
Abstract
Analyzing the roles of ecology and geography on speciation and lineage diversification can shed light on the processes that generate and maintain biodiversity. Additionally, lineages rapidly diversifying across unstable habitats provide substantial challenges for resolving evolutionary histories and delimiting species. Physaria is represented in South American by six species distributed from southern Bolivia to northern central Argentina and growing in highlands of the southern-central Andes, but also along the hills and lowlands of central-eastern Argentina. This biogeographical variability, not common among other South American crucifers (Brassicaceae), prompted us to conduct different climatic niche and geographical range comparisons to study the potential roles of ecology and geography through the diversification of the group. However, the remarkable similarity between these species, coupled with the continuous variability of the diagnostic morphological characters, blurs the species boundaries. Therefore, in order to identify independent evolving lineages, we first employed species delimitation methods together with the general lineage concept of species, and used molecular sequences from nuclear ribosomal and chloroplast loci. Secondly, and in the light of the lineages obtained, we explored the roles of geography and ecology on the diversification of South American Physaria and tested for presence of phylogenetic niche-conservatism or niche-divergence patterns, as well as potential ecological speciation. Lineages identified by these delimitation methods were highly congruent with described species; nevertheless, some morphospecies were included under the same independent evolutionary lineage. Our results suggest that the climatic niche divergence along the heterogeneous landscape apparently was a major factor promoting diversification of the South American Physaria. Divergence was registered mainly on the temperature dimension, which promoted shifts between cold-temperate habitats associated with the highlands of the central-southern Andes and warm lowlands from central-eastern Argentina, i.e., the Monte and Dry Chaco ecoregions. In addition, some degree of niche divergence along the precipitation gradient was also secondarily recovered. Allopatry and dispersal capabilities also seem to be associated with the diversification of the group, presumably through the Late Pliocene-Pleistocene, and promoted by glacial cycles and climatic oscillations during the Quaternary. Results of these analyses are also discussed in a general context, which will contribute to the understanding of the evolutionary and ecological patterns of South American Brassicaceae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A central question of evolutionary biology is to understand the relative roles of ecology and geography in lineage divergence and speciation (Coyne and Orr 2004; Nosil 2012), and molecular phylogenetic analyses, coupled with morphological, ecological, and geographical data, can reveal patterns of differentiation associated with underlying diversification processes (Barraclough and Vogler 2000; Crandall et al. 2000; Schluter 2001; Fitzpatrick and Turelli 2006). Environmental and geographical factors play a key role in shaping species diversification and biodiversity, acting directly on processes of speciation, dispersion, persistence, and extinction (Ricklefs 1987; Wiens 2012; Futuyma and Kirkpatrick 2017). Physical barriers, and geographic distances in organism with low dispersal capabilities, can restrict or reduce gene flow among populations, promoting speciation both via genetic drift (non-adaptative speciation) and/or local adaptation (Jordan 1905; Mayr 1959; Barraclough and Vogler 2000; Fitzpatrick and Turelli 2006; Kisel and Barraclough 2010; Phillimore 2014). Alternatively, ecological adaptation to local environments results in an important process promoting divergence in nature (Rundle and Nosil 2005; Schluter 2001; Nosil 2012; Pyron et al. 2015), and numerous studies reported its importance in plant diversification (Levin 2003; Givnish 2010; Schemske 2010; Ramsey 2011; Kolář et al. 2016; Nürk et al. 2018; Liu et al. 2020). In this context, divergent natural selection drives the reduction of gene flow between populations as a consequence of adaptation to different ecological niches (Schluter 2009; Rundle and Nosil 2005), which may eventually result in complete reproductive isolation (ecological speciation—Nosil and Harmon 2009; Pyron et al. 2015). Alternatively, adaptation to the ecological niche can also promote speciation by phylogenetic niche conservatism (PNC) (Wiens 2004a; Wiens and Graham 2005; Pyron et al. 2015; Kozak and Wiens 2006), defined as the tendency of closely related species to retain characteristics of their fundamental niche over time (Peterson et al. 1999; Peterson 2011; Wiens et al. 2010). In this case, ecological constraints and stabilizing selection play a role in speciation, particularly during periods of environmental change, limiting adaptation to new climatic conditions when ancestral distributional ranges become fragmented (e.g., along elevational gradients during periods of warming or orogeny), and incipient species fail to adapt to novel environmental conditions that would facilitate the maintenance of gene flow (Wiens 2004a, b; Pyron et al. 2015). Therefore, the relative contributions of geography, niche divergence, and niche conservatism during the speciation processes should consequently affect the diversification of lineages, especially in plants because they may be more affected by small-scale heterogeneity than mobile organisms, increasing the relative significance of geographic and environmental differentiation (Anacker and Strauss 2014). Refining our understanding of the phylogenetic relationships and the ecological-geographical boundaries of taxa provides an essential framework for other fields in the study of biodiversity, especially conservation (Moritz 1994; Riddle and Hafner 1999).
However, the study of patterns and processes associated with lineage diversification involves a critical step, the delimitation of the evolutionary units reflected in the species boundaries. Since species delimitation is inextricably linked to patterns of species diversity and diversification, the criteria used to delimit species have profound implications to understanding the dynamics of these processes (de Queiroz 1998). Consequently, species delimitation becomes a critical issue in evolutionary, ecological, and conservation studies (Riddle and Hafner 1999; Agapow et al. 2004; Isaac et al. 2004; Dayrat 2005; Sukumaran and Knowles 2017), particularly for taxonomically complex groups (Federici et al. 2013) with low levels of evolutionary and ecological differentiation. Recently, divergent species can rapidly adapt to local conditions, often resulting in significant ecological and morphological divergence, but with few neutral genetic changes (Simpson 1953; Schluter 2000; Sudhaus 2004). Conversely, species isolated for a long time by a barrier may accumulate genetic differences of neutral evolution, but as the environments may remain similar, few ecological or morphological differences may occur (Coyne and Orr 2004; Phillimore 2014). Therefore, species delimitation integrating multiple datasets is more desirable (Dayrat 2005; Carstens et al. 2013; Sukumaran and Knowles 2017). Although different concepts can be applied to delimit species boundaries (e.g., biological, evolutionary, ecological, phylogenetic), a unified species concept can be achieved through the General Lineage Concept (de Queiroz 1998, 1999, 2007) by treating species as separately evolving metapopulation lineages through time (Simpson 1951; Wiley 1978; de Queiroz 1998, 2005, 2007), and with the other secondary species criteria as complementary (but not exclusive) lines of evidence to support lineage separation. Thus, the primary objective to analyze patterns of diversification is to identify independent evolutionary lineages, also interpreted as evolutionary significant units (ESUs—Moritz 1994).
The mustard family (Brassicaceae) is well represented in South America by ca. 406 native species (ca. 10% of the family) distributed mainly along the Andes. These species inhabit a variety of different habitats along the biogeographical provinces of North Andean Paramo, Puna, Prepuna, Altoandina, Yungas, and Subandean Patagonia (Cabrera and Willink 1973; Morrone 2017). These regions, together with the Atacama-Sechura Desert, the Chilean Matorral, and the Patagonian Steppe, provide a high diversity of habitats for the diversification of numerous plant groups (Luebert and Weigend 2014), including several lineages of this family (e.g., Salariato et al. 2016). The genus Physaria (Nutt.) A. Gray (tribe Physarieae, Al-Shehbaz 2012a) includes ca. 105 species distributed primarily in western North America, and morphologically characterized by having stellate trichomes and silicles (Al-Shehbaz and O’Kane 2002). Of these, the six species endemic to South America grow in Argentina and neighboring Bolivia (Al-Shehbaz 2012b). A single species, P. mendocina (Phil.) O’Kane & Al-Shehbaz, was previously thought to be the only species growing in South America (Boelcke 1967; Boelcke and Romanezuk 1984); however, with a better understanding of the morphological variation in the genus and substantial fieldwork, the number of species was subsequently elevated to six (O’Kane and Al-Shehbaz 2004; Al-Shehbaz and Prina 2009) (Fig. 1). Like other Andean genera, some of these species, such as P. urbaniana (Muschl.) O’Kane & Al-Shehbaz and P. pygmaea O’Kane & Al-Shehbaz inhabit highlands of the central Andes up to 5000 m, but others, as P. mendocina or P. lateralis O’Kane & Al-Shehbaz, are also distributed in lowlands of central and eastern Argentina (Fig. 2), primarily along the biogeographical provinces of Chaco and Monte (Cabrera and Willink 1973), where crucifer biodiversity are notably less represented (Al-Shehbaz 2012b). Of the 164 native species in Argentina, approx. 80% are distributed along the Altoandina, Puna, Prepuna, and Patagonia biogeographical provinces, while the remaining 20% inhabit the Chaco, Espinal, Pampa, and Monte provinces (http://www.floraargentina.edu.ar/). Although PNC has been the predominant ecological pattern recovered for South American Brassicaceae (Salariato and Zuloaga 2017; Salariato et al. 2018, 2020), the diversity of environments where these species grow seems to suggest the potential presence of niche divergence during their diversification. Additionally, species of Physaria also exhibit considerable geographic-range asymmetry, as evident in the wide distribution range of P. mendocina (Fig. 2), which grows along the Dry Chaco, Low Monte, and Espinal ecoregions (Olson et al. 2001), and the micro-endemic distributions of P. okanensis Al-Shehbaz & Prina and P. crassistigma, which are restricted to the High Monte and the Southern Andean steppe ecoregions, respectively.

Representatives of South American Physaria. a–c P. crassistigma. a Plant with flowers. b Plant with fruits. c Detail of fruits. d–e P. lateralis. d Plant with flowers and fruits. e Detail of fruits. f–g P. mendocina. f Plant with flowers. g Plant with fruits. h–i P. pygmaea. h Plant with flowers and fruits. i Details of fruits. j–l P. urbaniana. j Plant with flowers. k Plant with fruits. l Detail of fruits. a–c from Salariato et al. 233 (SI), d–e from Zuloaga et al. 12408 (SI), f from Zuloaga et al. 15741 (SI), g from Deginani et al. 2173 (SI), h–i from Zuloaga et al. 13572 (SI), j–k from Zuloaga et al. 12956 (SI), l from Zanotti et al. 787 (SI). Photos by Diego L. Salariato (a–c), Fernando O. Zuloaga (d–f, h–k), Norma B. Deginani (g), Christian Zanotti (l)

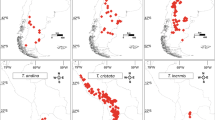
Distribution map of South American Physaria. Dots represent specimens of South American Physaria species. Blue: P. crassistigma, pink: P. lateralis, red: P. mendocina, black: P. okanensis, yellow: P. pygmaea, green: P. urbaniana. Labels associated with dots indicate sampled specimens used in the molecular analyses (see Appendix 1 for complete vouchers)
The above data prompted us to analyze patterns associated to the climatic-niche and geographical-range evolution within this group. However, the remarkable similarities between these species, coupled with the variability of the diagnostic morphological characters (O’Kane and Al-Shehbaz 2004; Al-Shehbaz 2012b) (Fig. 1), complicate the a priori delimitation of species in the sense of evolutionary significant units. Hence, the goal of this work is first to identify evolutionary independent lineages under the generalized lineage concept of species by using molecular sequences from nuclear ribosomal and chloroplast loci together with morphological, ecological, and geographic data. Secondly, and in the light of the lineages obtained, to explore the roles of geography and ecology on the diversification of South American Physaria by testing for presence of PNC or niche-divergence patterns and potential ecological speciation. Results of these analyses are also discussed in a general context to contribute to the understanding of the evolutionary and ecological patterns of South American Brassicaceae.
Materials and methods
Sampling and DNA sequencing
For the molecular analyses, we sampled 47 accessions representing the six currently accepted morphospecies of South American Physaria (O’Kane and Al-Shehbaz 2004; Al-Shehbaz and Prina 2009; Al-Shehbaz 2012b), covering all major geographical areas and morphological variation: P. crassistigma (6), P. lateralis (11), P. mendocina (16), P. okanensis (2), P. pygmaea (4), and P. urbaniana (8) (Fig. 2). To conduct species-delimitation analyses, we generated ITS (nrDNA), trnL-F, trnH-psbA, trnG intron, and trnS-trnG (cpDNA) sequences for all Physaria accessions. Voucher information and GenBank accession numbers for sequences used in this study are provided in Appendix S1. Total DNA was isolated from leaves (collected in the field and dried in silica gel) using a modified (CTAB) protocol by Doyle and Doyle (1987), or from herbarium material using a DNeasy plant mini kit (Qiagen, Hilden, Germany). The nuclear ribosomal ITS region (ITS1-5.8S-ITS2) was amplified by PCR in one or two fragments using the ITS2, ITS3, ITS4, and ITS5 primers of Baldwin (1992); the chloroplast trnL-F region (trnL intron/trnL-F spacer) was amplified in one or two fragments using primers C, D, and E of Taberlet et al. (1991), and Fdw (Salariato et al. 2013). Sequences for trnH-psbA spacer, trnG intron, and trnS-trnG spacer were amplified in one fragment using primers trnH (GUG)/psbA (Hamilton 1999), trnG-F/trnG-R (Tewes et al. 2018), and T1/T2 (Liu et al. 2011), respectively. The PCR reactions were performed in 25 μL final volumes with 50–100 ng of template DNA, 0.2 μM of each primer, 25 μM dNTP, 5 mM MgCl2, 1 Χ buffer, and 1.5 units of Taq polymerase provided by Invitrogen Life Technologies (São Paulo, Brazil). The PCR amplifications were set at the following conditions for most species: (ITS) 94 °C, 3 min; 35 × (94 °C, 30 s; 50 °C, 60 s; 72 °C, 90 s); 72 °C, 7 min; (trnL-F) 94 °C, 3 min; 35 × (94 °C, 30 s; 48 °C, 60 s; 72 °C, 90 s); 72 °C, 7 min; (trnH-psbA) 94 °C, 3 min, 35 × (94 °C, 30 s; 52 °C, 60 s; 72 °C, 90 s); 72 °C, 10 min; (trnG intron) 94 °C, 3 min; 35 × (94 °C, 30 s; 50 °C, 60 s; 72 °C, 90 s); 72 °C, 7 min; (trnS-trnG spacer) 94 °C, 3 min; 35 × (94 °C, 30 s; 54 °C, 60 s; 72 °C, 90 s); 72 °C, 7 min. Cleaning of PCR products was done by Macrogen, Inc. (Seoul, South Korea), using the Montage PCR purification kit from Millipore and following the manufacturer’s protocol. Sequencing reactions were also performed by Macrogen using the ABI PRISM BigDye Terminator cycle-sequencing kits with AmpliTaq DNA polymerase (Applied Biosystems, Seoul, South Korea) following the protocols supplied by the manufacturer. Sequences were assembled and edited using the program Chromas Pro 1.7.7 (Technelysium Pty Ltd., Brisbane, Australia), which was also used for checking the presence of single peaks in the chromatograms, especially in the ITS sequences. In total, 221 new sequences were obtained and submitted to GenBank (Appendix S1). Alignments were generated with MUSCLE 3.8.31 (Edgar 2004) using a first round of multiple alignments and posterior rounds of refinement under the default settings. The alignments obtained were then checked and improved manually where necessary using Bioedit 7.2.5 (Hall 1999). For the trnS-trnG alignment, a hyper variable poly-AT region of 140 bp was removed previous to analyses. Aligned matrices, and all other supplemental data, are available from the Supporting Information, TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S26615) and the Figshare Digital Repository (https://doi.org/10.6084/m9.figshare.12645218). Monophyly of South American Physaria was previously reported by O’Kane (2012), but it was also corroborated here, previous to the phylogenetic and species delimitation analyses using ITS sequences for representatives of South American Physaria species and a broad sampling of North American species. Because monophyly was confirmed (Fig. S1, Supplementary Material), posterior phylogenetic analyses were rooted using the North American species P. fendleri (A.Gray) O’Kane & Al-Shehbaz.
