
Abstract
Plasmodial slime molds are members of the class Amoebozoa forming elaborate fruit bodies releasing airborne spores. Two species concepts have been developed independently: a morphological relying on fruit body characters, and a biological relying on crossing studies of a few cultivable species. In an attempt to reconcile both concepts, we obtained for 198 specimens of the common species Trichia varia partial sequences of three independent markers (nuclear small-subunit (SSU) ribosomal RNA gene, extrachromosomal; elongation factor 1 alpha gene, chromosomal; cytochrome oxidase subunit 1 gene, mitochondrial). The resulting phylogeny revealed 21 three-marker genotypes clustering into three groups. Combinations of the single-marker genotypes occurred exclusively within these groups, called 1, 2a, and 2b. To examine the suitability of group I introns to monitor speciation events, complete SSU sequences were generated for 66 specimens, which revealed six positions that can carry group I introns. For each of the groups 1 and 2a, five of these positions were occupied by different intron genotypes; and no genotype was shared by the two groups. Group 2b was devoid of introns. Putatively functional or degenerated homing endonuclease genes were found at different positions in groups 1 and 2a. All observations (genotypic combinations of the three markers, signs of recombination, intron patterns) fit well into a pattern of three cryptic biological species that reproduce predominantly sexual but are reproductively isolated. The pattern of group I introns and inserted homing endonuclease genes mounts evidence that the Goddard-Burt intron life cycle model applies to naturally occurring myxomycete populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plasmodial slime molds (Myxomycetes, Myxogastria), sometimes called “sexual amoebae,” are the most species-rich monophyletic group of Amoebozoa (Adl et al. 2012; Lahr et al. 2011a; Smirnov et al. 2011). These free-living amoebae are abundant in soil (Urich et al. 2008) and multiply by binary fission. According to the life cycle found in many textbooks, haploid amoebae or amoeboflagellates undergo syngamy, and zygotes form a diploid, syncytially organized plasmodium as the second vegetative stage. Most of the plasmodial biomass converts into sporocarpic fruit bodies which release airborne meiospores like fungi. Myxomycetes are one of the few groups of simple eukaryotes that appear to be macroscopic in dimension, which has led to a considerable body of data on distribution and ecology (Stephenson et al. 2008). Only a limited number of species, mostly of the order Physarales, will complete their life cycle in culture, even if food organisms are supplied (Clark and Haskins 2010). Well known are Physarum polycephalum and Didymium iridis as model organisms in cell physiology, developmental studies (Goodman 1980; Sauer 1982), and systems biology (Rätzel et al. 2013).
Apart from the advanced knowledge we have about a few model species, myxomycetes in nature are poorly studied by molecular methods. Only recently were first phylogenies published (Fiore-Donno et al. 2010, 2012, 2013), which recovered two main clades: the bright-spored clade (spores lightly colored by various pigments; Trichia varia as the target species of this study belongs to this clade), and the dark-spored clade (spores dark colored by melanin, most of the cultivable species). Many taxa are in critical need of revision (Schnittler et al. 2012). First studies employing partial nuclear small-subunit (SSU) ribosomal RNA gene sequences as a barcode marker revealed that many morphospecies seem to be highly diverse (Novozhilov et al. 2013b), mounting evidence that the “everything is everywhere” hypothesis (Fenchel and Finlay 2004) being popular for microorganisms should be rejected, and cryptic speciation is likely to occur (Aguilar et al. 2013).
Still, the only species concept applicable for the group as a whole is entirely based on the morphology of the sporocarps (Schnittler and Mitchell 2000). However, culture experiments carried out with members of the Physarales pose a challenge for this morphospecies concept prevailing for nearly 200 years (Clark 2000). Most isolates of a morphospecies behave heterothallic: amoebal populations derived from a single spore will not sporulate if they do not come in contact with amoebae from a different clone (Collins 1981). Heterothallism is maintained via mating types which can be arranged in a single-locus multiallelic system (Didymium iridis; Betterley and Collins 1983; Clark 1993), or in three loci with multiple alleles (Physarum polycephalum; Clark and Haskins 2010; Collins 1975; Moriyama and Kawano 2010). Often, some but not all of the mating types in a morphologically defined species are compatible. Groups of compatible strains can be seen as biospecies (Clark 1995; Collins 1979), thus providing an alternative species concept. This was the first question of the study: Can we detect evidence for coexisting biospecies in nature?
If we assume that myxomycetes reproduce sexually (as it should be expected for Amoebozoa, Lahr et al. 2011b) with mating types ensuring outcrossing (Clark and Haskins 2010; Collins 1979), asexual lineages can develop via clonal populations of non-fruiting amoebae (Clark and Haskins 2011). However, in culture experiments involving multiple isolates, some strains behave non-heterothallic and form fruit bodies in single-spore cultures (Collins 1981). Automixis, i.e., incomplete meiosis and the formation of diploid instead of haploid meiospores, was invoked as the most likely reproductive system for such non-heterothallic, presumably asexual isolates (Clark and Haskins 2013). Such diploid spores should give rise to diploid amoebae which may complete the life cycle without a sexual event. If existing, these entirely asexual lineages should have an advantage over sexual lineages for the colonization of habitat islands like accumulations of plant debris, since a single spore colonizing a new habitat island can complete the entire life cycle and disperse anew by spores (Schnittler and Tesmer 2008). A first study on a wild population of Lamproderma spp. (Fiore-Donno et al. 2011) suggested the existence of asexual populations, since specimens from geographical independent accessions were found to be identical in three molecular markers: the first part of SSU, its internal transcribed spacer 1 (ITS1), and the first part of the elongation factor 1 alpha gene (EF1A). Therefore, sexual and asexual biospecies may coexist within one morphologically circumscribed species. This was the second question for our study: Can we find evidence for asexual strains in a natural population?
