127_2022_586_MOESM5_ESM.docx
Supplementary file5 Table S5. P-distance among and within the main lineages of A. lagotis for the mitochondrial genes and nuclear intron. (DOCX 14 KB)
13127_2022_586_MOESM6_ESM.tif
Supplementary file6 Figure S1. Statistical parsimony network of the mitochondrial concatenated genes of A. lagotis. Colors of the pie plots correspond to the identifications of the individuals as in Figs. 1 and 2 (TIF 4410 KB)
13127_2022_586_MOESM7_ESM.tif
Supplementary file7 Figure S2. Statistical parsimony network of the cox1 gene of A. lagotis. Colors of the pie plots correspond to the identifications of the individuals as in Figs. 1 and 2 (TIF 7361 KB)
13127_2022_586_MOESM8_ESM.tif
Supplementary file8 Figure S3. Summary species tree obtained with the species delimitation analyses in STACEY. Colors boxes indicate the clusters identified by STACEY. Bayesian posterior probability values are indicated above each node. The names of clades denote main lineages identified in the concatenated mitochondrial gene tree (TIF 8475 KB)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
González, M., Kacevas, N., Nori, J. et al. Not the same: phylogenetic relationships and ecological niche comparisons between two different forms of Aglaoctenus lagotis from Argentina and Uruguay. Org Divers Evol 23, 103–124 (2023). https://doi.org/10.1007/s13127-022-00586-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-022-00586-4