Species delimitation: discovery approaches
First, we explored relationships among DNA sequences within the nrDNA and the cpDNA datasets using median-joining networks (mjn) with the package pegas 0.13 (Paradis 2010) of R 3.5.2 (R Core Team 2018), also analyzing the distribution of haplotypes within morphospecies and ecoregions. Second, to investigate phylogenetic relationships among specimens of South American Physaria, individual and concatenated datasets (ITS, cpDNA, and ITS + cpDNA) were analyzed using maximum likelihood (ML) and Bayesian inference (BI). Best-fit models of nucleotide evolution were identified using the Akaike information criterion (AIC) implemented in jModeltest2 2.1.6 (Darriba et al. 2012): K80+G (ITS), TPM1uf+G (both trnL-F and trnG intron), F81+G (trnH-psbA), and F81+I (trnS-trnG). The ML analyses were conducted in RAxML 8.2.10 (Stamatakis 2014) using nonparametric bootstrap (BS) analysis and searches for the best-scoring ML tree in a single run (Stamatakis et al. 2008). We performed 1000 rapid bootstrap inferences and a thorough ML search under the GTRGAMMA (ITS, trnL-F, trnH-psbA, trnG intron) and GTRGAMMAI (trnS-trnG) models. Bayesian analyses were conducted using MrBayes 3.2.6 (Ronquist et al. 2012) setting the number of substitution types to “mixed” (which results in the Markov chain sampling over the space of all possible reversible substitution models), and rates = gamma (ITS, trnL-F, trnH-psbA, trnG intron) or propinv (trnS-trnG). Two simultaneous analyses, starting from different random trees and with four Markov Monte Carlo chains, were run for 40 million generations, sampling every 10,000 generations to ensure independence of the successive samples. The convergence and effective sample size were checked with the average standard deviation of split frequencies (ASDSF) < 0.01, the potential scale reduction factor (PSRF) ~ 1, and the effective sample size (ESS) for all parameters > 200. The first 1000 trees (25% of total trees) were discarded as burn-in, and the remaining samples of each run were combined and used to calculate the 50% majority rule consensus tree and the maximum clade credibility (MCC) tree, the latter estimated using TreeAnnotator 1.8.4 (Drummond et al. 2012) (http://beast.community/treeannotator). Trees obtained in ML and BI analyses are available from TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S26615) and the Figshare Digital Repository (https://doi.org/10.6084/m9.figshare.12645218). All RAxML and MrBayes analyses were conducted in the CIPRES Science Gateway 3.3 (http://www.phylo.org/) (Miller et al. 2010).
To address levels of discordance among nuclear ribosomal (ITS) and plastid (trnL-F, trnH-psbA, trnG intron, trnS-trnG), specimen trees, and their influence on the concatenated analyses, congruence among partitions was assessed using a Bayesian concordance analysis (BCA) (Ané et al. 2007; Baum 2007) implemented in the software BUCKy 1.4.4 (Larget et al. 2010). The BUCKy analysis was conducted using the posterior distribution of the ITS and cpDNA gene trees produced with MrBayes, with two runs, four chains, and one million generations following a burn-in of 100,000 (10%), while the discordance parameter (α), which represents the a priori expected level of discordance, was set to 1, 10, and 100. In addition to the concatenated and concordance analyses, incongruences between ITS and cpDNA data were also visualized in a filtered supernetwork calculated with SplitsTree 4.14.18 (Huson and Bryant 2006) using the Z-closure algorithm (Huson et al. 2004), 1000 Bayesian posterior trees of each nuclear and plastid dataset, and filtering the splits to show only those present in a minimum of 30% input trees.
For identification of putative independent evolutionary lineages, we also applied the Generalized Mixed Yule Coalescent (GMYC) method (Pons et al. 2006; Fujisawa and Barraclough 2013) implemented in the R package splits 1.0-19 (Ezard et al. 2009). The GMYC method uses a maximum likelihood framework to delimit species by fitting intraspecific (coalescence) and interspecific (yule) species branching models on ultrametric trees, estimating the transition point before which all nodes reflect species diversification events and after which all nodes represent a population coalescent process (Pons et al. 2006; Fujisawa and Barraclough 2013). This method was designed for the analysis of single-locus data, but is frequently applied to ultrametric trees from concatenated multilocus data by postulating a shared genealogical history (e.g., Arrigoni et al. 2016; Nieto-Montes de Oca et al. 2017; Renner et al. 2017). The GYMC analyses were conducted under the single-threshold model as recommend Fujisawa and Barraclough (2013), and with ultrametric trees obtained from ITS, cpDNA, and concatenated ITS + cpDNA data using BEAST 1.8.4 (Drummond et al. 2012). For tree estimation, we used four runs of 100 million generations sampling every 25,000, an uncorrelated lognormal clock model (UCLN), a yule process for the species tree prior, and the models of nucleotide substitution and relaxed clock unlinked across character partition. For estimation of relative divergence times, the most recent common ancestor (MRCA) of South American Physaria was calibrated under a normal distribution of mean=1 and sd=0.01. The first 25% of each run was discarded as burn-in, and ESS > 200 was checked in Tracer 1.7.1 (Rambaut et al. 2018). Replicates were combined using LogCombiner 1.8.4, and the MCC tree calculated with TreeAnnotator 1.8.4 (http://beast.community/treeannotator) was used in the GYMC analyses. Alternatively, assuming that discordance between partitions could be caused by incomplete lineage sorting, and in order to compare with the GMYC results obtained from the concatenated analyses, we also estimated specimen trees with the ITS + cpDNA data under the multispecies coalescent (MSC) model implemented in *BEAST extension (Heled and Drummond 2010), using all accessions as separate operational taxonomic units (OTUs). An UCLN clock model, the Yule tree prior, and the piecewise linear with constant root for the population size model were assigned to each sampled locus. Six runs were conducted in BEAST using 100 million generations and sampling every 25,000. After checking for convergence and ESS, and discarding the burn-in (25%), the MCC tree was calculated and used in the GYMC analyses. All BEAST analyses were conducted in the CIPRES Science Gateway v3.3 (www.phylo.org; Miller et al. 2010).
Finally, potential evolutionary independent lineages identified with molecular data were contrasted with morphospecies (sensu O’Kane and Al-Shehbaz 2004; Al-Shehbaz and Prina 2009; Al-Shehbaz 2012b) and ecoregions (sensu Olson et al. 2001) they inhabit, selecting the lineages (primary hypotheses) that maximize the congruence of all data sources. For morphological assignment, we examined 366 herbarium specimens mainly from BA, BAA, CORD, LIL, LPB, MERL, and SI (acronyms follow Thiers 2020) and fresh material collected during field trips along the Andes of Argentina and Bolivia (specimen vouchers in Table S1 of Supplementary Material). For ecoregion mapping, we used specimen occurrence data (see climatic niche analyses), extracting from a shapefile based on the ecoregions proposed by Olson et al. (2001) (https://c402277.ssl.cf1.rackcdn.com/publications/15/files/original/official_teow.zip?1349272619) the corresponding ecoregion for each occurrence point. Additionally, we also registered biogeographical provinces where specimens occur using the bioregionalization and shapefiles proposed by Morrone (2015), Romano (2017), and Arana et al. (2017) for the Andes and Argentina.
Species delimitation: validation approaches
Lineages identified with the discovery approaches were first tested using the multispecies coalescent (MSC) model implemented in BPP (Bayesian Phylogenetics and Phylogeography) 4.1.4 (Yang and Rannala 2014; Yang 2015; Flouri et al. 2018). BPP analyzes multiple loci under the MSC to compare different models of species delimitation and species phylogeny (Yang and Rannala 2014; Rannala and Yang 2017) in a Bayesian framework, estimating relative species-divergence times (τs) and population sizes for both modern and ancestral species (θs). BPP was shown to outperform other species delimitation methods across different speciation scenarios, generally producing fewer overestimates of the number of species than other methods (Luo et al. 2018). We conducted joint-lineage delimitation and species-tree estimation (algorithm A11—unguided species delimitation) (Yang and Rannala 2014), assigning specimens to the lineages previously delimited under the discovery approaches. To perform the analysis, we separated data in 2 partitions: ribosomal nuclear and concatenated plastid data. Analyses were conducted using the default prior for the species-tree models (speciesmodelprior = 1) assigning equal probabilities for the rooted trees (Yang and Rannala 2014). For priors of parameters τ0 and θ, we used inverse gamma distributions [IG(α, β)] considering α = 3 (diffuse prior) and values of β that cover different alternative scenarios for ancestral population size θ = IG(3, 0.002), IG(3, 0.02), IG(3, 0.2), and root age τ0 = IG(3, 0.004), IG(3, 0.2). Consequently, we analyzed a total of six different combinations of parameters, where θ = IG(3, 0.2) τ0 = IG(3, 0.004) (large population size and shallow divergence) was the most conservative speciation scenario, favoring models containing fewer species (Leaché and Fujita 2010). Each combination was run twice using different random seeds in order to check convergence and the MCMC was set to 500,000 samples with burn-in = 10,000 and sample frequency = 50.
Second, we employed the Bayes factor-delimitation approach (BFD) (Grummer et al. 2014) for lineage validation. Unlike BPP, which provides species delimitation probabilities, BFD provides a ranking of the species delimitation models that are tested. In BFD, marginal likelihood estimates (MLEs) of each competing species delimitation hypotheses are estimated and Bayes factors (BFs) are used to assess their support. Different hypotheses were tested using the MSC model in *BEAST, with the nucleotide substitution models selected in jModeltest2, an uncorrelated lognormal clock model (UCLN), a Yule process for the species tree prior, and the piecewise linear with constant root for the population size model assigned to each locus. For each hypothesis, four runs were conducted using 100 million generations and sampling every 10,000. The BF was calculated using MLE obtained both by path-sampling (PS; Lartillot and Philippe 2006) and stepping-stone sampling methods (Xie et al. 2011) with 100 steps of one million generations each and α = 0.3. All *BEAST analyses were conducted in the CIPRES Science Gateway v3.3 (www.phylo.org; Miller et al. 2010). The 2lnBF = 2[MLE (model 1)−MLE (model 2 )] was calculated to compare the competing hypotheses following criteria of Kass and Raftery (1995): 2lnBF = 0–2 “not worth more than a bare mention,” 2lnBF = 2–6 “positive” support, 2lnBF = 6–10 “strong” support, and 2lnBF > 10 “decisive” support for model 1 over 2. Finally, MCC species tree of the best ranked model was obtained with TreeAnnotator discarding the first 25% of each run.
Finally, we also evaluated lineage boundaries using the genealogical divergence index (gdi) (Jackson et al. 2017; Leaché et al. 2019) following the heuristic approach conducted in Chan and Grismer (2019). We performed the A00 analysis in BPP to generate posterior distributions for the parameters θ and τ. For prior assignment of these parameters, we used α = 3 for both θ and τ0, and empirical estimations of β adjusted as follows: (1) for θ using the mean (mθ) estimate of pairwise uncorrected p-distances within each lineage calculated with the R package ape 5.3. (Paradis and Schliep 2018), and (2) for τ0 using the mean (mτ) estimate of the MRCA height of South American Physaria from the species trees obtained in the BEAST analyses. Values of β were then calculated using the equation m = β/(α−1), for α > 2 (Flouri et al. 2018) (empirical values obtained: βθ = 0.018, βτ = 0.02). Two separate MCMC runs (1 million generations, sampling each 1000 generations, and with burn-in = 10,000) were performed to ensure convergence and combined to generate posterior distributions of θ and τ parameters that were subsequently used to calculate the gdi following the equation: gdi = 1 − e −2τ/θ (Jackson et al. 2017; Leaché et al. 2019), with population A distinguished from population B by 2τAB/θA. Lineages are considered distinct species when gdi values are > 0.7, while low gdi values < 0.2 indicate that populations belong to the same lineage. Values of 0.2 ≤ gdi ≤ 0.7 indicate ambiguous lineage status (Jackson et al. 2017).
Climatic niche analyses
Once evolutionary independent lineages and their boundaries were defined with the delimitation methods (see above), we conducted climatic-niche comparisons among them in order to detect PNC or niche-divergence patterns. We used occurrence data obtained from the examination of specimens deposited in different herbaria and field collections (specimen vouchers in Table S1, Supplementary Material). All records were previously mapped using QGIS v2.18.12 “Las Palmas” (Quantum GIS Development Team 2016) for visual inspection, and in cases of specimens with no GPS coordinates but exact locality names, records were georeferenced using Google Earth Pro v7.3.2.5776 (https://www.google.com/intl/en/earth/). After removing duplicates and occurrences closer to 30 arc-seconds (~ 1 km), we obtained a total of 312 data points corresponding to six evolutionary lineages, with an average of 52 data points per lineage and ranging from eight to 110 (crassistigma lineage: 63, northern lateralis lineage: 14, southern lateralis lineage: 13, mendocina lineage: 110, urbaniana lineage: 104, yungas lineage: 8; see results) (dataset available in Table S1, see supplemental material online). Because tests of niche overlap in the geographical space (G-space) (e.g., geographical projections derived from species distribution-modeling (SDM) techniques) are likely to vary depending on the extent and distribution of environmental gradients in the study area (Broennimann et al. 2012; Brown and Carnaval 2019), analyses were conducted along the environmental space (E-space). For this purpose, information on the current climatic conditions within the study area was extracted from the CHELSA 1.2 climatic dataset (Karger et al. 2017a, b) at a resolution of 30 arc-seconds (~ 1 km2). CHELSA data is suitable for studies in mountain areas because it incorporates orographic predictors as wind field and valley exposition, and thus increases accuracy of species-range prediction, principally for the precipitation pattern (Karger et al. 2017a). Values of all 19 bioclimatic variables were extracted from the area defined by a minimum convex polygon enclosing all specimen records with 50-km buffer zone (ca. 20°58′S–42°52′S lat, 71°06′W–38°49′W long; Fig. S2, see Supplementary Material online). Additionally, we also included data from the annual aridity index (AI) and potential evapotranspiration (PET) from the CGIARCS database 2 (Trabucco and Zomer 2019) at the same resolution. Data extraction and manipulation were done using the packages adehabitatHR 0.4.16 (Calenge 2006), raster 2.8.19 (Hijmans 2019), sp 1.3.1 (Bivand et al. 2013), and maptools 0.9.5 (Bivand and Lewin-Koh 2019). Dimensionality of the environmental space was reduced by performing a PCA-env approach (Broennimann et al. 2012) with the R package vegan 2.5-6 (Oksanen et al. 2019), in which a principal component (based on a correlation matrix) is calibrated on the entire environmental space (in our case, 19 bioclimatic variables from CHELSA+ IA + PET) included in the study area (the 50-km-buffered minimum convex polygon enclosing all specimen occurrences for this work, Fig. S2, Supplementary Material online). Numbers of statistically significant principal components (PCs) to include in the analyses were estimated using the broken-stick method (Jackson 1993) implemented in the R package BiodiversityR 2.11-3 (Kindt and Coe 2005), retaining the first three PCs, which accounted for 83.73% of the total niche variation (see Results). First, as a preliminary approximation, we conducted univariate analyses and the Kruskal-Wallis test together with the Bonferroni correction for multiple comparisons to assess for differences between Physaria lineages for the three first PCs, elevation, annual mean temperature (BIO1), and annual precipitation (BIO12). Furthermore, comparisons of environmental niche overlaps among lineages were also visualized using density profiles computed for each bioclimatic niche axis in the sm 2.2-5.6 package (Bowman and Azzalini 2018). Then, PCs were used to estimate the climatic niche of each species using the n-dimensional hypervolume (Blonder et al. 2014). The hypervolume method creates an n-dimensional space reflecting the Hutchinsonian niche (Hutchinson 1957) of species or clades (Blonder 2018; Blonder et al. 2014). The hypervolume was calculated using the R package hypervolume 2.0.11 (Blonder and Harris 2018) with the Gaussian kernel density-estimation method, bandwidth estimated from the data (Silverman estimator), quantile of 90%, and 10,000 replicates per data point. We first explored position of the climatic niches occupied by the Physaria lineages within the environmental hypervolume estimating centroid distances (Euclidean distance between centroids of two hypervolumes) because distance metrics are most appropriate for fully disjunct hypervolumes (Mammola 2019). To visualize niche position from centroid distances, we applied multidimensional scaling on the resulting dissimilarity matrix, and plotted the first two axes in a phylomorphospace plot using the estimated MCC species tree and the R packages ape and phytools 0.6.20 (Revell 2012). Alternatively, to analyze potential patterns of PNC or niche divergence between lineages, niche overlap among their hypervolumes was quantified with the Sørensen–Dice similarity index, and used to perform the equivalency and similarity tests (Warren et al. 2008). To conduct the equivalence test, which studies whether niches of two species are the same, occurrences of the lineages A and B were pooled and randomly assigned to one of two groups, and thus maintaining the original number of occurrences. Then, hypervolumes for each random group and the Sørensen–Dice similarity index were estimated using the same settings described above. Resampling was replicated 1000 times and the null distribution (niches are equivalent) was rejected if empirical niche overlap was lower than the 95% of simulated values (one-tail test). For similarity test, which studies whether niches of two species are more similar (conserved) or dissimilar (divergent) than expected by chance accounting for the differences in the surrounding environmental conditions, we estimated niche overlap between lineage A and the random sampling of the background space of lineage B, and vice versa. Resampling was replicated 1000 times and the null distribution (similarities or differences between niches are explained by chance) was rejected if empirical niche overlap was greater (similar) or lower (dissimilar) than the 95% of simulated values (two-tail test).