In this study, we analyze one species for the first time with three independent marker genes: SSU (in myxomycetes located in multiple extrachromosomal DNA molecules; Torres-Machorro et al. 2010), EF1A (chromosomal), and cytochrome oxidase subunit 1 gene (COI, mitochondrial). T. varia (Pers. ex J.F. Gmel.) Pers. is a common myxomycete described already by Persoon (1794) based on the Linnean name Stemonitis varia (Linne 1792). In contrast to all established model species in myxomycetes, T. varia belongs to the bright-spored clade of myxomycetes (Fiore-Donno et al. 2013) and is easy to recognize by elaters within the spore mass that show only two, not several, spiral bands like the other members of this genus. This might be the reason why the name remained valid until today, although the morphological variability of the species is reflected by a rather long list of heterotypic synonyms (Lado 2014), the last one dating from 1974.
Additionally, we studied group I introns in complete SSU sequences. Perhaps caused by the lifestyle of plasmodia, which prey on various microbes (besides bacteria, also yeasts and green algae may be utilized; Haskins and Wrigley de Basanta 2008; Lazo 1961), myxomycete sequences are rich in introns (Johansen et al. 1992; Lundblad et al. 2004). As for mating types, cultivable members of the Physarales were established as a model system for the study of group I introns (Nandipati et al. 2012; Wikmark et al. 2007a, b). Ten intron insertion positions are known for SSU of myxomycetes; four of these (positions 516, 911, 959, and 1199) were reported to occur in T. varia (Fiore-Donno et al. 2013). Two mechanisms for intron homing are known. As the most powerful mechanism, intron homing via an intron-coded homing endonuclease (HE) is assumed, but reverse splicing through an RNA intermediate was invoked as well (Haugen et al. 2005a; Lambowitz and Belfort 1993). Although, in a strict sense, every intron poses an additional burden to the DNA replication of its host and introns can thus be seen as parasitic genes, they are selectively neutral (Edgell et al. 2011; Haugen et al. 2005a). Consequently, mutations in homing endonuclease genes (HEGs) should slowly render these genes non-functional, and according to the life cycle model of group I introns proposed by Goddard and Burt (1999), after degeneration and complete loss of the intron at one position, a new intron homing event can take place, thus ensuring survival of the intron in the population via sexual transmission. In myxomycetes, transmission of introns can take place via syngamy and subsequent karyogamy of amoebae of different lineages (mating types) within the sexual cycle. Plasmodial fusion, controlled by a very complex set of gene loci (Clark and Haskins 2012), is limited to closely related strains and should not contribute to intron transmission, since karyogamy does not take place. Similar to findings in other groups of organisms, sexually reproducing populations of a myxomycete biospecies should display a mixture of group I introns in different stages of HEG degeneration, and their genotypes should differ from intron populations in related but reproductively isolated biospecies. As such, the intron distribution in wild populations of myxomycetes may effectively tell apart biospecies and reveal their mode of reproduction. This study presents first results on genetic variation in three independent marker genes and the distribution of group I introns for a naturally occurring metapopulation of a myxomycete.
Methods
Specimens
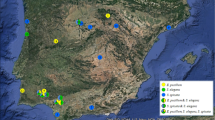
A total of 197 specimens of T. varia were collected throughout Eurasia, with two regions (southern Germany and southern Siberia) intensely studied. In addition, the single specimen of T. varia (AMFD451) used in the phylogeny of bright-spored myxomycetes (Fiore-Donno et al. 2013) was included in this study. Sequences of Perichaena depressa and Trichia scabra were used as outgroups in phylogenetic analyses. Specimens were deposited in private collections of Fiore-Donno AM, Germany (AMFD); van Hooff H, Netherlands (Hooff); Van Roy J, Belgium (JVR); de Haan M, Belgium (MdH); and Schnittler M, Germany (sc; to be deposited in M), and in the herbaria KRAM (Ronikier A, Poland) and LE (Novozhilov YK, Russia). Information about specimens and GenBank accession numbers (KM494990-KM495078) is listed in Supplementary Table S1 and a database on localities (Supplementary Database S1).
DNA Extraction, PCR, and sequencing
About 5–20 sporocarps from each specimen were used for DNA extraction. Sporocarps were pre-cooled at −80 °C and grounded using a ball mill (Retsch MM301) at 30 Hz for 80 s. Genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen) or the Invisorb Spin Food Kit II (Stratec). PCR amplifications on T. scabra and one specimen of T. varia (AMFD451) were conducted with the same DNA as used for the published phylogeny (Fiore-Donno et al. 2013).
Partial SSU sequences were obtained using the primers SFATri and SR4Bright (Fiore-Donno et al. 2013), which amplify the first fragment of the gene (5’-end up to the intron insertion site 516). Partial EF1A sequences covering the first part of the gene include a short spliceosomal intron; here the primers 1FCyt and E500R were used (Fiore-Donno AM, pers. comm.). For the amplification of the first half of COI, the primers COIF1 and COIR1 were designed within reported conservative regions (Walker et al. 2011) based on gene and transcript sequences of six myxomycete species (Gott et al. 1993; Horton and Landweber 2000; Traphagen et al. 2010).
For complete SSU sequences, a single PCR product covering nearly the complete gene was obtained using the primers NUSSUF3 and NUSSUR3, which are designed within conserved regions found in the SSU alignment for bright-spored species (Fiore-Donno et al. 2013). This product was sequenced in sections, using multiple primers designed in this study. In addition, some of the partial SSU sequences were obtained using the primers NUSSUF3 and NUSSUR4.
All primers used in this study are listed in Table 1. PCR reactions were performed using the MangoTaq Kit (Bioline). Cycle sequencing reactions were performed using the BigDye Kit (ABI). In addition, Q-Solution (Qiagen) and Triton X-100 (Omnilab) were applied for PCR reactions and cycle sequencing reactions to obtain complete SSU sequences of some specimens following online protocols (Lieb 2014).
PCR and sequencing of single spores
Single spores were isolated from T. varia specimens showing heterogeneities in complete SSU sequences. Under a dissecting microscope (Zeiss, Stemi SV6, magnification ×50), a single spore was separated with a needle from a group of spores scattered over a glass slide. Spore crushing and PCRs were conducted according to a reported protocol (Frommlet and Iglesias-Rodríguez 2008) with modifications. The single spore was transferred directly to a PCR tube containing 1 μl TE buffer (10 mM Tris-HCl, 1 mM EDTA; pH 8.0). The PCR tube for negative control contained 1 μl TE and a glass bead. Tubes were vortexed at 2700 rpm for 60 s; PCR reagents (MangoTaq Kit) were then added directly to the PCR tube. The first PCR was performed in a volume of 10 μl with the primer pair NUSSUF3 and NUSSUR3. After initial denaturing at 94 °C for 2 min, we run 45 cycles of denaturing at 94 °C for 30 s, annealing at 56 °C for 30 s, elongation at 72 °C for 2 min, and a final elongation at 72 °C for 5 min. The second PCR was performed using 2 μl from the first PCR as template, and two primer pairs (NUSSUF10 and NUSSUR6; NUSSUF16 and NUSSUR11) in separate reactions. PCR settings were identical except for a total volume of 25 μl; the elongation step was set at 72 °C for 1 min. Cycle sequencing reactions were performed with the reverse primer using the BigDye Kit.
Alignments and phylogenetic analyses of partial SSU, EF1A, and COI sequences
For a first data set, we generated 198 partial SSU sequences from T. varia and one sequence from Perichaena depressa. Together with one sequence from T. scabra (GenBank: JX481314; Fiore-Donno et al. 2013), they were aligned using the multiple sequence alignment software MUSCLE (Edgar 2004) implemented in MEGA 5.05 (Tamura et al. 2011) with default settings. The resulting alignment was manually added to that of the published phylogeny of bright-spored myxomycetes (Fiore-Donno et al. 2013), resulting in 430 sites of which 272 were used for the original phylogeny (Fiore-Donno et al. 2013) plus an additional 10 sites that could be unambiguously aligned by us.
Furthermore, we obtained 58 partial EF1A sequences for T. varia and one sequence from Perichaena depressa. These and two reported sequences (T. scabra, GenBank: JX481343; T. varia, GenBank: JX481344; Fiore-Donno et al. 2013) were first aligned by MUSCLE with default settings and then manually added to the existing alignment (Fiore-Donno et al. 2013), resulting in 322 sites of which the exon part (258 sites) was used in phylogenetic analyses.
Finally, all 198 T. varia specimens, one Perichaena depressa specimen, and one T. scabra specimen were sequenced to obtain partial COI sequences. Sequences were aligned by MUSCLE with default settings. An alignment of 537 sites was used for phylogenetic analysis.
Phylogenetic analyses on individual genes were conducted including all single-gene genotypes identified for T. varia. Selected in accordance with the phylogeny of bright-spored myxomycetes (Fiore-Donno et al. 2013), specimens from Perichaena depressa and T. scabra were used as outgroups (TreeBASE: M30972, M30973, M30974). Substitution models were selected by TOPALi v2 (Milne et al. 2009) under the Bayesian information criterion (BIC) for maximum likelihood (ML) analysis and Bayesian analysis. For T. varia alignments, the best models were TrN (ML analysis) and HKY+G (Bayesian analysis) for partial SSU, TrNef (ML analysis) and K80 (Bayesian analysis) for partial EF1A, and HKY+G for partial COI. For alignments including additionally Perichaena depressa and T. scabra, the best models were K80+I+G for partial SSU, JC for partial EF1A, and TIM+G (ML analysis) and GTR+G (Bayesian analysis) for partial COI. ML analyses for single genes were conducted using PhyML 2.4.5 (Guindon and Gascuel 2003) implemented in TOPALi with 1000 bootstrap replications. Bayesian analyses were carried out with MrBayes 3.1.1 (Ronquist and Huelsenbeck 2003) implemented in TOPALi with two runs, one million generations, and 50 % as burn-in, sampled every 100 generations.
Sequences from all three genes were concatenated to generate a combined three-gene alignment. ML analysis was conducted on the combined three-gene alignment using PhyML with model GTR+G and 1000 bootstrap replications; Bayesian analysis with data partitions was conducted using MrBayes 3.2.2 (Ronquist et al. 2012) with two runs, one million generations, and 50 % as burn-in, sampled every 100 generations, and with three partitions each using the best model for the respective gene.
To compare genetic and geographic distances for the 197 specimens (excluding one specimen containing polymorphic sites) with partial SSU and partial COI sequences, we computed pairwise genetic distances using the maximum composite likelihood model (Tamura et al. 2004) implemented in MEGA 5.2 (Tamura et al. 2011). Geographic distances between localities of specimens were calculated using an Excel macro for the Vincenty formula (Dalgleish 2014); a Mantel test including 999 iterations for the comparison of both matrices was performed with the ExtraStats function of PopTools 3.2.5 (Hood 2010).
Alignments and phylogenetic analyses of complete SSU sequences
Our second data set includes complete SSU sequences from single PCR products of 66 specimens of T. varia ranging from 1835 to 5660 bp in size. Sequences with polymorphic sites were recorded using the respective IUPAC symbols.
The reported complete SSU sequence of T. varia specimen AMFD451 (GenBank: JX481315; Fiore-Donno et al. 2013) was inconsistent with the sequences obtained in this study. From this reason, a single PCR product was obtained with the primers NUSSUF3 and NUSSUR3 from an aliquot of the DNA from AMFD451 used by these authors and sequenced in sections directly. Comparisons revealed that the original sequence GenBank: JX481315 is most likely an artificial chimera of two origins with a breaking point near the end of the first exon. Therefore, the corrected sequence of the same specimen (GenBank: KM495017) was used for this study.
Complete SSU sequences were aligned using MUSCLE with the option “gap open −550; gap extend −3,” with manual adjustment on intron locations and within-intron alignments, resulting in a total of 9797 sites (TreeBASE: M30975). For three specimens, gaps due to incomplete sequences were recorded as missing data. The complete SSU alignment of T. varia was added as a block to the reported SSU alignment (supplementary alignment “journal.pone.0062586.s007” in Fiore-Donno et al. 2013, with updates based on GenBank accessions) with congruent delimitation of exon and intron borders between the two alignments. For phylogenetic analysis, the same set of 1325 sites as used by these authors was extracted. All T. varia specimens representing distinct genotypes within these 1325 sites were considered for a phylogeny using Ceratiomyxa fruticulosa as outgroup (TreeBASE: M30970). The best substitution model under the criterion BIC was SYM+I+G. Both ML (1000 bootstrap replications) and Bayesian analyses (two runs, four million generations, 50 % as burn-in, sampled every 100 generations) were conducted using PhyML and MrBayes, respectively.