Geographical-range overlap
Because patterns of geographical-range overlap and niche similarity between closely related species are complementary to delineate speciation processes (Lynch 1989; Chesser and Zink 1994; Barraclough and Vogler 2000; Fitzpatrick and Turelli 2006; Kozak and Wiens 2006; Anacker and Strauss 2014; Grossenbacher et al. 2014; Spalink et al. 2016), we calculated the species-range overlap among Physaria lineages using binary presence/absence maps of each lineage derived from species distribution-modeling (SDM) predictions. To model distribution of lineages, we used the maximum entropy algorithm implemented in Maxent 3.4.1 (Phillips et al. 2017). Because inclusion of the 19 bioclimatic variables in the SDM can be problematic due to high degrees of collinearity among predictors, we performed initial analyses on all 19 CHELSA variables and then chose climatic variables that contribute most in the Maxent models using jackknife test, and with a Spearman’s rank correlation coefficient (ρ) < 0.7. Seven variables were selected for the SDM analyses: annual mean temperature (BIO1), mean diurnal range (BIO2), isothermality (BIO3), temperature seasonality (BIO4), mean temperature of driest quarter (BIO9), annual precipitation (BIO12), and precipitation of driest quarter (BIO17). Maxent analyses were performed using ten cross validation runs with maximum iterations of 1000, logistic output, and all other options left as default (convergence threshold of 1×10−5, 1×104 background points, regularization multiplier of 1, default prevalence of 0.5, and autofeatures). The area under the receiver operating-characteristic curve (AUC) was used as a measure of model performance, and variable contribution to SDM was evaluated through both permutation importance and jackknife tests. Since the choice of a threshold is a topic of ongoing debate, we used the threshold indicating maximum training sensitivity plus specificity (MTSS), which is considered a more robust approach (Liu et al. 2005, 2013). The degree of range overlap between two species was estimated as the ratio of the area shared by both species to the area of the smaller ranged species (Barraclough and Vogler 2000). In this way, if one distribution range is contained within another (sympatric distribution), overlap is one (100%). Alternatively, because Last Glacial Maximum (LGM) is one of the episodes that had a major effect on the evolutionary history of extant species, with climate promoting fragmentation, connectivity, extinction, or population expansion (Hewitt 2004; Peterson and Lieberman 2012), we also estimated range overlap among lineages projecting the species distribution models on climatic data from CHELSA corresponding to the LGM (~ 21 kya) (CCSM4 model), using the MTSS logistic threshold to establish areas where the lineage is likely to have been present or absent.
Results
Delimitation of independent evolving lineages
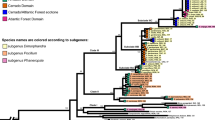
Features of nrDNA (ITS) and cpDNA (trnL-F, trnH-psbA, trnG, and trnS-trnG) alignments are summarized in Table S2 (see supplemental material online). We obtained 29 and 43 haplotypes for the ITS and cpDNA datasets, respectively. Physaria mendocina, the morphospecies with the widest geographical range (Fig. 2), also presented the highest number of haplotypes (13 for ITS and 16 for cpDNA). Median-joining networks from nrDNA and cpDNA show similar relationships, and groups with the same or closely related haplotypes included specimens mostly of one or more morphospecies, which were also associated with the different environments in which they inhabit: group A—P. urbaniana, P. pygmaea, and P. okanensis from the Puna of central Andes; group B—most P. crassistigma from highlands of southern Andes; group C—most P. lateralis from lowlands of the Dry Chaco and Low Monte; group D—specimens of P. urbaniana from Yungas of central Andes; and group E—specimens of P. mendocina from central and eastern Argentina, mostly distributed along the Dry Chaco and Low Monte (Fig. 3). Phylogenetic analyses using ITS, cpDNA, and concatenated ITS+cpDNA also recovered similar groups (Figs. 4 and S3, S4, Supplementary Material) with clade A “urbaniana lineage” (PP ITS: 0.52, cpDNA: 1.00, concatenated: 1.00, coalescence: 0.97) composed by specimens of P. urbaniana, P. pygmaea, and P. okanensis inhabiting highlands of the central Andes in northern Argentina and southern Bolivia (Central Andean Puna and High Monte ecoregions); clade B “crassistigma lineage” (PP ITS: 0.79, cpDNA: 0.85, concatenated: 0.90, coalescence: 0.64) including P. crassistigma and some specimens of P. mendocina and P. lateralis, all growing on highlands of the southern Andes in the Mendoza Province (High Monte and Southern Andean Steppe ecoregions); clade C “lateralis lineage” (PP ITS: <0.5, cpDNA: <0.5, concatenated: 0.92) including P. lateralis from lowlands of Dry Chaco and Low Monte ecoregions; clade D “yungas lineage” (PP ITS: 1.00, cpDNA: 1.00, concatenated: 1.00, coalescence: 0.98) grouping specimens of P. urbaniana from the Yungas of the central Andes; and clade E “mendocina lineage” (PP ITS: <0.5, cpDNA: 0.54, concatenated: 0.52, coalescence: 0.50) including specimens of P. mendocina from lowlands of the Dry Chaco and Low Monte ecoregions. Additionally, under cpDNA, concatenated ITS+cpDNA, and coalescence analyses, clade C was split in subclade C1 “northern lateralis lineage” (PP cpDNA: 1.00; concatenated: 1.00; coalescence: 0.95) with specimens of P. lateralis from the Comechingones biogeographical province in San Luis Province, and C2 “southern lateralis lineage” (PP ITS: 0.79; cpDNA: 1.00; concatenated: 1.00; coalescence: 0.87) including specimens of P. lateralis from the Low Monte biogeographical province in Mendoza province. Subclade C2 was recovered with ITS data (PP ITS: 0.79) but within the C1 subclade. When ITS and cpDNA data were analyzed using concordance analyses, variation of the discordance prior (α) had no effect on topology or concordance, and the primary concordance tree produced by BUCKy also recovered the main lineages present in concatenated analyses (Fig. S5a, Supplementary Material). Alternatively, when differences between the ITS and cpDNA trees were represented graphically by a filtered supernetwork, specimens were largely grouped in these lineages, despite the evidence of incongruences within them (Fig. S5b, Supplementary Material). Results from the specimen-tree analysis under the multispecies-coalescent model were congruent with the concatenated analyses, recovering same lineages, excepting for the lateralis lineage (clade C), for which the monophyly between northern lateralis (C1) and southern lateralis (C2) subclades was not obtained (Fig. 4(d)). The single threshold GMYC analyses delimited three lineages with the ITS and concatenated ITS+cpDNA trees (urbaniana, crassistigma, and the “lateralis+yungas+mendocina” lineages), and six using the cpDNA and coalescence ITS-cpDNA trees (all main lineages identified in the phylogenetic analyses and the lateralis lineage splits into the southern lateralis and northern lateralis lineages) (Fig. 5(a)). Main lineages, identified with molecular data and the discovery approaches, were morphologically characterized and proved to be generally congruent with one or more morphospecies. The urbaniana lineage included specimens of P. okanensis, P. pygmaea, and P. urbaniana, all of them easily distinguishable by their ellipsoid to ovoid and angustiseptate fruits. On the other hand, the crassistigma lineage mostly included specimens of P. crassistigma, characterized by its fruits with a short style (0.7–1.5 mm long), together with some specimens with styles slightly longer than 1.5 mm, but with fruits wider than long, subinflated, and with the stigma broadly capitate; all these features are characteristic of P. crassistigma. The lateralis lineage included specimens with the typical morphology of P. lateralis (procumbent to decumbent stems with entire leaves and globose-terete fruits), while the mendocina lineage included specimens associated with the morphology of P. mendocina (erect to ascending stems with dentate leaves and globose-terete fruits). Finally, specimens included in the yungas lineage have a vegetative morphology similar to P. urbaniana, but they were clearly differentiated by presenting terete-globose fruits, similar to those of P. mendocina and P. lateralis, instead of the ellipsoid-ovoid and angustiseptate fruits characteristic of species of the urbaniana lineage. Based on results obtained with the discovery approaches, we decided to use as primary hypothesis to be tested by the validation approaches, the six main clades reported above, since each of these lineages was characterized by different morphospecies and by the ecoregions/biogeographical provinces in which they inhabit.

Median-joining networks of a, nrITS dataset; b, cpDNA dataset (trnLF, trnH-psbA, trnG intron, trnS-trnG spacer). Six morphologically defined species are distinguished by different colors: blue, P. crassistigma; pink, P. lateralis; red, P. mendocina; black, P. okanensis; yellow, P. pygmaea; green: P. urbaniana. Intermediate (unobserved) haplotypes are distinguished by small gray circles. Circle sizes correspond to relative numbers of individuals sharing a particular haplotype

Phylogenetic placement of sampled specimens of South American Physaria. (a–c) Maximum clade credibility (MCC) tree generated by Bayesian inference with BEAST 1.8.4. (a) nrITS dataset. (b) cpDNA dataset (trnL-F/trnH-psbA/trnG intron/trnS-trnG). (c) Concatenated ITS + cpDNA datasets. (d) MCC tree estimated from ITS and cpDNA datasets using the multispecies coalescent method implemented in *BEAST v.1.8.4. The small circles on nodes indicate posterior probability (pp): black circles pp≥0.9, gray circles 0.9>pp≥0.7, white circles 0.7>pp≥0.5. Letters on the branches show delimited lineages: A, urbaniana lineage (green); B, crassistigma lineage (blue); C, lateralis lineage; C1, northern lateralis lineage (purple); C2, southern lateralis lineage (pink); D, mendocina lineage (red); E, yungas lineage (yellow). For tip labels: CRA, P. crassistigma; LAT, P. lateralis; MEN, P. mendocina; OKA, P. okanensis; PYG, P. pygmaea; URB, P. urbaniana. In all cases, the geographical location of each specimen was plotted

Results of species delimitation analyses. (a) Results from GMYC (discovery approach), BPP, and BFD (validation approaches) plotted onto the MCC tree obtained with the concatenated ITS+cpDNA dataset. GMYC analyses were conducted using MCC trees obtained with nrITS, cpDNA, concatenated nrITS+cpDNA, and coalescence nrITS–cpDNA analyses. BPP analyses were conducted using six different combinations of Θ and τ0 priors. For Bayes factor delimitation analyses (BFD), only the most supported hypothesis is shown. (b) gdi values for each delimited lineage. Numbers correspond to the median and 95th percentile. Dashed lines indicate gdi values of 0.2 and 0.7. Image: flower and fruit of P. mendocina
Results from the validation approaches using the BPP analyses with joint-lineage delimitation and species-tree estimation highly supported the six lineage model for all prior combinations (PP >0.99), except for the most conservative scenario (β = 0.2 for Θ) (Fig. 5(a), Table 1), in which a five-lineage model with the “northern lateralis + southern lateralis” lineage was favored with low support (PP =0.50 and 0.56 for β τ0 = 0.004 and 0.2, respectively). Under β = 0.002 and 0.02 for Θ prior posterior probabilities for each of the six candidate lineages were > 0.99, while with β = 0.2 only the urbaniana and mendocina lineages received PP > 0.90 (Table 1). Alternatively, different values of τ0 prior had no influence on results obtained.
BFD analyses favored models with 5 or 6 lineages over models with 1–4 lineages (Table 2). The most supported model included five lineages with the “northern lateralis + southern lateralis” lineage, followed by the model with the six candidate groups. The MCC species trees from these models recovered the yungas lineage sister to the mendocina lineage (Fig. 6(a)), while in the six-lineage tree the northern lateralis lineage was sister to southern lateraris lineage (Fig. 6(a)). Finally, gdi index only present conclusive support (gdi > 0.7) for differentiation of the urbaniana lineage (Fig. 5(b)), while for the remaining candidate lineages status was ambiguous (0.2 < gdi < 0.7) (Fig. 5(b)).

Geographic and climatic niche distribution for delimited lineages of South American Physaria. (a) Maximum clade credibility (MCC) species tree estimated from ITS and cpDNA datasets using the multispecies coalescent method implemented in *BEAST 1.8.4 and the hypothesis of six independently evolving lineages. Numbers on branches correspond to posterior probability. (b) Geographic distribution of lineages: green, urbaniana lineage; blue, crassistigma lineage; purple: northern lateralis lineage; pink, southern lateralis lineage; red, mendocina lineage; yellow, yungas lineage. (c–e) Distribution of lineages along the environmental space (PCA-env). (c) PC1 vs PC2. (d) PC1 vs PC3. (e) PC2 vs PC3. (f) Violin plot of elevation for delimited lineages. Points on plots indicate median values
Because results supported differentiation of the urbaniana, crassistigma, lateralis, yungas, and mendocina lineages, we included them in the ecological and geographical analyses. For the northern and southern lateralis lineages, the evidence suggest that they could be part of the same species. However, on the basis that (1) different genetic structure was recovered for both groups, (2) one-lineage model was only slightly favored over two-lineages models in the BFD analyses, (3) both groups exhibit an allopatric distribution and inhabit different ecoregions/biogeographical provinces, and (4) the aim of this work is to study ecological and geographical patterns in separately evolving metapopulation lineages, rather than determining the taxonomic status of the groups over the extended process of speciation, we decided to analyze the niche and geographic range of these two lineages separately. Thus, climatic and geographic patterns were studied for the six identified South American Physaria lineages.
Climatic niche comparisons
Eigenvalues and variable loadings for the PCA-env approach are shown in Table S2 (see Supplementary Material online). Using the broken-stick method, we selected the first three PCs, accounting for 83.72% of the niche variation (39.32%, 24.87%, and 19.52%, respectively). Variable loadings (Fig. S6, Table S3, Supplementary Material) showed that the first component was primarily influenced by the minimum temperature of coldest month (BIO6), the mean temperature of coldest quarter (BIO11), and the annual mean temperature (BIO1); the second component by the temperature seasonality (temperature change over the course of the year—BIO4), the temperature annual range (BIO7), and the precipitation of wettest month/quarter (BIO13/BIO16); and the third component by the precipitation of driest quarter/month (BIO17/BIO14), the potential evapotranspiration (PET—ability of the atmosphere to remove water through evapotranspiration processes), and the precipitation of coldest quarter (BIO19).
Climatic space occupied by the Physaria lineages along the different components of the environmental space shows that occurrences are extended along almost all the background (Fig. 6(c–d)). Univariate analyses found significant differences in all PCs, elevation, annual mean temperature (BIO1), and annual precipitation (BIO12) (Fig. S7, Supplementary Material). Most differentiated lineages were the urbaniana lineage for elevation (growing at higher elevation, Fig. 6(f)), the mendocina lineage for PC1 (with higher annual and winter temperatures), the urbaniana and yungas lineages for PC2 (with lower temperature seasonality, lower temperature annual range, and higher precipitation in the rainy season), and the yungas lineage for PC3 (with the lower precipitation in the dry season and the higher PET) (Fig. S7, Supplementary Material). The northern lateralis and mendocina lineages, which grow at a lower elevation, had the highest mean annual temperatures, while the crassistigma lineage presented the lowest mean annual precipitations (Fig. S7, Supplementary Material).
Hypervolumes estimated for the different lineages using the first three PCs are shown in Fig. 7(a–c). The urbaniana and mendocina lineages exhibit the largest volumes (greater niche breadth), while the yungas and northern lateralis lineages presented the smaller ones. Multidimensional scaling using centroid distances show that related lineages were highly differentiated in their niche positions (Fig. 7(d)). In addition, values of niche overlap (quantified using the Sørensen-Dice similarity index) were generally low for all lineages (Table 3), with exception of the crassistigma-southern lateralis pair (= 0.58). According to these results, the niche equivalency test recovered significant differentiation (non-equivalency) for all niche pairs (p<0.05) (Table 3), except for the crassistigma-southern lateralis lineages (p = 0.139). Similarity test shows niche divergence for most lineage pairs, especially when closely related lineages were compared. Niches in sister groups yungas-mendocina lineages and northern lateralis-southern lateralis lineages were always divergent, while lineages, in which niches presented significant similarity when the background space was considered, were not closely related in the phylogenetic analyses (Table 3, Fig. 6(a)), thus reinforcing the idea of niche divergence across the evolution of these taxa.

Climatic niche comparisons along the environmental space using hypervolumes for delimited lineages of South American Physaria. (a–c) Hypervolumes (point density and alpha-hull contour boundary) for delimited lineages of South American Physaria representing their climatic niches, and estimated using the values extracted from the components of the PCA-env (first three components). (a) PCenv1 vs PCenv2. (b) PCenv1 vs PCenv3. (c) PCenv2 vs PCenv3. (d) Phylomorphospace plot showing niche position between delimited lineages obtained using centroid distances between each pair of hypervolumes and multidimensional scaling
Spatial patterns
When specimen distribution was analyzed, all lineages were characterized by inhabiting a different combination of ecoregions/biogeographical provinces (Fig. 6(a)). This pattern was most pronounced between closely related lineages, in line with the results from the climatic-niche comparisons. Geographic-range estimations (from current projections using SDMs and the maximum-training sensitivity plus specificity as threshold) showed that the largest ranges were obtained for the mendocina and urbaniana lineages, while the northern lateralis and yungas lineages presented the smallest ones (Figs. 8 and S8, Supplementary Material). Range overlap was close to zero for most lineages (Table 3), except between the non-closely related lineages urbaniana-yungas and mendocina-northern lateralis (65% and 100%, respectively). Sister lineages (yungas-mendocina and northern lateralis-southern lateralis) exhibited a strong allopatric pattern (overlap=0%, Table 3). When geographic ranges were calculated over the LGM projections, low-range overlap continued to be predominant, but for sister northern and southern lateralis lineages overlap changed to 100% (Table 3) (northern lateralis was included within the southern lateralis range, Fig. S9 of Supplementary Material). Finally, closely related mendocina-yungas lineages conserved their allopatric distribution (overlap=0%).