Exon sequences were extracted from the T. varia complete SSU alignment, yielding a total of 1825 exon sites. The best substitution model for phylogenetic analyses under the criteria BIC was TrN+G for PhyML and GTR+G for MrBayes. Settings for the analyses were 1000 bootstrap replications for PhyML, and two runs, three million generations, 50 % as burn-in, and sampling every 100 generations for MrBayes. On the alignment excluding sequences containing polymorphic sites, overall mean distance was estimated by MEGA with the Tamura three-parameter substitution model and omitting gaps and missing data. Detection of recombination was performed by the pairwise homoplasy index (PHI) test (Bruen et al. 2006) implemented in SplitsTree4 (Huson and Bryant 2006).
Analyses on intron parts of SSU sequences
Intron sequences, excluding two containing polymorphic sites, were extracted from the T. varia complete SSU alignment. The overall mean distance of distinct intron sequences at each insertion position was estimated by MEGA with the Tamura three-parameter substitution model, applying complete deletion of gaps and missing data. The PHI test was conducted for intron sequences at each insertion position. For the association analysis of introns across different positions, each distinct intron sequence and the state of intron-lacking were coded by numbers. Association indices I A S (LIAN 3.6; Haubold and Hudson 2000) and rBarD (MultiLocus 1.2.2; Agapow and Burt 2001) were computed from intron profiles, with simulated significance calculated from 10000 randomized data sets. All extrachromosomal copies of SSU in one specimen were assumed to be identical and were treated as one copy.
Known group I intron sequences of other myxomycete species were extracted from published SSU sequences (Fiore-Donno et al. 2005, 2008, 2010, 2011, 2012, 2013; Nandipati et al. 2012). The GenBank accession DQ903669 (Meriderma aggregatum) was replaced by the sequence named “Meriderma_aggregatum_DQ903669” in the supplementary alignment “journal.pone.0035359.s001” in Fiore-Donno et al. (2012) due to the inconsistency between sequences in GenBank and the publication (deletion at site 1085 in GenBank: DQ903669; sites 2280 to 5529 in Meriderma_aggregatum_DQ903669 were absent in GenBank: DQ903669).
Group I intron sequences with known assignment to an intron class (classes IA, IB, IC, and ID, Michel and Westhof 1990; class IE, Li and Zhang 2005) were downloaded from the Comparative RNA Web Site (CRW; Cannone et al. 2002) and the Group I Intron Sequence and Structure Database (GISSD; Zhou et al. 2008). The reliability of intron class assignments performed with BLASTN 2.2.24 (Altschul et al. 1997) used in this study was tested by searching one of the two databases (CRW or GISSD) against the other database or the database itself, setting an Expect value less than 0.1, and eliminating self-matching intron sequences. The results showed that regions matched by BLASTN lie within conserved regions in group I introns (Burke et al. 1987; Li and Zhang 2005; Michel and Westhof 1990), and BLASTN could be applied in group I intron class assignment. Intron classes were assigned to introns of T. varia and other myxomycetes by BLASTN searches against introns from databases CRW and GISSD. Direct inspection of an alignment of S956 introns in myxomycetes automatically generated by MAFFT 7.158 (Katoh and Standley 2013) with the option “E-INS-i” detected two sets of sequences within S956 introns corresponding with the initial assignment to intron classes IC1 (a subclass of IC; Michel and Westhof 1990) and IE by BLASTN searches. Multiple conserved regions were identified in the MAFFT alignment of S956 introns. The conserved regions in S956 introns identified by BLASTN and MAFFT were compared with the structural alignments from GISSD, and the identified P7:P7’ pairing regions were consistent with those of IC1 and IE introns, respectively. Classifications for some introns of myxomycete species not investigated by us were adopted from the literature (Lundblad et al. 2004; Wikmark et al. 2007b).
Analyses of intron-encoded HEGs
BLASTX 2.2.24 searches were conducted on group I introns of T. varia and other myxomycetes against annotated HE protein sequences containing the motif His-Cys box found in nuclear ribosomal genes (Brown et al. 2012; Elde et al. 1999; Haugen et al. 1999, 2002, 2003, 2004, 2005b; Johansen et al. 1997; Machouart et al. 2004; Muscarella et al. 1990; Müller et al. 2001; Tanabe et al. 2002; Xu et al. 2013; Yokoyama et al. 2002). Intron sequences with matches to HE proteins were analyzed using StarORF 1.0 (STAR 2014). Similar to published sequences (Haugen et al. 2004), discontinued open reading frames (ORFs) were connected manually by introducing frameshifts or ignoring regions with premature stop codons. Due to low sequence similarity outside the His-Cys Box (Johansen et al. 1993), the start codon was located at the first codon for methionine in the ORF.
Eighteen putative HE protein sequences from introns of myxomycetes identified within this study, three known HE protein sequences from myxomycetes (Physarum polycephalum, I-PpoI, GenBank: AAB81102; Didymium iridis, I-DirI, GenBank: CAI83766; Didymium iridis, I-DirII, GenBank: CAI83767), and one HE protein sequence from the amoeboflagellate Naegleria jamiesoni (Heterolobosea, I-NjaI, GenBank: AAB71747) were aligned by MAFFT 7.220 with the option E-INS-i. The region from residue C131 to residue H134 of I-PpoI (Flick et al. 1998) was manually adjusted based on the HE protein alignment (pfam05551, corresponding to the zinc-binding loop region of HE) from the Conserved Domain Database (Marchler-Bauer et al. 2013), and published HE protein alignments (Flick et al. 1998; Haugen et al. 2004, 2005b; Johansen et al. 1993). Thirty-seven sites spanning residues S97–H134 of I-PpoI (Flick et al. 1998), all not affected by frameshifts, were included in phylogenetic analyses with I-PpoI as the outgroup (TreeBASE: M30976). The model WAG+I+G was selected as the best substitution model under the criterion BIC for phylogenetic analyses using PhyML (performed with 1000 bootstrap replications ) and MrBayes (two runs, four million generations, 50 % as burn-in, sampling every 100 generations).
Results
Intraspecific diversity of T. varia genotypes revealed by partial genes
Our first data set comprised partial sequences of three independent marker genes for 198 specimens of T. varia collected throughout Eurasia (Supplementary Table S1). We found 12 genotypes for partial SSU, 3 for partial EF1A, and 12 for partial COI sequences (TreeBASE: M30972, M30973, M30974). The spliceosomal intron in the first part of EF1A (sites 241–304 in the alignment TreeBASE: M30973) was 57 to 60 bp in length and distinguished three genotypes by two indels of 1 and 3 bp in length, respectively. Four genotypes contained polymorphic single nucleotides showing up as double peaks in sequence chromatograms, one in partial SSU and three in the spliceosomal intron of EF1A. If this spliceosomal intron is excluded, the heterozygous partial EF1A genotypes merged with the other homozygous genotypes into only three genotypes. The single partial SSU genotype with polymorphic single nucleotides was not treated as a separate genotype, as it comprises both nucleotides showing up in the two homogeneous genotypes. Combining the partial sequences of the three genes, 21 three-gene genotypes were identified in T. varia (see Supplementary Table S1 for genotype assignment and Fig. 1a for genotypic combinations of partial SSU and partial COI). Unrooted ML and Bayesian trees generated for each of the three genes separately recovered always three major clades arranged in two subsequent bifurcations (data not shown). The same clades showed up in a phylogeny constructed with the concatenated partial sequences of the three genes (Fig. 1b) and were designated as groups 1, 2a, and 2b, comprising 9, 9, and 3 three-gene genotypes, respectively.

Three-gene phylogeny of Trichia varia. a Associations between partial SSU and partial COI genotypes within the three groups (1, 2a, 2b). Dotted lines indicate associations found in one specimen, dashed lines in two to five specimens, and solid lines more than five specimens. b Bayesian majority-rule consensus tree of combined partial sequences of SSU, EF1A, and COI of T. varia. Shown is one representative specimen per three-gene genotype. c Bayesian majority-rule consensus tree of combined partial sequences rooted with Perichaena depressa and Trichia scabra. Bayesian posterior probabilities >0.70 and bootstrap replicates >50 are indicated
Various associations (1:1, 1:n, and n:n) of partial SSU and partial COI genotypes were observed within but never among these three groups (Fig. 1a). In contrast, partial EF1A sequences exhibited only one genotype per group if three specimens with single-nucleotide polymorphisms in the splicesomal intron are neglected. Combining the three partial sequences to an alignment of 1282 sites, a fine-scale phylogenetic structure emerged within 21 three-gene genotypes (Fig. 1b). This structure was recovered as well in a rooted tree with Perichaena depressa and T. scabra as outgroup, where 1077 sites could be aligned (Fig. 1c). The bifurcation between groups 1 and 2a/2b is well resolved in both unrooted and rooted trees. Groups 2a and 2b receive high support only for the unrooted trees but show lower support values or unresolved structures for rooted Bayesian/ML trees (0.52/not resolved for group 2a, 1.00/100 for group 2b), most likely due to the reason that only a lower number of phylogenetically informative sites could be retained for the rooted trees.
Geographic structure within T. varia three-gene genotypes
Correlation between genetic and geographic distances was extremely weak for partial SSU (r = −0.009), partial COI (r = 0.001), and combined SSU and COI sequences (r = −0.005), and these figures were always within the 95 % confidence intervals calculated for 999 matrix permutations. However, specimens of groups 1, 2a, and 2b where unevenly distributed among the major collection localities (Germany: Greifswald: 20/1/1; Hainich 25/0/3; Bavarian Forest 33/39/4; Russia: Caucasus, Teberda: 8/1/8; Novosibirsk 7/0/0; and Altai Mts. 12/3/3 specimens, see Supplementary Database S1). The three groups differed as well in mean elevation of the specimens (group 1, 429 m above sea level (a.s.l.), n = 123; 2a, 820 m, n = 49; 2b, 748 m, n = 25), and the differences were significant between group 1 and the two other groups (p = 0.05; unpaired non-tailed Student’s t test).
Phylogenetic position of T. varia within bright-spored myxomycetes revealed by complete SSU
Our second data set, comprising 66 complete SSU sequences (7, 35, and 24 for specimens belonging to groups 1, 2a, and 2b, respectively), was used to ascertain the position of the three groups within the phylogeny of Fiore-Donno et al. (2013) constructed for bright-spored myxomycetes. This phylogeny takes only 1325 exon sites into account which are alignable over the sampling of bright-spored myxomycete species and resulted in a fully supported monophyletic clade with seven different exon genotypes for the 66 specimens of T. varia (TreeBASE: M30970; three, three, and one genotype for groups 1, 2a, and 2b, respectively). Both ML and Bayesian trees showed identical topology but slightly different levels of support for each internal node (Supplementary Fig. S1). T. varia appeared in the clade “Trichia and allied genera” identified by Fiore-Donno et al. (2013) which is now better resolved than in the original phylogeny for both ML and Bayesian trees. The well-supported subclade uniting all sequences of T. varia was placed at the basal position, being sister to the other members of this clade which received moderate or high level of support in ML and Bayesian trees, respectively. Within the subclade T. varia, two sister clades were identified with high level of support, corresponding to groups 1 and 2a/2b in the three-gene phylogeny constructed with the first data set.
Polymorphisms within complete SSU sequences of T. varia
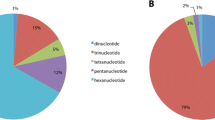
Considering the complete SSU sequences of the 66 specimens from T. varia, we observed 33–34 genotypes (the uncertainty is due to a part of the sequence we could not obtain for three specimens of group 1) for the exon parts, but 54 genotypes if both exon and intron parts are considered (Fig. 2). Eight genotypes were represented by more than one specimen, often from different localities (Supplementary Table S1).