Results from the species distribution modeling (SDM) for delimited lineages of South American Physaria. Predicted suitable climatic conditions (logistic output) from the MaxEnt model for the present time and Last Glacial Maximum (LGM) (~ 21 kya) for each delimited lineage. Binary (presence/absence) distributions maps obtained from these predictions using the maximum training sensitivity plus specificity as threshold are shown in Figs. S8 and S9 of supplementary material
Discussion
In this study, we investigated ecological and geographical patterns associated with the diversification of South American Physaria. However, instead of using predefined OTUs based on a particular species concept, we attempted to identify independently evolving metapopulation lineages in the light of the general lineage concept of species (de Queiroz 1998). Under this unified species concept, properties from other criteria (morphological differentiation, reciprocal monophyly, reproductive isolation, ecological divergence, etc.) are reinterpreted as non-excluding properties of the species category. Instead, they are considered contingent properties which may or may not be acquired through the species formation process (de Queiroz 2007). Identification of these “independently evolving lineages” or “evolutionarily significant units” (populations with ecological and genetic variation of adaptive significance and unique evolutionary histories; Ryder 1986; Moritz 1994; Crandall et al. 2000) is a fundamental step for the subsequent inclusion of the OTUs in evolutionary, macroecological, and conservation studies (Riddle and Hafner 1999; Isaac et al. 2004; Agapow et al. 2004; Sukumaran and Knowles 2017), especially for groups where phenotypic variation does not seem to be sufficient (either because it is highly conserved or highly variable) for a robust delimitation (Dayrat 2005). Nevertheless, delimited lineages should be interpreted as tentative hypotheses based on the data used, and to be confirmed or rejected by subsequent analyses with additional evidences (see Carstens et al. 2013). Species delimitation can be obscured by several process in addition to the incomplete lineage sorting, as introgression between the established species (Naciri and Linder 2015) or gene flow between the divergent metapopulations (migration) (Jackson et al. 2017). As a consequence, we used the identified evolutionary independent lineages in order to test the role of climatic niche and geography on the diversification of the group regardless whether they represent true established species or metapopulations of incipient divergence through a protracted process of speciation. We prefer not to introduce taxonomic changes or establish species ranks to the lineages obtained until we generated more data, for example using RAD-Seq data analyses.
South American Physaria represents a group with remarkable similarity and low morphological differentiation, being the six species basically indistinguishable in trichome type and density, shape of basal and cauline leaves, flower size and color, fruit indumentum, number of ovules/seeds per ovary/fruit, seed shape and size, and cotyledonary position (O’Kane and Al-Shehbaz 2004; Al-Shehbaz and Prina 2009; Al-Shehbaz 2012b). Diagnostic characters in the group are mainly related to the style length, fruit flattening (terete vs. laterally compressed), leaf margins, and plant habit (rosulate vs. pulvinate) (for a complete discussion about the morphological variation in the group, see O’Kane and Al-Shehbaz 2004; Al-Shehbaz 2012b). Although these diagnostic morphological characters exhibit a continuous variation that often diffuses the species boundaries, lineages identified here were generally associated with one or more morphospecies. Of these, three main lineages were exclusively associated with the mountain areas of the Andes. The urbaniana lineage included all morphospecies distributed along the highlands of the Argentinean and Bolivian Central Andes (P. urbaniana, P. pygmaea, and P. okanensis), growing these species in the Central Andean Puna and the High Monte ecoregions (21°24′S–29°20′S lat) (Fig. 6(a)), mainly between 2000 and 4500 m. The Central Andean Puna ecoregion is a high-elevation montane grassland in the High central Andes, and it is characterized by aridity and cold temperatures. The annual mean temperature ranges between 8.5 and 9.5 °C, while precipitation is seasonal and varies between 250 and 500 mm per year (Cabrera and Willink 1973). The isothermality is also pronounced, with drastic temperature changes between day and night. Similarly, the High Monte ecoregion includes dry grasslands and shrublands restricted to highlands of the pre-Andean region of western Argentina, and its climate is also temperate-arid with very little rainfall (Cabrera 1976).
The crassistigma lineage includes P. crassistigma, which is characterized by its short style (0.7–1.5 mm), together with specimens with styles more than 1.5 mm long, and classified as P. lateralis or P. mendocina, but with fruits wider than long and subinflated as in P. crassistigma (Al-Shehbaz 2012b). All members of this lineage are also characterized by growing along the southern Andean slopes in the Mendoza province, in the high Monte and Southern Andean steppe ecoregions (32°S–36°S lat) (Fig. 6) mainly between 2000 and 3500 m. The Southern Andean steppe ecoregion extends along the high elevations of the southern Andes of central Argentina. This corresponds to the Cuyan high Andean Province of Morrone (2018) and the Cuyan High Andean district of the Altoandina phytogeographical province of Cabrera (1976). Climate of this area is dry and very cold at high elevations, seasonality is pronounced, and frosts are frequent all year round. Mean annual precipitations vary from north to south, from approximately 200 to 400 mm (Cabrera 1976), and plants generally show adaptations to extreme dry conditions, cold, and wind. In these areas, aridity and seasonality are greater than those in the geographical range where the urbaniana lineage lives.
The remaining Andean group, the yungas lineage, included specimens with similar morphology to P. urbaniana, but growing in the humid slopes along the transition zone between the Yungas and the Central Andean Puna at Salta Province (ca. 25°S lat). Members of this group also inhabit high elevation areas (3200–3700 m); however, unlike the urbaniana lineage, they live in areas with greater temperature and precipitation (annual mean precipitations between 700 and 900 mm). Highlands of the Southern Andean Yungas essentially form a mesic habitat between two much drier regions, the dry Chaco to the east, and the higher Central Puna to the west (Olson et al. 2001). Climate is wet and humid due to both rain and mountain fogs, with precipitations exceeding 2500 mm/year (Cabrera 1976). Nonetheless, ecological transition zones harbor a higher number of species as a result of a mixture between floras of different ecoregions (Gaston et al. 2001). Specifically, the transition zone between the Central Andean Puna-Southern Andean Yungas ecoregions, where plants of the yungas lineage inhabit, exhibits a high number of endemic species (Godoy-Bürki et al. 2014). Since results from molecular data showed that this group is related to the mendocina but not the urbaniana lineage, possible independent colonization of this habitat from the lowlands of central Argentina is a likely scenario. Furthermore, the high asymmetry recovered both for the geographical range and for the niche breadth between the yungas-mendoncina lineage pair could suggest a pattern of peripatric speciation (Mayr 1954; Barraclough and Vogler 2000; Losos and Glor 2003). In the peripatric model of speciation, a small population colonizes a novel habitat and becomes reproductively isolated from the larger-ranged species (the mendocina lineage in our case). Isolation was initially thought to be driven by extreme founder effects and subsequent genetic drift (Mayr 1954, 1982); however, in heterogeneous landscapes, as the transition zone “Central Puna-Yungas-Chaco,” habitat-mediated reproductive isolation and strong divergent selection between immigrants can reduce gene flow and facilitate speciation, occupying the newly formed small-ranged species (the yungas lineage in our case) a distinct realized niche when compared to their large-ranged sister (Barton and Charlesworth 1984; Coyne 1992; Baldwin 2005; Grossenbacher et al. 2014).
Of the three groups inhabiting lowland regions outside the Andes, both northern and southern lateralis lineages include specimens of P. lateralis. The northern group, which includes the type locality of the species, is endemic to the Comechingones biogeographical province. This biogeographical province includes moderate-altitude grasslands located in the Dry Chaco ecoregion, but presenting high number of endemics to deserve its own biogeographical categorization (Martínez et al. 2017). The Southern group includes specimens of P. lateralis from the Low Monte ecoregion in the Mendoza province. The Low Monte ecoregion is a temperate-warm scrub desert that extends primarily between the Puna, Patagonia, and Dry Chaco ecoregions. The climate is temperate-arid with very low rainfall (between 80 and 250 mm/year) and annual mean temperature between 13 and 15 °C (Cabrera and Willink 1973). These two lineages were closely related in the molecular analyses, and some delimitation approaches (especially under a conservative scenario assuming large population sizes) suggested that they may represent populations of the same species. This result is also reinforced by the sympatric distribution of both lineages obtained in the SDM analysis for the Last Glacial Maximum (LGM). However, both lineages currently present an allopatric distribution, inhabit different ecoregions and biomes (tropical and subtropical grasslands for the northern group, and temperate grasslands for the southern group), and their ecological niches proved to be significantly divergent. The Monte greatest floristic similarity is to its neighbor, the Chaco biogeographical province, with which it shares 60% of its species (Solbrig et al. 1977). Nevertheless, the Monte is a well-defined biogeographical and ecological area, with at least 30% of its biota endemic and exhibiting greater aridity than the Chaco (Roig et al. 2009). Specimens of the southern group proved to grow in substantially colder and drier areas than those of the northern group. Then, these two populations/metapopulations, with their own genetic structure and ecological divergence, could be evolving under divergent climatic adaptive optima. Finally, the mendocina lineage includes specimens from P. mendocina from lowlands mainly of the Dry Chaco and Low Monte ecoregions, and secondary also of the Espinal and Pampas ecoregions in eastern Argentina. In the Dry Chaco ecoregion, climate is arid, with an annual rainfall of 350–650 mm, but also considerably warmer than in the Andean ecoregions, with an average temperature of 12–28 °C (Cabrera and Willink 1973). Similarly, the Espinal ecoregion is located in lowlands of central Argentina, and in its southern portion, where the range of P. mendocina is extended, the climate is temperate to dry (Cabrera 1976). This lineage presented the greatest geographic range, including both the temperate and the tropical-subtropical grasslands biomes; and although it did not present significant differences along the precipitation axis, its niche shows the highest temperatures of all South American Physaria lineages.
The climatic niche, defined as the set of environmental conditions associated with the occurrence of a given species (Grinnellian niche; Hutchinson 1957; Soberón 2007), and resulting from the cumulative effects of the physiological tolerance in response to climate (Ackerly 2003), has long been a central concept in ecology. Uncovering how climate niche dimensions vary among closely related species across the geographic space can help us to understand the processes that underlie lineage divergence and species diversification (Nürk et al. 2015, 2018; Kolanowska et al. 2017; Jezkova and Wiens 2018). Results obtained in this study show that the niche divergence is the dominant pattern throughout the diversification of South American Physaria. Most niche comparison resulted in niche differentiation for the equivalence test and niche divergence for the similarity test. Significant resemblance was obtained in the similarity test only for non-closely related lineages such as the pairs northern lateralis-mendocina and urbaniana-yungas. These results reinforce the differentiation of ecoregions and biomes presented by the lineages, also reflected in the general allopatric pattern obtained for the groups, in which only sympatric and parapatric distributions were registered for the non-closely related lateralis-mendocina and urbaniana-yungas. Cold to temperate climates characterized the crassistigma, urbaniana, southern lateralis, and yungas lineages, while northern lateralis and mendocina lineages were defined by warm climates. Alternatively, niches of crassistigma and southern lateralis lineages were the most arid of the group, while the yungas and northern lateralis lineages are the ones with the highest precipitation.
Divergence of the climatic niche is also represented by the wide niche breadth exhibited by the group as a whole. Although most of the cruciferous species present in the southern cone of South America are associated with the central and southern Andes (Al-Shehbaz 2008, 2012b), the environmental space occupied by the South American Physaria includes both high mountain environments, such as the Puna, the Yungas, and the Cuyan highlands in the Southern Andes, but also warm lowlands from central and eastern Argentina represented mainly by the Monte desert and the Dry Chaco regions. Brassicaceae taxa distributed in these latter regions are much less frequent and are represented mainly by species of Lepidium (Tribe Lepidieae), Rorippa and Cardamine (Cardamineae), and some species of Descurainia (Descuraineae), Exhalimolobos (Halimolobeae), and Mostacillastrum (Thelypodieae) (Al-Shehbaz 2012b). Previous studies on genera of tribes Cremolobeae (Salariato and Zuloaga 2017, 2020) and Eudemeae (Salariato et al. 2015, 2018), which are endemic to South America and largely associated with the Andes, reported the phylogenetic-niche conservatism as the predominant pattern through their diversification. Contrasting with these groups, within South American Physaria, niche divergence is present between highland and lowland habitats, through changes primarily in the gradient of temperature, represented in our study mainly by the cold-temperate habitats of Andean lineages vs. the warm environments of lineages distributed along central-eastern Argentina. Regarding precipitation, while the group presented a common adaptation to arid environments, the most significant shift along this niche dimension was exhibited by the northern lateralis and yungas lineages, growing on mesic habitats. Under ecological speciation, reproductive isolation between two populations evolves by divergent ecologically based selection (Rundle and Nosil 2005; Schluter 2009; Nosil 2012). Thus, divergent selection acting on the climatic niche (mainly on temperature and secondarily on precipitation) seems to be one of the factors associated with the diversification of the group, promoting, together with the dispersal capabilities, the colonization of both Andean environments as the Puna or the Yungas, and warm-dry grasslands and shrublands of central-eastern Argentina as the Monte and the Dry Chaco.
An additional factor that could play an important role in the diversification of South American Physaria is the occurrence of hybridization and polyploid speciation. This mechanism of speciation has been reported as particularly common in plants, since polyploids frequently exhibit ecological differentiation, local dispersal, high fecundity, perennial life history, and self-fertilization (Mallet 2007; Rieseberg and Willis 2007). In this way, new hybrids can colonize unfilled ecological niches or adaptative peaks, reducing the gene flow with their parent species (Mallet 2007), being this phenomenon widespread in many genera of Brassicaceae (see Marhold and Lihová 2006; Koch and Grosser 2017; Marhold et al. 2018; Mandáková et al. 2019). Additionally, Mandakova et al. (2017) reported an ancestral mesopolyploid whole-genome duplication (WGD) event (specifically a mesohexaploid WGDs) followed by subsequent genome diploidization for Physaria. Variable chromosome numbers (n = 4, 5, 6, 7, 8, 9, 10, 12 and higher counts; Warwick and Al-Shehbaz 2006) have been reported for North American Physaria, but polyploidy in South American species is unknown and no chromosome numbers were reported for these species. Future studies using for example sequences from low-copy nuclear genes, genomic in situ hybridization (GISH), and comparative chromosome painting (CCP) (Koch 2015; Mandáková et al. 2019) are needed to analyze the role of these processes through the diversification of the South American lineage.
South American terrestrial ecosystems experienced drastic transformations in the Cenozoic, during which events occurred in the Neogene, and later by the Pleistocene, had enormous effects on the diversification of the local biodiversity (Hoorn et al. 2010; Antonelli and Sanmartín 2011; Rull 2011; Hazzi et al. 2018). The Neogene presented strong climatic, tectonic, and geographical changes, mainly related to events such as the middle Miocene climatic optimum (Zachos et al. 2001), the uplift of the Andes (Jordan et al. 2001; Blisniuk et al. 2005; Graham 2009; Encinas et al. 2013: Gregory-Wodzicki 2000; Graham et al. 2001; Garzione et al. 2008; Leier et al. 2013), marine ingressions into the continent (Del Río et al. 2013; Uba et al. 2009), and changes in the Amazonian drainage system (Hoorn et al. 2010). In particular, Andean Mountain uplift caused a decrease in the precipitation of the western regions of Southern South America as the Central Andean Puna and the High Monte ecoregions, generating semi-arid and arid conditions that, together with inter Andean valleys isolated from each other, promote diversification in numerous plant lineages (Young et al. 2002; Antonelli et al. 2009; Roig et al. 2009; Luebert and Weigend 2014). On the other hand, the Pleistocene also presented significant environmental alterations due to climatic oscillations (Zachos et al. 2001), glacial-interglacial cycles (Rabassa 2008; Rutter et al. 2012), and changes in the sea level and the seashores (Ponce et al. 2011; Rabassa and Ponce 2013).