Variations in 54 genotypes of the complete SSU sequences derived from 66 specimens of Trichia varia. The first six columns indicate collection number, locality code, assignment to groups 1, 2a, and 2b, the SSU genotype (numbered 1–54), the exon genotype (1–33), and the number of specimens per SSU genotype (for genotypes represented by multiple specimens, see Supplementary Table S1 for all collection numbers and localities). Exon positions showing within-group variation are annotated. Intron groups are color-coded for the six insertion positions (letters A–F) and intron genotypes (numbers 1–14), with zero indicating the absence of an intron. Positions of the intron were numbered in accordance with the Escherichia coli 16S ribosomal RNA gene. Numbers in the table head indicate positions according to the alignment TreeBASE: M30975. Introns occupy the following positions: S516, 579–1727; S529, 1741–3046; S911, 3700–4986; S956, 5032–6313; S1199, 6624–7885; and S1389, 8081–9656. The symbols filled square and open square indicate introns with full or with remnants of putative homing endonuclease genes. Comments: aheterogeneous state, bincomplete due to missing data, cpresence/absence not certain due to missing data, dabbreviation of location shown in Supplementary Table S1, and enumber of specimens with identical SSU genotype
Exon genotypic diversity differs strongly within groups; the relation of specimens to exon genotypes is 7:5–6 for group 1 (three sequences incomplete), 35:10 for group 2a, and 24:18 for group 2b. If introns are considered, these relations increase to 7:6, 35:30, and 24:18, respectively. Figure 2 shows within-group variations in exon sequences. Genetic distances between exons from distinct SSU genotypes are much smaller in groups 1 and 2a than in group 2b, and traces of recombination events were detected for the latter group only (Table 2). ML and Bayesian trees constructed using exon parts (Supplementary Fig. S2) recovered the same three groups (1, 2a, 2b) found for the phylogeny of three partial genes constructed with the first data set.
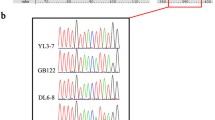
Four specimens displayed polymorphisms in their SSU sequences. To verify their source and to exclude the possibility of DNA contaminations caused by spores of other T. varia specimens, a nested PCR was performed from a single spore; with the first reaction targeting the complete SSU region and a second targeting two partial regions covering the polymorphic sites. This confirmed the heterogeneous state of SSU sequences for one specimen (sc27667c1) from group 2a in both exon and intron parts and three specimens (sc22413, sc22503, sc27657) from group 2b in exon parts: The partial sequences derived from single spores still displayed the same polymorphic patterns (double peaks in chromatograms) as seen in complete sequences from sporocarps. Nucleotides found for polymorphic sites were always identical with those occurring at corresponding sites of other homogeneous sequences from the same group (Fig. 2). In addition, homogeneous SSU sequences were obtained by single-spore PCR conducted in parallel from three of these specimens (except for sc22503).
Introns in the SSU sequence of T. varia
Each investigated specimen of groups 1 and 2a contained at least one putative group I intron (Fig. 2). Altogether, six insertion positions were found (516, 529, 911, 956, 1199, and 1389; positions numbered in accordance with the Escherichia coli 16S ribosomal RNA gene). In group 1, introns can occur at all positions except for 1199; in group 2a, all positions except for 529 may be occupied. Group 2b was devoid of introns. PCR reactions with the primers NUSSUF6 and NUSSUR11 targeting the potential insertion position 1199 were conducted for 18 additional specimens from group 1, and gel band patterns confirmed that S1199 introns were consistently absent in this group (data not shown). Altogether, 48 distinct intron sequences could be distinguished in T. varia: 12 in group 1 and 36 in group 2a.
Pairwise genetic distances between S911 and S956 introns from group 2a were higher than those for the other three positions. Traces from recombination events were detected in S911 and S956 introns but not in introns at other positions (Table 2). A set of specimens showing identical intron sequences at one position always differed in intron sequences at other positions. For 25 specimens of group 2a (excluding specimen sc27667c1 with heterogeneous intron sequences at positions S911 and S1199, and excluding specimens sc27737, sc27772c2, LE254838, and LE256579 with long introns), figures for two indices of association I A S and rBarD (both ranging from 0 to 1) were 0.0638 and 0.0631, respectively. The null hypothesis, assuming a complete random association, was nevertheless rejected with p values of 0.0087 and 0.0073, respectively.
Similarity searches on conserved motifs were used to assign group I introns in SSU of T. varia to classes IC1 and IE. S529 introns from group 1 could not be sequenced completely and were thus not classified. For intron-carrying groups of T. varia, the following pattern (group/intron class) emerged: S516: 1/IC1, 2a/IC1; S529: 1/?, 2a/none; S911: 1/IC1, 2a/IC1; S956: 1/IC1, 2a/IE; S1199: 1/none, 2a/IC1; and S1389: 1/IC1, 2a/IC1. In addition, we found both putatively functional and degenerated homing endonuclease genes at all insertion positions except for position 529 (Fig. 3).

Intron types found in 66 specimens of Trichia varia at six insertion positions. For each of the phylogroups 1 and 2a, introns are drawn to scale as horizontal bars. White sections denote intron sequences and gray sections homing endonuclease genes. Red/solid and blue/broken vertical lines indicate the approximate location of sites with point mutations and one-base indels, respectively. Columns at the right indicate the phylogroup, the intron genotype according to the codes used in Fig. 2, and the number of specimens sharing it (Color figure online)
Remarkable is the bipartite distribution of S956 introns, which was found as well for numerous species of both bright- and dark-spored myxomycetes (Fig. 4 and Supplementary Table S2). These two major phylogenetic groups possess each introns assigned to classes IC1 and IE, and their distribution does not coincide with myxomycete phylogeny. Within several genera (e.g., Cribraria, Colloderma, Diachea, Lamproderma, and Lepidoderma), IC1 and IE introns coexist. Similar to T. varia showing class IC1 introns in group 1 but class IE introns in group 2a, both intron classes were as well represented by two specimens of the dark-spored myxomycete Lamproderma puncticulatum (Fiore-Donno et al. 2011).

Simplified SSU phylogeny showing distribution of IC1 and IE class introns at position S956 in myxomycetes. Tree topology was derived from published SSU phylogenies (Fiore-Donno et al. 2010, 2012, 2013) and the tree presented in Supplementary Fig. S1 of this study. Trichia varia group 1 is represented by a specimen with an intron lacking an HEG; group 2a is represented by a specimen with a HEG-containing intron. Taxa printed in red and marked with “IC1” carry IC1 introns; those printed in blue and marked with “IE” carry IE introns. Nodes marked by dots received support of both Bayesian posterior probability >0.70 and bootstrap replicates >50 (Color figure online)
Overall genetic variation in T. varia
From the two data sets depicting genetic variation in T. varia, the first (three partial genes, 21 genotypes among 198 specimens) displays by far a lower resolution than the second (complete SSU sequences with their introns, 54 genotypes among 66 specimens). Combining the two data sets does only marginally enhance resolution: only two specimens sharing a complete SSU genotype (17) differ in their COI genotypes (5 and 7, see Supplementary Table S1 and Supplementary Database S1); these are 500 km apart (French Jura/Bavarian Forest).
In seven cases, all investigated markers were identical for multiple specimens (Supplementary Table S1; Supplementary Database S1). In five cases, the two to five specimens came from the same collecting site and are separated over distances of less than 0.6 km (three cases) and 6 km (two cases). However, in two cases, identical genotypes were found over distances of 604 km (France/southern Germany) and 605 km (Netherlands/northern Germany).
Intron-encoded homing endonuclease genes in T. varia and other myxomycetes
All group I introns at the six insertion positions identified for T. varia can carry putative full-length HEGs (Figs. 2 and 3); we only failed to detect them at position S529 (uncertain due to incomplete SSU sequences). Within introns of T. varia, remnants of ORF were identified in a 21-bp region of S516 introns (sites 597–618 in the alignment TreeBASE: M30975) from one specimen of group 1 and in a 47-bp region of S956 introns (sites 5089–5135 in the alignment TreeBASE: M30975) from 22 specimens of group 2a (Fig. 2). Both ORF remnants are homologous to the first part of the corresponding HEGs found at these positions in other specimens of T. varia.
Using all available data, we assembled an HE protein alignment from 22 putative HEGs of Amoebozoa (Supplementary Alignment S6). We included nine HEGs found in published SSU sequences of myxomycetes (Fiore-Donno et al. 2005, 2008, 2012, 2013; Nandipati et al. 2012). Seven new sequences for HEGs were found in T. varia, of which the two HEGs found in S516 introns of group 1 differ by one nucleotide which translates into a different amino acid (Table 3; Supplementary Alignment S6). Existing literature reports one HEG inserted in the S1199 intron of Diderma niveum (Hedberg and Johansen 2013), and one in the S516 intron of Echinostelium coelocephalum (Brown et al. 2012). Four HEGs were already described and tested for functionality (two in the S956 intron of Didymium iridis, I-DirI and I-DirII, Haugen et al. 2005b; Johansen et al. 1997; one in the group I intron L1925 from the nuclear large subunit ribosomal RNA gene of Physarum polycephalum, I-PpoI, Muscarella et al. 1990; one in the S516 intron of N. jamiesoni, I-NjaI, Johansen et al. 1993). Two putative HEGs were not included in our analysis, since the high level of variation in sequences did not allow to align them. These are HEGs in the S529 intron of Diderma niveum (Hedberg and Johansen 2013) and in the S788 intron of Meriderma fuscatum (Fiore-Donno et al. 2008).
Except for HEG from L1925 of Physarum polycephalum and HEG from S516 of N. jamiesoni, 20 HEGs from SSU sequences of myxomycetes remain. Of these, 4 (all in S956 introns) were found in antisense strands while the other 16 (various insertion positions) were detected in sense strands. Their host introns classify as both IC1 (16) and IE (4). The latter were all inserted in sense strands at position 956 (see Fig. 5 for intron directions and classes). Frameshifts were observed in ten HEGs (see Table 3 for range of ORFs). Three HEGs found at position 956 (Diderma meyerae, Diachea subsessilis, Elaeomyxa cerifera) have frameshifts located at the same region where in I-DirI and I-DirII nested spliceosomal introns were found (Haugen et al. 2005b; Vader et al. 1999) suggesting that spliceosomal introns might as well be nested in these HEGs. These regions were not included in protein sequences translated from ORFs, which were used for the phylogenetic analysis.

Bayesian majority-rule consensus tree of protein sequences from homing endonuclease genes of myxomycetes. The tree is rooted with I-PpoI from the L1925 intron of Physarum polycephalum. Support values are indicated for nodes with either Bayesian posterior probability >0.50 or bootstrap replicates >50, with the symbol “-” indicating topologies with branches deviating between Bayesian and ML trees. Taxa printed in red carry IC1, and those printed in blue IE introns as HEG hosts. Additional symbols: IC1 = IC1 introns as HEG hosts, IE = IE introns as HEG hosts, rightward arrow sense strand, leftward arrow antisense strand, A = protein with experimentally confirmed enzymatic activity, F = frameshift, open circle bright-spored myxomycetes, filled circle dark-spored myxomycetes (Color figure online)
Using I-PpoI from L1925 of Physarum polycephalum as the outgroup, we constructed a phylogeny of the 22 alignable HE protein sequences (Fig. 5). Both the ML tree and the Bayesian tree revealed similar topologies. HEGs from antisense strands (S956 introns, class IC1) and HEGs from sense strands form two separate, strongly supported clades. HEGs from S956 introns are located within two separate clades, consistent with their differences in sense/antisense insertion and classes of the host intron (IC1/IE). Within the clade uniting all sense-inserted HEGs, two subclades are resolved with high support in the Bayesian tree but not in the ML tree, and the single HEG from S1065 shifted its position between Bayesian and ML trees. Usually, HEGs coming from one insertion position cluster together, but this is not the case, if the respective hosts come from different groups of myxomycetes (dark- or bright-spored).
Discussion
Cryptic speciation in T. varia
Based on its unique capillitium morphology, T. varia was always seen as a well-defined taxon. Although the taxonomic database Nomenmyx (Lado 2005–2014) lists not less than 13 heterotypic synonyms (all but one described before 1930), based on sporocarp morphology no intraspecific taxa are currently accepted. All recent monographs (Martin and Alexopoulos 1969; Nannenga-Bremekamp 1991; Neubert et al. 1993; Poulain et al. 2011) treat T. varia as a single, yet variable species. This was confirmed using 1325 exon sites of complete SSU sequences (Supplementary Fig. S1): All investigated specimens of T. varia were united within a single clade of the phylogeny of bright-spored myxomycetes. The detailed analysis resolved three phylogenetic groups (1, 2a, 2b) occurring in both data sets assembled by us: the three-gene phylogeny of partial SSU, partial EF1A, and partial COI (Fig. 1), as well as the exon parts of complete SSU sequences (1825 sites; Supplementary Fig. S2).