Glaciations in South America began in the Southern Andes in the late Miocene–early Pliocene (approx. 7–4.4 Mya), followed by successive expansion/retraction events (Rutter et al. 2012). During the Late Pliocene (~ 3.5–2.79 Mya), extra-Andean ice sheets formed in Patagonia, with the major expansions of the ice from the Andes, the Great Patagonian Glaciation (GPG), during the Pleistocene (~ 1.5–1 Mya) (Rabassa 2008; Rabassa et al. 2011; Rutter et al. 2012). Evidence of early Pleistocene ice sheet expansions (approx. 2.6 Ma) has also been recovered in the central Andes of Bolivia (Rutter et al. 2012). Finally, the Last Glacial Maximum (LGM) in the South America ranges from ~ 48 to 25 Kya (Rabassa 2008; Rutter et al. 2012), with the ice sheets reaching their maximum near 26.5 Kya, and deglaciation starting ~19 Kya (Clark et al. 2009), initiating the current interglacial period about 11.7 Kya. During the LGM, precipitation decreased due to lower temperatures and lower atmospheric humidity (Ortiz-Jaureguizar and Cladera 2006). Icefields in the Southern Andes were most expansive when global temperature and sea level were lowest. Furthermore, South American rivers were affected primarily by changes in climate and global sea level. Low-gradient river networks became reduced and incised as global sea level fell during the LGM. Reduction of water-tables also impacted the forest cover, enhancing the drying influence of the lower sea surface temperature and atmospheric humidity (Clapperton 1993). This increasing aridity fragmented and reduced the extent of forest and grassland. Under these conditions, Central Andean Puna became a cold montane grassland and scrub with glaciers and ice caps, the Monte ecoregion turned into a sand desert, and the Dry Chaco became a xeric shrubland (Clapperton 1993), the latter region being particularly climatically unstable and a more extensive area during the Pleistocene glacial/interglacial periods compared to the present time (Ab’Saber 2000; Pennington et al. 2004). Although to date there are no estimates of divergence times for South American Physaria, Huang et al. (2020) in their study on diversification rates of Brassicaceae reported a crown node age for Physaria mainly on the early-mid Pliocene (~4.7 Mya). These estimations suggest that diversification of the South American group could be associated to Late Pliocene–Quaternary glaciations and the successive glacial expansions and retreats. These Quaternary events have been identified as important driving forces in the evolution of the southern/central Andean lineages promoting aridification and geographical range fragmentation (Luebert and Weigend 2014).
Alternatively, results from geographical-range overlap among the South American Physaria favored an allopatric pattern for lineage divergence. The presence of allopatric ranges between young sister lineages (mendocina-yungas and northern-southern lateralis lineages) seems to support the predominance of diversification with dispersion and substantial geographical isolation. However, since species ranges can expand, contract, and change considerably through time (Losos and Glor 2003; Peterson and Lieberman 2012), the current geographical range of a species is not necessarily an indicator of its historical distribution. Allopatric conditions were also largely recovered with the LGM projections, with the exception of the northern-southern lateralis group, for which a sympatric range was retrieved. These results seem to suggest that the LGM and the subsequent warming and retraction of ice sheets in the current interglacial period have been major factors in shaping the present-day genetic and geographical differentiation only for the lateralis lineage. Thus, although glaciation cycles might have been important in the diversification and differentiation of lineages, data show a minor role of the Last Glacial Maximum in the diversification of the whole South American group, in accordance with Collevatti et al. (2020) that LGM have had a more local and regional effect on the differentiation among populations and patterns of genetic diversity. In particular for the lateralis lineage, the increase of geographic overlap between the northern and southern lineages at the LGM is explained by the greater geographic range of the southern group, which is in line with reports that during dry periods, the Monte was larger than it is today and included current areas of the Chaco (Mares et al. 1985). Hence, these results could suggest a recent vicariant isolation between the northern and southern lateralis lineages due to the climatic change in the warming of the last 21ky.
Along with the six species that comprise the South American lineage, Physaria includes about 105 species, most of these perennial herbs with caudice (rarely annual or biennial species), which are distributed mainly in western USA and northern Mexico, with a few species growing in Canada, and one species in northwestern Russia (Rollins 1939; Al-Shehbaz and O’Kane 2002). This high species diversity seems to be the product of a rapid and recent diversification, and Physaria is one of the 12 cruciferous taxa in which shifts in the speciation rates were detected (Huang et al. 2020). Based in the monophyly of the South American Physaria and the highest number of North American species (ca. 99 spp., more than 80%), it seems likely that the colonization of South America occurred by migration of a common North American ancestor. However, how much niches among South and North American groups differ, and if the colonization of South America implied a significant change in the fundamental niche of the genus, is still a question to be studied. Climatic-niche estimations for North American species, together with molecular phylogenies including all members of the genus, should be used to define potential divergence or PNC patterns throughout its diversification, as well as changes in the niche position and niche breadth, tempo and mode of climatic-niche evolution (rate shifts and evolutionary models), and potential morphological/functional factors associated to these changes.
In this work, we used species delimitation methods and the general lineage concept of species to identify independent evolving lineages (i.e., evolutionary significant units), which can be used as study units to identify evolutionary patterns and processes, but which can also be used for other ecological studies and conservation initiatives. Our results suggest that, rather than the phylogenetic-niche conservatism, which is the most common pattern among South American crucifers, climatic-niche divergence appears to have been a major factor promoting diversification of the South America Physaria. Divergence was registered mainly on the temperature axis, promoting shifts between cold-temperate habitats associated to the Andes, and warm lowlands from central and eastern Argentina as the Monte and Dry Chaco ecoregions. However, although the group is mostly adapted to arid climate, secondary changes in the rainfall axis were also recovered. Allopatry and dispersal capabilities also seem to be associated with the diversification of the group, presumably through the Late Pliocene-Pleistocene and promoted by glacial cycles and climatic oscillations during the Quaternary. Our study contributes to understand the evolutionary history of South American Physaria in particular, and the southern and central Andean Brassicaceae in general. New analyses including all members of the genus will help to clarify the role of ecological-niche evolution on its diversification.
Data availability
Data generated or analyzed in this paper are included in the electronic supplementary material, the TreeBase repository (http://purl.org/phylo/treebase/phylows/study/TB2:S26615), and the Figshare repository (https://doi.org/10.6084/m9.figshare.12645218). New DNA sequences generated for this study are available in the GenBank repository (see corresponding GenBank numbers and vouchers in Appendix 1).
References
Ab’saber, A. N. (2000). Spaces occupied by the expansion of dry climates in South America during the Quaternary ice ages. Revista do Instituto Geológico, 21(1–2), 71–78. https://doi.org/10.5935/0100-929X.20000006.
Ackerly, D. (2003). Community assembly, niche conservatism, and adaptative evolution in changing environments. International Journal of Plant Sciences, 164, S165–S184. https://doi.org/10.1086/368401.
Agapow, P. M., Bininda-Emonds, O. R., Crandall, K. A., Gittleman, J. L., Mace, G. M., Marshall, J. C., & Purvis, A. (2004). The impact of species concept on biodiversity studies. The Quarterly Review of Biology, 79(2), 161–179. https://doi.org/10.1086/383542.
Al-Shehbaz, I. A. (2008). Brassicaceae. In F. O. Zuloaga, O. Morrone, & M. J. Belgrano (Eds.), Catalogue of the vascular plants of the southern cone (Argentina, southern Brazil, Chile, Paraguay and Uruguay), Dicotyledoneae: Acanthaceae-Fabaceae (Abarema-Schizolobium) (Vol. 2, pp. 1663–1709). Missouri: Missouri Botanical Garden Press.
Al-Shehbaz, I. A. (2012a). A generic and tribal synopsis of the Brassicaceae (Cruciferae). Taxon, 61, 931–954. https://doi.org/10.1002/tax.615002.
Al-Shehbaz, I. A. (2012b). Brassicaceae. In M. A. Anton & F. O. Zuloaga (Eds.), Flora Argentina (Vol. 8, pp. 1–270). Buenos Aires: Editorial Sigma.
Al-Shehbaz, I. A., & O’Kane Jr., S. L. (2002). Lesquerella is united with Physaria (Brassicaceae). Novon, 12(3), 319–329.
Al-Shehbaz, I. A., & Prina, A. O. (2009). Physaria okanensis (Brassicaceae), a new species from Catamarca, Argentina. Novon, 19(3), 284–285.
Anacker, B. L., & Strauss, S. Y. (2014). The geography and ecology of plant speciation: range overlap and niche divergence in sister species. Proceedings of the Royal Society of London B: Biological Sciences, 281(1778), 20132980. https://doi.org/10.1098/rspb.2013.2980.
Ané, C., Larget, B., Baum, D. A., Smith, S. D., & Rokas, A. (2007). Bayesian estimation of concordance among gene trees. Molecular Biology and Evolution, 24, 412–426. https://doi.org/10.1093/molbev/msl170.
Antonelli, A., & Sanmartín, I. (2011). Why are there so many plant species in the Neotropics? Taxon, 60(2), 403–414. https://doi.org/10.1126/science.1194585.
Antonelli, A., Nylander, J. A., Persson, C., & Sanmartín, I. (2009). Tracing the impact of the Andean uplift on Neotropical plant evolution. Proceedings of the National Academy of Sciences, 106(24), 9749–9754. https://doi.org/10.1073/pnas.0811421106.
Arana, M. D., Martinez, G. A., Oggero, A. J., Natale, E. S., & Morrone, J. J. (2017). Map and shapefile of the biogeographic provinces of Argentina. Zootaxa, 4341(3), 420–422. https://doi.org/10.11646/zootaxa.4341.3.6.
Arrigoni, R., Berumen, M. L., Chen, C. A., Terraneo, T. I., Baird, A. H., Payri, C., & Benzoni, F. (2016). Species delimitation in the reef coral genera Echinophyllia and Oxypora (Scleractinia, Lobophylliidae) with a description of two new species. Molecular Phylogenetic and Evolution, 105, 146–159. https://doi.org/10.1016/j.ympev.2016.08.023.
Baldwin, B. G. (1992). Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: an example from the Compositae. Molecular Phylogenetics and Evolution, 1, 3–16. https://doi.org/10.1016/10557903(92)90030-K.
Baldwin, B. G. (2005). Origin of the serpentine-endemic herb Layia discoidea from the widespread L. glandulosa (Compositae). Evolution, 59, 2473–2479. https://doi.org/10.1554/05-147.1.
Barraclough, T. G., & Vogler, A. P. (2000). Detecting the geographical pattern of speciation from species-level phylogenies. The American Naturalist, 155(4), 419–434. https://doi.org/10.1086/303332.
Barton, N. H., & Charlesworth, B. (1984). Genetic revolutions, founder effects, and speciation. Annual Review of Ecology, Evolution, and Systematics, 15, 133–164. https://doi.org/10.1146/annurev.es.15.110184.001025.
Baum, D. A. (2007). Concordance trees, concordance factors, and the exploration of reticulate genealogy. Taxon, 56, 417–426. https://doi.org/10.1002/tax.562013.
Bivand R, & Lewin-Koh, N. (2019). maptools: tools for handling spatial objects. R package version 0.9-5. https://CRAN.R-project.org/package=maptools. Accessed March 2019
Bivand, R. S., Pebesma, E., & Gomez-Rubio, V. (2013). Applied spatial data analysis with R (2nd ed.). New York: Springer.
Blisniuk, P. M., Stern, L. A., Chamberlain, C. P., Idleman, B., & Zeitler, P. K. (2005). Climatic and ecologic changes during Miocene surface uplift in the southern Patagonian Andes. Earth and Planetary Science Letters, 230, 125–142. https://doi.org/10.1016/j.Epsl.2004.11.015.
Blonder, B. (2018). Hypervolume concepts in niche-and trait-based ecology. Ecography, 41, 1441–1455. https://doi.org/10.1111/ecog.03187.
Blonder, B., & Harris, D. J. (2018). hypervolume: high dimensional geometry and set operations using kernel density estimation, support vector machines, and convex hulls. R package version 2.0.11. https://CRAN.R-project.org/package=hypervolume. Accessed March 2019
Blonder, B., Lamanna, C., Violle, C., & Enquist, B. J. (2014). The n-dimensional hypervolume. Global Ecology and Biogeography, 23, 595–609. https://doi.org/10.1111/geb.12146.
Boelcke, O. (1967). Cruciferae. In A. L. Cabrera (Ed.), Flora de la Provincia de Buenos Aires, Vol. 4, pt. 3 (pp. 281–371). Buenos Aires: Instituto Nacional de Tecnología Agropecuaria (INTA).
Boelcke, O., & Romanezuk, C. (1984). Cruciferae. In M. N. Correa (Ed.), Flora Patagonica (Vol. 4A, pp. 373–544). Buenos Aires: Instituto Nacional de Tecnología Agropecuaria (INTA).
Bowman, A. W. & Azzalini, A. (2018). R package ‘sm’: nonparametric smoothing methods (version 2.2-5.6). http://www.stats.gla.ac.uk/~adrian/sm. Accessed November 2019
Broennimann, O., Fitzpatrick, M. C., Pearman, P. B., Petitpierre, B., Pellissier, L., Yoccoz, N. G., Thuiller, W., Fortin, M., Randin, C., Zimmermann, N. E., Graham, C. H., & Guisan, A. (2012). Measuring ecological niche overlap from occurrence and spatial environmental data. Global Ecology and Biogeography, 21, 481–497. https://doi.org/10.1111/j.1466-8238.2011.00698.x.
Brown, J. L., & Carnaval, A. C. (2019). A tale of two niches: methods, concepts, and evolution. Frontiers of Biogeography, 11(4), e44158. https://doi.org/10.21425/F5FBG44158.
Cabrera, A. L. (1976). Regiones fitogeográficas Argentinas. Enciclopedia Argentina de Agricultura y Jardinería (Vol. II, Second ed.). Buenos Aires: Editorial ACME.
Cabrera, A., & Willink, A. (1973). Biogeografía de América Latina. Washington: Monografías OEA.
Calenge, C. (2006). The package adehabitat for the R software: a tool for the analysis of space and habitat use by animals. Ecological Modelling, 197, 516–519. https://doi.org/10.1016/j.ecolmodel.2006.03.017.
Carstens, B. C., Pelletier, T. A., Reid, N. M., & Satler, J. D. (2013). How to fail at species delimitation. Molecular Ecology, 22(17), 4369–4383. https://doi.org/10.1111/mec.12413.
Chan, K. O., & Grismer, L. L. (2019). To split or not to split? Multilocus phylogeny and molecular species delimitation of southeast Asian toads (family: Bufonidae). BMC Evolutionary Biology, 19(1), 95. https://doi.org/10.1186/s12862-019-1422-3.
Chesser, R. T., & Zink, R. M. (1994). Modes of speciation in birds: a test of Lynch’s method. Evolution, 48(2), 490–497. https://doi.org/10.1111/j.1558-5646.1994.tb01326.x.
Clapperton, C. M. (1993). Nature of environmental changes in South America at the Last Glacial Maximum. Palaeogeography, Palaeoclimatology, Palaeoecology, 101(3–4), 189–208. https://doi.org/10.1016/0031-0182(93)90012-8.
Clark, P. U., Dyke, A. S., Shakun, J. D., Carlson, A. E., Clark, J., Wohlfarth, B., Mitrovica, J. X., Hostetler, S. W., & McCabe, A. M. (2009). The Last Glacial Maximum. Science, 325, 710–714. https://doi.org/10.1126/science.1172873.
Collevatti, R. G., Lima, N. E., & Vitorino, L. C. (2020). The diversification of extant angiosperms in the South America dry diagonal. In V. Rull & A. C. Carnaval (Eds.), Neotropical Diversification: Patterns and Processes (pp. 547–568). Cham: Springer Nature.
Coyne, J. A. (1992). Genetics and speciation. Nature, 355, 511–515. https://doi.org/10.1038/355511a0.
Coyne, J. A., & Orr, A. H. (2004). Speciation. Massachusetts: Ed. Sunderland, Sinauer Associates, Inc..
Crandall, K. A., Bininda-Emonds, O. R., Mace, G. M., & Wayne, R. K. (2000). Considering evolutionary processes in conservation biology. Trends in Ecology & Evolution, 15(7), 290–295. https://doi.org/10.1016/S0169-5347(00)01876-0.
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). jModelTest2: more models, new heuristics and parallel computing. Nature Methods, 9, 772–772. https://doi.org/10.1038/nmeth.2109.
Dayrat, B. (2005). Towards integrative taxonomy. Biological Journal of the Linnean Society, 85(3), 407–417. https://doi.org/10.1111/j.1095-8312.2005.00503.x.
De Queiroz, K. (1998). The general lineage concept of species, species criteria, and the process of speciation and terminological recommendations. In D. J. Howard & H. S. Berlocher (Eds.), Endless Forms: Species and Speciation (pp. 57–75). Oxford: Oxford University Press.
De Queiroz, K. (1999). The general lineage concept of species and the defining properties of the species. In R. Wilson (Ed.), Species: new interdisciplinary essays (pp. 49–89). Cambridge, MA: MIT Press.
De Queiroz, K. (2005). Different species problems and their resolution. BioEssays, 27(12), 1263–1269. https://doi.org/10.1002/bies.20325.
De Queiroz, K. (2007). Species concepts and species delimitation. Systematic Biology, 56(6), 879–886. https://doi.org/10.1080/10635150701701083.
Del Río, C. J., Griffin, M., McArthur, J. M., Martínez, S., & Thirlwall, M. F. (2013). Evidence for early Pliocene and late Miocene transgressions in southern Patagonia (Argentina): 87Sr/86Sr ages of the pectinid “Chlamys” actinodes (Sowerby). Journal of South American Earth Sciences, 47, 220–229. https://doi.org/10.1016/j.jsames.2013.08.004.
Doyle, J. J., & Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin, 19, 11–15.
Drummond, A. J., Suchard, M. A., Xie, D., & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. https://doi.org/10.1093/molbev/mss075.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. https://doi.org/10.1093/nar/gkh340.
Encinas, A., Zambrano, P. A., Finger, K. L., Valencia, V., Buatois, L. A., & Duhart, P. (2013). Implications of deep-marine Miocene deposits on the evolution of the north Patagonian Andes. The Journal of Geology, 121, 215–238. https://doi.org/10.1086/66997.
Ezard, T., Fujisawa, T., & Barraclough, T. G. (2009). Splits: species’ limits by threshold statistics. R package version, 1.0-19 1(11), r29. http://r-forge.r-project.org/projects/splits/. Accessed January 2020
Federici, S., Galimberti, A., Bartolucci, F., Bruni, I., De Mattia, F., Cortis, P., & Labra, M. (2013). DNA barcoding to analyse taxonomically complex groups in plants: the case of Thymus (Lamiaceae). Botanical Journal of the Linnean Society, 171(4), 687–699. https://doi.org/10.1111/boj.12034.
Fitzpatrick, B. M., & Turelli, M. (2006). The geography of mammalian speciation: mixed signals from phylogenies and range maps. Evolution, 60(3), 601–615. https://doi.org/10.1554/05-453.1.
Flouri, T., Jiao, X., Rannala, B., & Yang, Z. (2018). Species tree inference with BPP using genomic sequences and the multispecies coalescent. Molecular Biology and Evolution, 35(10), 2585–2593. https://doi.org/10.1093/molbev/msy147.
Fujisawa, T., & Barraclough, T. G. (2013). Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: a revised method and evaluation on simulated data sets. Systematic Biology, 62(5), 707–724. https://doi.org/10.1093/sysbio/syt033.
Futuyma, D. J., & Kirkpatrick, M. (2017). Evolution (4th ed.). Sunderland, MA: Sinauer Associates.
Garzione, C. N., Hoke, G. D., Libarkin, J. C., Withers, S., MacFadden, B., Eiler, J., Ghosh, P., & Mulch, A. (2008). Rise of the Andes. Science, 320, 1304–1307. https://doi.org/10.1126/science.1148615.
Gaston, K. J., Rodrigues, A. S. L., Van Rensburg, B. J., Koleff, P., & Chown, S. L. (2001). Complementary representation and zones of ecological transition. Ecology Letter, 4, 4–9. https://doi.org/10.1046/j.1461-0248.2001.00196.x.
Givnish, T. J. (2010). Ecology of plant speciation. Taxon, 59(5), 1326–1366.
Godoy-Bürki, A. C., Ortega-Baes, P., Sajama, J. M., & Aagesen, L. (2014). Conservation priorities in the Southern Central Andes: mismatch between endemism and diversity hotspots in the regional flora. Biodiversity and conservation, 23(1), 81–107. https://doi.org/10.1007/s10531-013-0586-1.
Graham, A. (2009). The Andes: a geological overview from a biological perspective. Annals of the Missouri Botanical Garden, 96, 371–385. https://doi.org/10.3417/2007146.
Graham, A., Gregory-Wodzicki, K. M., & Wright, K. L. (2001). Studies in Neotropical paleobotany. XV. A Mio-Pliocene palynoflora from the Eastern Cordillera, Bolivia: Implications for the uplift history of the Central Andes. American Journal of Botany, 88, 1545–1557. https://doi.org/10.2307/3558398.
Gregory-Wodzicki, K. M. (2000). Uplift history of the Central and Northern Andes: a review. Geological Society of America Bulletin, 112, 1091–1105. https://doi.org/10.1130/00167606(2000)112%3C1091:UHOTCA%3E2.0.CO;2.
Grossenbacher, D. L., Veloz, S. D., & Sexton, J. P. (2014). Niche and range size patterns suggest that speciation begins in small, ecologically diverged populations in North American monkey-flowers (Mimulus spp.). Evolution, 68(5), 1270–1280. https://doi.org/10.1111/evo.12355.
Grummer, J. A., Bryson Jr., R. W., & Reeder, T. W. (2014). Species delimitation using Bayes factors: simulations and application to the Sceloporus scalaris species group (Squamata: Phrynosomatidae). Systematic Biology, 63(2), 119–133. https://doi.org/10.1093/sysbio/syt069.
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hamilton, M. B. (1999). Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Molecular Ecology, 8, 521–523.
Hazzi, N. A., Moreno, J. S., Ortiz-Movliav, C., & Palacio, R. D. (2018). Biogeographic regions and events of isolation and diversification of the endemic biota of the tropical Andes. Proceedings of the National Academy of Sciences of United States of America, 115(31), 7985–7990. https://doi.org/10.1073/pnas.1803908115.
Heled, J., & Drummond, A. J. (2010). Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution, 27, 570–580. https://doi.org/10.1093/molbev/msp274.
Hewitt, G. M. (2004). Genetic consequences of climatic oscillations in the Quaternary. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 359(1442), 183–195. https://doi.org/10.1098/rstb.2003.1388.
Hijmans, R. J. (2019). raster: geographic data analysis and modeling. R package version 2.8-19. https://CRAN.R-project.org/package=raster. Accessed March 2019
Hoorn, C., Wesselingh, F. P., Steege, T., Bermudez, H., Mora, M. A., Sevink, A., Sanmartín, I., Sanchez-Meseguer, A., Anderson, C. L., Figueiredo, J. P., Jaramillo, C., Riff, D., Negri, F. R., Hooghiemstra, H., Lundberg, J., Stadler, T., Särkinen, T., & Antonelli, A. (2010). Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science, 330, 927–931. https://doi.org/10.1126/science.1194585.
Huang, X. C., German, D. A., & Koch, M. A. (2020). Temporal patterns of diversification in Brassicaceae demonstrate decoupling of rate shifts and mesopolyploidization events. Annals of botany, 125(1), 29–47. https://doi.org/10.1093/aob/mcz123.
Huson, D. H., & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254–267. https://doi.org/10.1093/molbev/msj030.
Huson, D. H., Dezulian, T., Klopper, T., & Steel, M. A. (2004). Phylogenetic super-networks from partial trees. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 1, 151–158.
Hutchinson, G. (1957). Concluding remarks. Cold Spring Harbor Symposia on Quantitative Biology, 22, 415–427. https://doi.org/10.1101/SQB.1957.022.01.039.
Isaac, N. J., Mallet, J., & Mace, G. M. (2004). Taxonomic inflation: its influence on macroecology and conservation. Trends in Ecology & Evolution, 19(9), 464–469. https://doi.org/10.1016/j.tree.2004.06.004.
Jackson, D. A. (1993). Stopping rules in principal components analysis: a comparison of heuristical and statistical approaches. Ecology, 74, 2204–2214.
Jackson, N. D., Carstens, B. C., Morales, A. E., & O’Meara, B. C. (2017). Species delimitation with gene flow. Systematic Biology, 66(5), 799–812. https://doi.org/10.1093/sysbio/syw117.
Jezkova, T., & Wiens, J. J. (2018). Testing the role of climate in speciation: new methods and applications to squamate reptiles (lizards and snakes). Molecular Ecology, 27(12), 2754–2769. https://doi.org/10.1111/mec.14717.
Jordan, D. S. (1905). The origin of species through isolation. Science, 22(566), 545–562.
Jordan, T. E., Burns, W. M., Veiga, R., Pangaro, F., Copeland, P., Kelley, S., & Mpodozis, C. (2001). Extension and basin formation in the Southern Andes caused by increased convergence rate: a mid-Cenozoic trigger for the Andes. Tectonics, 20, 308–324. https://doi.org/10.1029/1999TC001181.
Karger, D. N., Conrad, O., Böhner, J., Kawohl, T., Kreft, H., Soria-Auza, R. W., Zimmermann, N. E., & Kessler, M. (2017a). Climatologies at high resolution for the earth’s land surface areas. Scientific data, 4, 170122. https://doi.org/10.1038/sdata.2017.122.
Karger, D. N., Conrad, O., Böhner, J., Kawohl, T., Kreft, H., Soria-Auza, R. W., Zimmermann, N. E., Linder, H. P., & Kessler, M. (2017b). Data from: climatologies at high resolution for the earth’s land surface areas. Dryad Digital Repository. https://doi.org/10.5061/dryad.kd1d4.
Kass, R. E., & Raftery, A. E. (1995). Bayes factors. Journal of the American Statistical Association, 90, 773–795.
Kindt, R., & Coe, R. (2005). In World Agroforestry Centre (ICRAF) (Ed.), Tree diversity analysis. A manual and software for common statistical methods for ecological and biodiversity studies. Nairobi: http://www.worldagroforestry.org/output/tree-diversity-analysis. Accessed January 2020
Kisel, Y., & Barraclough, T. G. (2010). Speciation has a spatial scale that depends on levels of gene flow. The American Naturalist, 175(3), 316–334. https://doi.org/10.1086/650369.
Koch, M. A. (2015). A new chromosome was born: comparative chromosome painting in Boechera. Trends in Plant Science, 20(9), 533–535. https://doi.org/10.1016/j.tplants.2015.07.001.
Koch, M. A., & Grosser, J. (2017). East Asian Arabis species (Brassicaceae) exemplify past hybridization and subsequent emergence of three main evolutionary lineages in East Asia, America and the amphi-Beringian region. Botanical Journal of the Linnean Society, 184(2), 224–237. https://doi.org/10.1093/botlinnean/box020.
Kolanowska, M., Grochocka, E., & Konowalik, K. (2017). Phylogenetic climatic niche conservatism and evolution of climatic suitability in Neotropical Angraecinae (Vandeae, Orchidaceae) and their closest African relatives. PeerJ, 5, e3328. https://doi.org/10.7717/peerj.3328.
Kolář, F., Dušková, E., & Sklenář, P. (2016). Niche shifts and range expansions along cordilleras drove diversification in a high-elevation endemic plant genus in the tropical Andes. Molecular ecology, 25(18), 4593–4610. https://doi.org/10.1111/mec.13788.
Kozak, K. H., & Wiens, J. J. (2006). Does niche conservatism promote speciation? A case study in North American salamanders. Evolution, 60(12), 2604–2621. https://doi.org/10.1554/06-334.1.
Larget, B. R., Kotha, S. K., Dewey, C. N., & Ané, C. (2010). BUCKy: Gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics, 26, 2910–2911. https://doi.org/10.1093/bioinformatics/btq539.
Lartillot, N., & Philippe, H. (2006). Computing Bayes factors using thermodynamic integration. Systematic Biology, 55, 195–207. https://doi.org/10.1080/10635150500433722.
Leaché, A. D., & Fujita, M. K. (2010). Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proceedings of the Royal Society B: Biological Sciences, 277(1697), 3071–3077. https://doi.org/10.1098/rspb.2010.0662.
Leaché, A. D., Zhu, T., Rannala, B., & Yang, Z. (2019). The spectre of too many species. Systematic Biology, 68(1), 168–181. https://doi.org/10.1093/sysbio/syy051.
Leier, A., McQuarrie, N., Garzione, C., & Eiler, J. (2013). Stable isotope evidence for multiple pulses of rapid surface uplift in the Central Andes, Bolivia. Earth and Planetary Science Letters, 371, 49–58. https://doi.org/10.1016/j.epsl.2013.04.025.
Levin, D. A. (2003). Ecological speciation: lessons from invasive species. Systematic Botany, 28(4), 643–650. https://doi.org/10.1043/02-70.1.
Liu, C., Berry, P. M., Dawson, T. P., & Pearson, R. G. (2005). Selecting thresholds of occurrence in the prediction of species distributions. Ecography, 28, 385–393. https://doi.org/10.1111/j.0906-7590.2005.03957.x.
Liu, L., Zhao, B., Tan, D., & Wang, J. (2011). Phylogenetic relationships of Brassicaceae in China: insights from a non-coding chloroplast, mitochondrial, and nuclear DNA data set. Biochemical Systematics and Ecology, 39(4–6), 600–608. https://doi.org/10.1016/j.bse.2011.05.003.
Liu, C., White, M., & Newell, G. (2013). Selecting thresholds for the prediction of species occurrence with presence-only data. Journal of Biogeography, 40, 778–789. https://doi.org/10.1111/jbi.12058.
Liu, H., Ye, Q., & Wiens, J. J. (2020). Climatic-niche evolution follows similar rules in plants and animals. Nature Ecology & Evolution, 4(5), 753–763. https://doi.org/10.1038/s41559-020-1158-x.
Losos, J. B., & Glor, R. E. (2003). Phylogenetic comparative methods and the geography of speciation. Trends in Ecology & Evolution, 18(5), 220–227. https://doi.org/10.1016/S0169-5347(03)00037-5.
Luebert, F., & Weigend, M. (2014). Phylogenetic insights into Andean plant diversification. Frontiers in Ecology and Evolution, 2, 27. https://doi.org/10.3389/fevo.2014.00027.
Luo, A., Ling, C., Ho, S. Y., & Zhu, C. D. (2018). Comparison of methods for molecular species delimitation across a range of speciation scenarios. Systematic Biology, 67(5), 830–846. https://doi.org/10.1093/sysbio/syy011.
Lynch, J. D. (1989). The gauge of speciation: on the frequency of modes of speciation. In D. Otte & J. A. Endler (Eds.), Speciation and its consequences (pp. 527–553). Sunderland: Sinauer Associates.
Mallet, J. (2007). Hybrid speciation. Nature, 446(7133), 279–283. https://doi.org/10.1038/nature05706.
Mammola, S. (2019). Assessing similarity of n-dimensional hypervolumes: which metric to use? Journal of Biogeography, 46, 2012–2023. https://doi.org/10.1111/jbi.13618.
Mandakova, T., Li, Z., Barker, M. S., & Lysak, M. A. (2017). Diverse genome organization following 13 independent mesopolyploid events in Brassicaceae contrasts with convergent patterns of gene retention. The Plant Journal, 91(1), 3–21. https://doi.org/10.1111/tpj.13553.
Mandáková, T., Zozomová-Lihová, J., Kudoh, H., Zhao, Y., Lysak, M. A., & Marhold, K. (2019). The story of promiscuous crucifers: origin and genome evolution of an invasive species, Cardamine occulta (Brassicaceae), and its relatives. Annals of Botany, 124(2), 209–220. https://doi.org/10.1093/aob/mcz019.
Mares, M. A., Morello, J., & Goldstein, G. (1985). The Monte Desert and other subtropical semi and arid biomes of Argentina, with comments on their relationships to north American arid areas. In M. Evenari, I. N. Meir, & D. Goodall (Eds.), Hot Deserts and Arid Shrublands (pp. 203–236). Amsterdam: Elsevier.
Marhold, K., & Lihová, J. (2006). Polyploidy, hybridization and reticulate evolution: lessons from the Brassicaceae. Plant Systematics and Evolution, 259(2–4), 143–174. https://doi.org/10.1007/s00606-006-0417-x.
Marhold, K., Šlenker, M., & Zozomová-Lihová, J. (2018). Polyploidy and hybridization in the Mediterranean and neighbouring northern areas: examples from the genus Cardamine (Brassicaceae). Biologia Serbica, 40, 47–59. https://doi.org/10.5281/zenodo.1424339.
Martínez, G. A., Arana, M. D., Oggero, A. J., & Natale, E. S. (2017). Biogeographical relationships and new regionalisation of high-altitude grasslands and woodlands of the central Pampean ranges (Argentina), based on vascular plants and vertebrates. Australian Systematic Botany, 29(6), 473–488. https://doi.org/10.1071/SB16046.
Mayr, E. (1954). Change of genetic environment and evolution. In J. Huxley, A. C. Hardy, & E. B. Ford (Eds.), Evolution as a process (pp. 157–180). New York: Collier Books.
Mayr, E. (1959). Isolation as an evolutionary factor. Proceedings of the American Philosophical Society, 103(2), 221–230.
Mayr, E. (1982). Processes of speciation in animals. In C. Barigozzi (Ed.), Mechanisms of speciation (pp. 1–19). New York: Alan R. Liss.
Miller, M. A., Pfeiffer, W., & Schwartz, T. (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE) (pp. 1–8). New Orleans: IEEE.
Moritz, C. (1994). Defining ‘evolutionarily significant units’ for conservation. Trends in Ecology & Evolution, 9, 373–375. https://doi.org/10.1016/0169-5347(94)90057-4.
Morrone, J. J. (2015). Biogeographical regionalisation of the Andean región. Zootaxa, 3936, 207–236. https://doi.org/10.11646/zootaxa.3936.2.3.
Morrone, J. J. (2017). Neotropical biogeography: regionalization and evolution, CRC biogeography series. Boca Raton: CRC Press.
Morrone, J. J. (2018). Evolutionary biogeography of the Andean region, CRC biogeography series. Boca Raton: CRC Press.
Naciri, Y., & Linder, H. P. (2015). Species delimitation and relationships: the dance of the seven veils. Taxon, 64(1), 3–16. https://doi.org/10.12705/641.24.
Nieto-Montes de Oca, A., Barley, A. J., Meza-Lázaro, R. N., García-Vázquez, U. O., Zamora-Abrego, J. G., Thomson, R. C., & Leaché, A. D. (2017). Phylogenomics and species delimitation in the knob-scaled lizards of the genus Xenosaurus (Squamata: Xenosauridae) using ddRADseq data reveal a substantial underestimation of diversity. Molecular Phylogenetics and Evolution, 106, 241–253. https://doi.org/10.1016/j.ympev.2016.09.001.
Nosil, P. (2012). Ecological speciation. New York: Oxford University Press.
Nosil, P., & Harmon, L. (2009). Niche dimensionality and ecological speciation. In R. Butlin, J. Bridle, & D. Schluter (Eds.), Speciation and patterns of diversity (pp. 127–154). New York: Cambridge University Press.
Nürk, N. M., Uribe-Convers, S., Gehrke, B., Tank, D. C., & Blattner, F. R. (2015). Oligocene niche shift, Miocene diversification–cold tolerance and accelerated speciation rates in the St. John’s Worts (Hypericum, Hypericaceae). BMC Evolutionary Biology, 15(1), 80. https://doi.org/10.1186/s12862-015-0359-4.
Nürk, N. M., Michling, F., & Linder, H. P. (2018). Are the radiations of temperate lineages in tropical alpine ecosystems pre-adapted? Global Ecology and Biogeography, 27(3), 334–345. https://doi.org/10.1111/geb.12699.
O’Kane Jr., S. L. (2012). Evolution and classification of Physaria (Brassicaceae). In Principles of Crucifer Evolution, Botany 2012 Symposium. Columbus: Botanical Society of America.
O’Kane Jr., S. L., & Al-Shehbaz, I. A. (2004). The genus Physaria (Brassicaceae) in South America. Novon, 196–205.
Oksanen J., Blanchet F. G., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P. R., O’Hara R. B., Simpson G. L., Solymos P., Stevens M. H., Szoecs E., Wagner H. (2019). vegan: community ecology package. R package version 2.5-6. https://CRAN.R-project.org/package=vega. Accessed December 2019
Olson, D. M., Dinerstein, E., Wikramanayake, E. D., Burgess, N. D., Powell, G. V. N., Underwood, E. C., D’amico, J. A., Itoua, I., Strand, H. E., Morrison, J. C., Loucks, C. J., Allnutt, T. F., Ricketts, T. H., Kura, Y., Lamoreux, J. F., Wettengel, W. W., Hedao, P., & Kassem, K. R. (2001). Terrestrial ecoregions of the world: a new map of life on Earth. BioScience, 51, 933–938. https://doi.org/10.1641/0006-3568(2001)051[0933:TEOTWA]2.0.CO;2.
Ortiz-Jaureguizar, E., & Cladera, G. A. (2006). Paleoenvironmental evolution of southern South America during the Cenozoic. Journal of Arid Environments, 66(3), 498–532. https://doi.org/10.1016/j.jaridenv.2006.01.007.
Paradis, E. (2010). Pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics, 26, 419–420. https://doi.org/10.1093/bioinformatics/btp696.
Paradis, E., & Schliep, K. (2018). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35, 526–528. https://doi.org/10.1093/bioinformatics/bty633.
Pennington, R. T., Lavin, M., Prado, D. E., Pendry, C. A., Pell, S. K., & Butterworth, C. A. (2004). Historical climate change and speciation: Neotropical seasonally dry forest plants show patterns of both Tertiary and Quaternary diversification. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 359(1443), 515–538. https://doi.org/10.1098/rstb.2003.1435.
Peterson, A. T. (2011). Ecological niche conservatism: a time-structured review of evidence. Journal of Biogeography, 38(5), 817–827. https://doi.org/10.1111/j.1365-2699.2010.02456.x.
Peterson, A. T., & Lieberman, B. S. (2012). Species’ geographic distributions through time: playing catch-up with changing climates. Evolution: Education and Outreach, 5(4), 569. https://doi.org/10.1007/s12052-012-0385-2.
Peterson, A. T., Soberón, J., & Sánchez-Cordero, V. (1999). Conservatism of ecological niches in evolutionary time. Science, 285, 1265–1267. https://doi.org/10.1126/science.285.5431.1265.
Phillimore, A. (2014). Geography, range evolution, and speciation. In J. Losos, D. Baum, D. Futuyma, H. Hoekstra, R. Lenski, A. Moore, C. L. Peichel, D. Schluter, & M. Whitlock (Eds.), The Princeton guide to evolution (pp. 50–511). New Jersey: Princeton University Press.
Phillips, S. J., Anderson, R. P., Dudík, M., Schapire, R. E., & Blair, M. E. (2017). Opening the black box: an open-source release of Maxent. Ecography, 40, 887–893. https://doi.org/10.1111/ecog.03049.
Ponce, J. F., Rabassa, J., Coronato, A., & Borromei, A. M. (2011). Palaeogeographical evolution of the Atlantic coast of Pampa and Patagonia from the last glacial maximum to the Middle Holocene. Biological Journal of the Linnean Society, 103(2), 363–379. https://doi.org/10.1111/j.1095-8312.2011.01653.x.
Pons, J., Barraclough, T. G., Gomez-Zurita, J., Cardoso, A., Duran, D. P., Hazell, S., Kamoun, S., Sumlin, W. D., & Vogler, A. P. (2006). Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology, 55(4), 595–609. https://doi.org/10.1080/10635150600852011.
Pyron, R. A., Costa, G. C., Patten, M. A., & Burbrink, F. T. (2015). Phylogenetic niche conservatism and the evolutionary basis of ecological speciation. Biological Reviews, 90(4), 1248–1262. https://doi.org/10.1111/brv.12154.
Quantum GIS Development Team. (2016). QGIS Geographic Information System. Open Source Geospatial Foundation. http://qgis.org. Accessed December 2017
R Core Team. (2018). R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing https://www.R-project.org. Accessed Jan 2019
Rabassa, J. (2008). Late Cenozoic glaciations in Patagonia and Tierra del Fuego. Developments in Quaternary Sciences, 11, 151–204. https://doi.org/10.1016/S1571-0866(07)10008-7.
Rabassa, J., & Ponce, J. F. (2013). The Heinrich and Dansgaard-Oeschger climatic events during Marine Isotopic Stage 3: searching for appropriate times for human colonization of the Americas. Quaternary International, 299, 94–105.
Rabassa, J., Coronato, A., & Martinez, O. (2011). Late Cenozoic glaciations in Patagonia and Tierra del Fuego: an updated review. Biological Journal of the Linnean Society, 103(2), 316–335. https://doi.org/10.1111/j.1095-8312.2011.01681.x.
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., & Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67, 901–904. https://doi.org/10.1093/sysbio/syy032.
Ramsey, J. (2011). Polyploidy and ecological adaptation in wild yarrow. Proceedings of the National Academy of Sciences of United States of America, 108(17), 7096–7101. https://doi.org/10.1073/pnas.1016631108.
Rannala, B., & Yang, Z. (2017). Efficient Bayesian species tree inference under the multispecies coalescent. Systematic Biology, 66(5), 823–842. https://doi.org/10.1093/sysbio/syw119.
Renner, M. A., Heslewood, M. M., Patzak, S. D., Schäfer-Verwimp, A., & Heinrichs, J. (2017). By how much do we underestimate species diversity of liverworts using morphological evidence? An example from Australasian Plagiochila (Plagiochilaceae: Jungermanniopsida). Molecular Phylogenetics and Evolution, 107, 576–593. https://doi.org/10.1016/j.ympev.2016.12.018.
Revell, L. J. (2012). phytools: an R package for phylogenetic comparative biology (and other things). Methods in Ecology and Evolution, 3, 217–223. https://doi.org/10.1111/j.2041-210X.2011.00169.x.
Ricklefs, R. E. (1987). Community diversity: relative roles of local and regional processes. Science, 235(4785), 167–171. https://doi.org/10.1126/science.235.4785.167.
Riddle, B. R., & Hafner, D. J. (1999). Species as units of analysis in ecology and biogeography: time to take the blinders off. Global Ecology and Biogeography, 8(6), 433–441. https://doi.org/10.1046/j.1365-2699.1999.00170.x.
Rieseberg, L. H., & Willis, J. H. (2007). Plant speciation. Science, 317(5840), 910–914. https://doi.org/10.1126/science.1137729.
Roig, F. A., Roig-Juñent, S., & Corbalán, V. (2009). Biogeography of the Monte desert. Journal of Arid Environments, 73(2), 164–172. https://doi.org/10.1016/j.jaridenv.2008.07.016.
Rollins, R. C. (1939). The cruciferous genus Physaria. Contributions from the Gray Herbarium of Harvard University, 126, 392–415.
Romano, G. M. (2017). A high resolution shapefile of the Andean biogeographical region. Data in Brief, 13, 230–232. https://doi.org/10.1016/j.dib.2017.05.039.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., Larget, B., Liu, L., Suchard, M. A., & Huelsenbeck, J. P. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a Large Model Space. Systematic Biology, 61, 539–542. https://doi.org/10.1093/sysbio/sys029.
Rull, V. (2011). Neotropical biodiversity: timing and potential drivers. Trends in Ecology & Evolution, 26(10), 508–513. https://doi.org/10.1016/j.tree.2011.05.011.
Rundle, H. D., & Nosil, P. (2005). Ecological speciation. Ecology Letters, 8(3), 336–352. https://doi.org/10.1111/j.1461-0248.2004.00715.x.
Rutter, N., Coronato, A., Helmens, K., Rabassa, J., & Zárate, M. (2012). Glaciations in North and South America from the Miocene to the Last Glacial Maximum. Comparisons, linkages and uncertainties. New York: Springer. https://doi.org/10.1007/978-94-007-4399-1.
Ryder, O. A. (1986). Species conservation and systematics: the dilemma of subspecies. Trends in Ecology & Evolution, 1, 9–10.
Salariato, D. L., & Zuloaga, F. O. (2017). Climatic niche evolution in the Andean genus Menonvillea (Cremolobeae: Brassicaceae). Organisms Diversity & Evolution, 17, 11–28. https://doi.org/10.1007/s13127-016-0291-5.
Salariato, D. L., Zuloaga, F. O., & Al-Shehbaz, I. A. (2013). Revision and tribal placement of the Argentinean genus Parodiodoxa (Brassicaceae). Plant Systematics and Evolution, 299, 305–316. https://doi.org/10.1007/s00606012-0722-5.
Salariato, D. L., Zuloaga, F. O., Cano, A., & Al-Shehbaz, I. A. (2015). Molecular phylogenetics of tribe Eudemeae (Brassicaceae) and implications for its morphology and distribution. Molecular Phylogenetics and Evolution, 82, 43–59. https://doi.org/10.1016/j.ympev.2014.09.030.
Salariato, D. L., Zuloaga, F. O., Franzke, A., Mummenhoff, K., & Al-Shehbaz, I. A. (2016). Diversification patterns in the CES clade (Brassicaceae tribes Cremolobeae, Eudemeae, Schizopetaleae) in Andean South America. Botanical Journal of the Linnean Society, 181, 543–566. https://doi.org/10.1111/boj.12430.
Salariato, D. L., Al-Shehbaz, I. A., & Zuloaga, F. O. (2018). Reinstatement of the southern Andean genus Stenodraba (Brassicaceae) based on molecular data and insights from its environmental and geographic distribution. Systematic Botany, 43, 35–52. https://doi.org/10.1600/036364418X697085.
Salariato, D. L., Cano, A., Zuloaga, F. O., & Al-Shehbaz, I. A. (2020). Molecular phylogeny of Cremolobus (Brassicaceae) supports the recognition of the new genus Yunkia and demonstrates the high habitat diversity of tribe Cremolobeae. Systematics and Biodiversity, 18, 295–314. https://doi.org/10.1080/14772000.2020.1739777.
Schemske, D. W. (2010). Adaptation and the origin of species. The American Naturalist, 176(S1), S4–S25. https://doi.org/10.1086/657060.
Schluter, D. (2000). The Ecology of Adaptive Radiation. Oxford: Oxford University Press.
Schluter, D. (2001). Ecology and the origin of species. Trends in Ecology & Evolution, 16(7), 372–380. https://doi.org/10.1016/S0169-5347(01)02198-X.
Schluter, D. (2009). Evidence for ecological speciation and its alternative. Science, 323(5915), 737–741. https://doi.org/10.1126/science.1160006.
Simpson, G. G. (1951). The species concept. Evolution, 5, 285–298. https://doi.org/10.1111/j.1558-5646.1951.tb02788.x.
Simpson, G. G. (1953). The major features of evolution. New York: Columbia University Press.
Soberón, J. (2007). Grinnellian and Eltonian niches and geographic distributions of species. Ecology Letters, 10(12), 1115–1123. https://doi.org/10.1111/j.1461-0248.2007.01107.x.
Solbrig, O. T., Blair, W. F., Enders, F. A., Hulse, A. C., Hunt, J. H., Mares, M. A., Neff, J., Otte, D., Simpson, B. B., & Tomoff, C. S. (1977). The biota, the dependent variable. In G. H. Orians & O. T. Solbrig (Eds.), Convergent Evolution in Warm Deserts, an Examination of Strategies and Patterns in Deserts of Argentina and the United States (pp. 50–66). Pennsylvannia: Dowden, Hutchinson & Ross Inc..
Spalink, D., Drew, B. T., Pace, M. C., Zaborsky, J. G., Li, P., Cameron, K. M., Givnish, T. J., & Sytsma, K. J. (2016). Evolution of geographical place and niche space: Patterns of diversification in the North American sedge (Cyperaceae) flora. Molecular Phylogenetics and Evolution, 95, 183–195. https://doi.org/10.1016/j.ympev.2015.09.028.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033.
Stamatakis, A., Hoover, P., & Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web-servers. Systematic Biology, 57, 758–771. https://doi.org/10.1080/10635150802429642.
Sudhaus, W. (2004). Radiation within the framework of evolutionary ecology. Organisms Diversity & Evolution, 4(3), 127–134. https://doi.org/10.1016/j.ode.2004.04.001.
Sukumaran, J., & Knowles, L. L. (2017). Multispecies coalescent delimits structure, not species. Proceedings of the National Academy of Sciences, 114(7), 1607–1612. https://doi.org/10.1073/pnas.1607921114.
Taberlet, P., Gielly, L., Pautou, G., & Bouvet, J. (1991). Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Molecular Biology, 17, 1105–1109. https://doi.org/10.1007/BF00037152.
Tewes, L. J., Michling, F., Koch, M. A., & Müller, C. (2018). Intracontinental plant invader shows matching genetic and chemical profiles and might benefit from high defence variation within populations. Journal of Ecology, 106(2), 714–726. https://doi.org/10.1111/1365-2745.12869.
Thiers B. (2020). [continuously updated]. Index Herbariorum: a global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium. http://sweetgum.nybg.org/science/ih/. Accessed March 2020.
Trabucco, A., & Zomer, R. (2019). Global Aridity Index and Potential Evapotranspiration (ET0) Climate Database v2. figshare. Dataset. https://doi.org/10.6084/m9.figshare.7504448.v3. Accessed July 2019
Uba, C. E., Hasler, C. A., Buatois, L. A., Schmitt, A. K., & Plessen, B. (2009). Isotopic, paleontologic, and ichnologic evidence for late Miocene pulses of marine incursions in the central Andes. Geology, 37, 827–830. https://doi.org/10.1130/G30014A.1.
Warren, D. L., Glor, R. E., & Turelli, M. (2008). Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution, 62, 2868–2883. https://doi.org/10.1111/j.1558-5646.2008.00482.x.
Warwick, S. I., & Al-Shehbaz, I. A. (2006). Brassicaceae: chromosome number index and database on CD-Rom. Plant Systematics and Evolution, 259, 237–248. https://doi.org/10.1007/s00606-006-0421-17.
Wiens, J. J. (2004a). Speciation and ecology revisited: phylogenetic niche conservatism and the origin of species. Evolution, 58(1), 193–197. https://doi.org/10.1554/03-447.
Wiens, J. J. (2004b). What is speciation and how should we study it? The American Naturalist, 163(6), 914–923. https://doi.org/10.1086/386552.
Wiens, J. J. (2012). Perspective: why biogeography matters: historical biogeography vs. phylogeography and community phylogenetics for inferring ecological and evolutionary processes. Frontiers of Biogeography, 4(3), 128–135.
Wiens, J. J., & Graham, C. H. (2005). Niche conservatism: integrating evolution, ecology, and conservation biology. Annual Review of Ecology, Evolution, and Systematics, 36, 519–539. https://doi.org/10.1146/annurev.ecolsys.36.102803.095431.
Wiens, J. J., Ackerly, D. D., Allen, A. P., Anacker, B. L., Buckley, L. B., Cornell, H. V., Ellen, I., Damschen, E. I., Davies, T. J., Grytnes, J. A., Harrison, S. P., Hawkins, B. A., Holt, R. D., McCain, C. M., & Stephens, P. R. (2010). Niche conservatism as an emerging principle in ecology and conservation biology. Ecology Letters, 13(10), 1310–1324. https://doi.org/10.1111/j.1461-0248.2010.01515.x.
Wiley, E. O. (1978). The evolutionary species concept reconsidered. Systematic Zoology, 27, 17–26.
Xie, W., Lewis, P. O., Fan, Y., Kuo, L., & Chen, M. H. (2011). Improving marginal likelihood estimation for Bayesian phylogenetic model selection. Systematic Biology, 60, 150–160. https://doi.org/10.1093/sysbio/syq085.
Yang, Z. (2015). The BPP program for species tree estimation and species delimitation. Current Zoology, 61(5), 854–865. https://doi.org/10.1093/czoolo/61.5.854.
Yang, Z., & Rannala, B. (2014). Unguided species delimitation using DNA sequence data from multiple loci. Molecular Biology and Evolution, 31(12), 3125–3135. https://doi.org/10.1093/molbev/msu279.
Young, K. R., Ulloa Ulloa, C., Luteyn, J. L., & Knapp, S. (2002). Plant evolution and endemism in Andean South America: an introduction. The Botanical Review, 68, 4–21. https://doi.org/10.1663/0006-8101(2002)068[0004:PEAEIA]2.0.CO;2.
Zachos, J., Pagani, M., Sloan, L., Thomas, E., & Billups, K. (2001). Trends, rhythms, and aberrations in global climate 65 Ma to present. Science, 292, 686–693. https://doi.org/10.1126/science.1059412.
Acknowledgements
Our deep gratitude goes to Dr. Ihsan A. Al-Shehbaz for the critical review of this work and his valuable support, guidance, and suggestions in the study of South American Brassicaceae over the years. We thank the directors, curators, and collection managers of the herbaria listed. Special thanks are to Lone Aagesen, Juan M. Acosta, and Ariel Lliully Aguilar for their help in the fieldtrips. We especially appreciate the help of editors and two anonymous reviewers who provided useful suggestions to improve the early version of this paper. Botanical collections associated to this project were conducted under the permits: APN No. 1103 and 1546 (Argentina) and VMABCC No. 026/09 (Bolivia).
Funding
This work was funded by ANPCyT (Agencia Nacional de Promoción Científica y Tecnológica) grant PICT-2016-0096, CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas) grant PIP-112-201301-00124CO, and the National Geographic Society grant #9841-16, for which we are profoundly grateful.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not use animals and does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Fig. S1
Maximum likelihood tree generated with nrITS sequences and RAxML 8.2.10 showing the monophyly of South American Physaria. Values on branches correspond to Bootstrap support ≥ 50%. Units of branch length are proportional to nucleotide substitutions per site. (PDF 256 kb)
Fig. S2
Distribution map of South American Physaria and delimited area corresponding to the 50 km1423 buffered minimum convex polygon (study area) used in the niche analyses and geographic range comparisons. (PDF 1266 kb)
Fig. S3
Maximum likelihood trees generated with RAxML 8.2.10 showing the phylogenetic placement of sampled specimens of South American Physaria. a, nrITS dataset. b, cpDNA dataset (trnL-F/trnH-psbA/trnG intron/trnS-trnG). c, concatenated nrITS+cpDNA dataset. Values on branches correspond to Bootstrap support ≥ 50%. Units of branch length are proportional to nucleotide substitutions per site. Boxes to the rigth show delimited lineages. For tip labels: CRA, P. crassistigma, LAT, P. lateralis; MEN, P. mendocina; OKA, P. okanensis; PYG, P. pygmaea; URB: P. urbaniana. Image: Flower and fruit of P. mendocina (PDF 793 kb)
Fig. S4
Maximum clade credibility trees obtained from the analyses with MrBayes 3.2.6 showing the phylogenetic placement of sampled specimens of South American Physaria. a, nrITS dataset. b, cpDNA dataset (trnL-F/trnH-psbA/trnG intron/trnS-trnG). c, concatenated n rITS+cpDNA dataset. Circles on branches correspond to posterior probabilities (PP) ≥ 50%. Units of branch length are proportional to nucleotide substitutions per site. Boxes to the rigth show delimited lineages. For tip labels: CRA, P. crassistigma, LAT, P. lateralis; MEN, P. mendocina; OKA, P. okanensis; PYG, P. pygmaea; URB: P. urbaniana. Image: flower and fruit of P. crassistigma. (PDF 937 kb)
Fig. S5
Phylogenetic placement of sampled specimens of South American Physaria. a, Primary concordance tree from the BUCKy analyses using the 6002 trees from the nrITS and the cpDNA datasets obtained in the MrBayes analyses and under α = 1. Concordance factor values (CF) ≥ 0.5 are shown on branches. Units of branch length are proportional to the concordance factor. Results from concordance analyses varying the discordance prior (α) had no effect on topology or concordance. Stars on nodes indicate delimited lineages of South American Physaria. b, filtered supernetwork generated in SplitsTree 4.14.8 using 1000 Bayesian posterior trees per each nrITS and cpDNA dataset with the Z-closure algorithm, and filtering the splits to show only those present in a minimum of 30% input trees. Shades groups represent delimited lineages. (PDF 537 kb)
Fig. S6
Results from the climatic niche analyses in the environmental space obtained using the PCA env method. a–c, Correlation circles showing the contribution of environmental variables on the first three axes of the PCA-env and the percentage of inertia explained. a, PC1 vs PC2. b, PC1 vs PC3. c, PC2 vs PC3. d, Bar plot from the broken-stick analysis. PCA-env axes with larger percentages of accumulated inertia than the broken-stick variances are significant (three in this case). (PDF 31 kb)
Fig. S7
Univariate climatic niche comparisons in the environmental space among delimited lineages of South American Physaria. a, density plots computed for each independently evolving lineage using the PCenv1, PCenv2, PCenv3, elevation, annual mean temperature (BIO1), and annual mean precipitation (BIO12). b, p-values from Kruskal-Wallis test together with the Bonferroni correction for multiple comparisons to assess for differences between Physaria lineages for the three first PCs, elevation, BIO1, and BIO12. Values marked with an asterisk (red cells) are rejected at α=0.05. Labels: CRA, crassistigma lineage; NLA, northern lateralis lineage; SLA, southern lateralis lineage; MEN, mendocina lineage; URB, urbaniana lineage; YUN, yungas lineage. (PDF 645 kb)
Fig. S8
Geographic range overlap of delimited lineages of South American Physaria for the present time. Binary (presence/absence) distributions maps for delimited lineages derived from the species distribution modelling (SDM) in MaxEnt using the maximum training sensitivity plus specificity as threshold. Blue and green cells indicate presence of only one lineage, while red cells indicate presence of both species (sympatric cells). (PDF 1635 kb)
Fig. S9
Geographic range overlap of delimited lineages of South American Physaria for the Last Glacial Maximum (LGM) (~ 21 kya). Binary (presence/absence) distributions maps for delimited lineages derived from the species distribution modelling (SDM) in MaxEnt using the maximum training sensitivity plus specificity as threshold. Blue and green cells indicate presence of only one lineage, while red cells indicate presence of both species (sympatric cells). (PDF 2255 kb)
Table S1
(DOCX 13 kb)
Table S2
(DOCX 5 kb)
Table S3
(DOCX 10 kb)
Table S4
(DOCX 11 kb)
ESM 14
ITS alignment for the South American Physaria dataset used for phylogenetic analyses in this study. (NEX 32 kb)
ESM 15
trnL-F alignment for the South American Physaria dataset used for phylogenetic analyses in this study. (NEX 39 kb)
ESM 16
trnH-psbA alignment for the South American Physaria dataset used for phylogenetic analyses in this study. (NEX 15 kb)
ESM 17
trnG intron alignment for the South American Physaria dataset used for phylogenetic analyses in this study. (NEX 31 kb)
ESM 18
trnS-trnG alignment for the South American Physaria dataset used for phylogenetic analyses in this study. (NEX 26 kb)
Appendix 1
Appendix 1
Taxa and GenBank accession numbers for the ITS, trnH-psbA , trnL-F, trnG intron, and trnS-trnG sequences used in the molecular analyses.
Physaria fendleri (A. Gray) O'Kane & Al-Shehbaz (outgroup) (AF055198, -, AF055266, -). Physaria crassistigma O'Kane & Al-Shehbaz. ARGENTINA. Mendoza. Las Heras: Above Villavicencio on road to Paramillo de Uspallata, roadside, open areas among shrubs, ca. 44 km before Uspallata, Al-Shehbaz et al. 808 (SI) (CRA6) (MW366047, MW366135, MW366090, MW366180, MW366223). Luján de Cuyo: Centro de Ski Vallecitos, ladera derecha de cerros subiendo por la pista de ski, Salariato et al. 252 (SI) (CRA3) (MW366023, MW366110, MW366068, MW366157, MW366201). Tunuyán: RP94, Camino al Portillo Argentino, 0.5 km de las Llaretas desde el puesto de gendarmeria hacia el portillo, Salariato et al. 207 (SI) (CRA1) (MW366021, MW366108, MW366066, MW366155, MW366199); camino del Portillo Argentino, desde Las Llaretas hacia el hotel abandonado, ca. 3 km del desvío a Valle de Manantiales, Salariato et al. 233 (SI) (CRA2) (MW366022, MW366109, MW366067, MW366156, MW366200); Ruta Provincial 94 de Manzano Histórico a Portillo Argentino, Zuloaga et al. 5292 (SI) (CRA5) (MW366040, MW366128, MW366083, MW366173, MW366216). Tupungato: Ruta provincial n° 94: entre Los Manantiales y Real de Piedras Coloradas, Cocucci & Sérsic 2253 (CORD, SI) (CRA4) (MW366037, MW366125, MW366080, MW366170, MW366214). Physaria lateralis O'Kane & Al-Shehbaz. ARGENTINA. Mendoza. Las Heras: Ruta Provincial 13, alrededor de Monolito, Giussani et al. 602 (SI) (LAT8) (MW366048, MW366137, MW366092, MW366182, MW366225). Malargüe: Base del Cerro Nevado por Ruta Prov. 180, Prina et al. 2630 (SI) (LAT6) (MW366038, MW366126, MW366081, MW366171, -). San Carlos: Refugio militar Gral. Alvarado, entrada a la Reserva Laguna del Diamante, Al-Shehbaz et al. 813 (SI) (LAT10) (MW366050, MW366139, -, MW366184, MW366226). San Rafael: Cerro Nevado, Prina, et al. 2917 (SRFA, SI) (LAT5) (MW366036, MW366124, MW366079, MW366169, -); de Termas del Sosneado a Laguna El Sosneado, Zuloaga et al. 12408 (SI) (LAT9) (MW366049, MW366138, MW366093, MW366183, -); Ruta Nacional 144, cuesta de los Terneros hacia 25 de Mayo, Zuloaga et al. 15277 (SI) (LAT11) (MW366042, MW366130, MW366085, MW366175, MW366218). San Luis. Coronel Pringles: Ruta Provincial 9, entre Trapiche y La Carolina, Aagensen 26 (SI) (LAT1) (MW366015, MW366102, MW366060, MW366149, MW366193); Ruta provincial 9, Monumento y Museo Lafinur, pasando La Carolina, Biganzoli 2034 (SI) (LAT7) (MW366039, MW366127, MW366082, MW366172, MW366215); Alrededores de la Gruta de Intihuasi, Cerana 1266 (CORD) (LAT4) (MW366033, MW366121, MW366076, MW366166, MW366211); Camino a San Francisco 10-15 km de La Carolina, Cerana 1330 (CORD) (LAT3) (MW366028, MW366115, -, -, -); Cañada Honda, Cerana 1360 (CORD) (LAT2) (MW366027, MW366114, -, MW366161, MW366205). Physaria mendocina (Phil.) O'Kane & Al-Shehbaz. ARGENTINA. Córdoba. Calamuchita: San Agustin (Cerro La Cruz), Cantero 6107 (CORD) (MEN4) (MW366020, MW366107, MW366065, MW366154, MW366198). Colón: Candonga, Cantero 5897 (CORD) (MEN8) (MW366029, MW366116, MW366072, MW366162, MW366206). Punilla: Quilpo (Canteras), Cantero 5928 (CORD) (MEN7) (MW366026, MW366113, MW366071, MW366160, MW366204); Valle Hermoso, Cantero 6039 (CORD) (MEN6) (MW366025, MW366112, MW366070, MW366159, MW366203). Rio Cuarto: Alta Gracia, Cantero 5834 (CORD) (MEN9) (MW366030, MW366117, MW366073, MW366163, MW366207); Achiras (Monte Guazú), Cantero 6291 (CORD) (MEN3) (MW366019, MW366106, MW366064, MW366153, MW366197); Cuchi Yaco (Cerca Salsacate), Cantero 5656 (CORD) (MEN10) (MW366031, MW366118, MW366074, MW366164, MW366208); El Saucesito, Cantero 5565 (CORD) (MEN11) (MW366032, MW366119, MW366075, MW366165, MW366209); Iguazu, Cantero 5550 (CORD) (MEN2) (MW366018, MW366105, MW366063, MW366152, MW366196); Mesa de la Argentina, Cantero 5636 (CORD) (MEN5) (MW366024, MW366111, MW366069, MW366158, MW366202). La Pampa. Lihué Calel: Parque Nacional Lihue Calel, camino interior hacia cerros más bajos y pinturas rupestres, Zuloaga & Salariato 16073 (SI) (MEN1) (MW366016, MW366103, MW366061, MW366150, MW366194). Mendoza. Las Heras: de la RN 14 hacia los Berros, RN 153, Zuloaga et al. 15741 (SI) (MEN14) (-, MW366136, MW366091, MW366181, MW366224). Tunuyán: Manzano Histórico, Deginani et al. 2173 (SI) (MEN16) (MW366043, MW366131, MW366086, MW366176, MW366219). Tupungato: Bajando del Cerro Cristo Rey, Deginani et al. 2295 (SI) (MEN15) (MW366052, MW366141, MW366095, MW366186, MW366228). Río Negro. Valcheta: En km 50, cerros basálticos, Kiesling 9923 (SI) (MEN13) (MW366035, MW366123, MW366078, MW366168, MW366213). San Juan. Angaco: Pie de Palo, Kiesling et al. 9384 (SI) (MEN12) (MW366034, MW366122, MW366077, MW366167, MW366212). Physaria okanensis Al-Shehbaz & Prina. ARGENTINA. Catamarca. Ambato: al norte de cumbres de cerro el manchao, Halloy 809 (LIL) (OKA2) (-, MW366120, -, -, MW366210); subiendo hacia el manchao desde el rodeo, en el sitio Falda del Momo, Cantero et al. 7254 (CORD) (OKA1) (MW366017, MW366104, MW366062, MW366151, MW366195). Physaria pygmaea O'Kane & Al-Shehbaz. ARGENTINA. Catamarca. Andalgalá: Capillitas, alrededores del globo meteorológico, Acosta & von Mering 624 (SI) (PYG1) (MW366055, MW366144, MW366098, MW366189, MW366231); cerca de Mina Capillitas. subida al globo meteorológico, Salomón et al. 234 (SI) (PYG3) (MW366044, MW366132, MW366087, MW366177, MW366220). Jujuy. Humahuaca: Mina Aguilar, Espinazo del Diablo, Zuloaga et al. 13572 (SI) (PYG4) (MW366046, MW366134, MW366089, MW366179, MW366222). La Rioja. Famatina: Pampa de Tambería, Barboza et al. 3359 (CORD) (PYG2) (MW366058, MW366147, -, MW366191, MW366234). Physaria urbaniana (Muschl.) O'Kane & Al-Shehbaz. ARGENTINA. La Rioja. Famatina: Entre la bifurcación hacia Tres Piedras y Los Tambitos, Barboza et al. 2875 (CORD) (URB4) (MW366057, MW366146, MW366100, MW366190, MW366233); Pampa de Achavil, pasando el Río Achavil rumbo a Tres Piedras, en la pampa del "Ajenjo", Barboza et al. 2854 (CORD) (URB3) (MW366056, MW366145, MW366099, -, MW366232). Salta. Cachi: Cuesta de obispo, 200 mts pasando piedra del Molino, Zuloaga et al. 12956 (SI) (URB6) (MW366041, MW366129, MW366084, MW366174, MW366217). Cachi: Parque Nacional Los Cardones, pasando el Cerro Negro o Malacanta. en curso de agua, Zanotti et al. 787 (SI) (URB2) (MW366053, MW366142, MW366096, MW366187, MW366229); RP 33, Cuesta del Obispo, Zanotti et al. 800 (SI) (URB1) (MW366051, MW366140, MW366094, MW366185, MW366227); RP 33, Cuesta del Obispo, Piedra del Molino, Acosta & von Mering 607 (SI) (URB7) (MW366054, MW366143, MW366097, MW366188, MW366230). Tucumán. Tafí: RP 307, Abra el Infiernillo, Zanotti et al. 860 (SI) (URB8) (MW366045, MW366133, MW366088, MW366178, MW366221). BOLIVIA. Tarija. Yunchara: al S. de Copacabana, Beck et al. 23756 (LPB) (URB5) (MW366059, MW366148, MW366101, MW366192, MW366235).
Rights and permissions
About this article

Cite this article
Salariato, D.L., Zuloaga, F.O. Ecological and spatial patterns associated with diversification of South American Physaria (Brassicaceae) through the general concept of species. Org Divers Evol 21, 161–188 (2021). https://doi.org/10.1007/s13127-021-00486-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-021-00486-z