A subsample of 21 specimens from all three groups revealed no significant differences in stalk length (reaching from sessile to shortly stalked sporocarps), spore ornamentation and size, and capillitium diameter (data not shown). In-depth morphological investigations, including analyses of spore and capillitium ornamentation with scanning electron microscopy, will be needed to test for morphological differences between the three taxa, which we see as a prerequisite for their formal description.
These phylogenetic and morphological results are explained best by viewing the phylogenetic groups 1, 2a, and 2b as cryptic taxa within the morphospecies T. varia, a hypothesis unknowingly suggested in the choice of the basionym by Persoon (1794). Infraspecific genetic structure was found as well for north- and south-hemispheric populations of Badhamia melanospora (Aguilar et al. 2013), although a tree constructed of partial SSU sequences only did not resolve the respective clades. This dark-spored myxomycete occurs nearly exclusively on decayed tissues of succulent plants. For the three gene markers applied to the less specialized T. varia, we can clearly demonstrate cryptic speciation but did not find a clear-cut geographical structure in the investigated metapopulation. Specimens of group 1 were rarely found at higher elevations, but all three groups may share the same microhabitat: moderately to well-decayed white rotten logs, preferentially of deciduous trees. In three cases, specimens from different groups were collected on the same log (sc27648c1/2a and sc27648c2/1; sc27664c1/2a and sc27664c3/1; and sc27667c1/2a, sc27667c2/2a, and sc27667c3/1).
Sexual biospecies in T. varia
For T. varia, we employed three virtually independent markers for the first data set of 198 specimens. SSU genes are organized in extrachromosomal units (Torres-Machorro et al. 2010); crosses between strains show a progressive increase of one parental type within the vegetative phase of the growing plasmodium (Ferris et al. 1983). The inheritance of mitochondrial DNA of Physarum polycephalum is mostly uniparental with the choice of donor and recipient determined by a dominance hierarchy depending on the mating type locus (Moriyama and Kawano 2010). For the nuclear single-copy gene EF1A, we assume Mendelian inheritance. Given similar relationships in T. varia, the three chosen markers should be independently inherited except for the point that nuclear genes expressing the mating types have some control over COI inheritance.
For partial sequences of SSU, COI, and EF1A, we recorded 5/4/3, 4/6/2, and 1/1/1 genotypes for the three groups 1, 2a, and 2b, respectively. The three groups share none of these genotypes, indicating missing gene flow and thus reproductive isolation. If association between two partial genes mediated by sexual reproduction would not be involved, mutation alone is unlikely to create the recurrent n:n association between the most common two partial SSU genotypes and two partial COI genotypes in group 1 (Fig. 1a). Only EF1A displays one genotype per group (not considering single-base heterozygosities within the short spliceosomal intron in three specimens which are likely to result from point mutations). We thus can assume that sexual reproduction is responsible for the exchange of single-marker genotypes between individuals of the same group.
The second data set with complete SSU sequences lends as well evidence for sexual reproduction. We found four heterogeneous sequences, and as well as their homogenous counterparts, which is easier to explain by recombination between multiple genotypes mediated by sexual reproduction than by assuming simultaneous mutations. This was confirmed by single-spore PCR. Even if we assume some investigated spores to be multinucleated as recently reported for Physarum pseudonotabile (Novozhilov et al. 2013a), different SSU genotypes must have existed in the plasmodium giving rise to these spores. Three of the four heterogeneous specimens displayed additionally homogeneous SSU sequences in different PCRs from single spores. Although PCR conditions occasionally favor one SSU genotype over the other, a model of selective replication of a single SSU genotype is likely to be the best explanation for the coexistence of spores with heterogeneous and homogeneous SSU sequences in T. varia. This selection process takes place in each individual nucleus of the growing plasmodium but continues during meiosis (where only one of the four nuclei survives; Clark and Haskins 2013; Gray and Alexopoulos 1968), and even in germinating spores (Ferris et al. 1983). Our results of single-spore PCR in T. varia were not in conflict with this model and suggested further that one parental genotype seems to be completely eliminated in spores showing homogeneous sequences. In spores displaying heterogeneous sequences, elimination still continues. Individual spores of one specimen are not synchronized regarding this elimination process. Therefore, the occurrence of heterogeneous SSU sequences can be seen as a trace of recent sexual events.
In addition, variation in both group I introns and exon parts of complete SSU sequences points toward sexual reproduction within each of three phylogroups of T. varia. The seven specimens of group 1 constitute a small sample not sufficient to test for recombination, although the introns appear to be randomly distributed (Fig. 2). For the 35 specimens from group 2a, the near-random association of HEG-lacking introns in group 2a specimens (low values of both the I A S and rBarD indices) indicates that within-group homologous recombination between extrachromosomal ribosomal DNAs mediated by sexual reproduction effectively breaks down associations among intron sequences. Another possible cause for the near-random association of HEG-lacking introns is intron homing via reverse splicing, which as well requires different SSU genotypes being brought together by sexual reproduction. Signals of recombination were detected in the entire introns of this group (positions 911 and 956), indicating that homologous recombination between extrachromosomal ribosomal DNAs mediated by sexual reproduction had occurred. Specimens of group 2b uniformely lack introns, but exon regions of the complete SSU show as well signals of recombination (Table 2).
Furthermore, patterns of SSU group I intron distribution provide convincing evidence for reproductive isolation between the three groups. Although all groups possess identical recognition sequences for intron insertion at all six insertion positions, groups 1 and 2a lack introns at different positions (529 vs 1199), none of the intron sequences found at the commonly occupied positions (516, 911, 956, and 1389) occurs in both groups, and group 2b is devoid of any introns (Fig. 2). The HEGs in the single position (1389) common to both intron-bearing groups of T. varia cluster as well together but are highly divergent from each other (Fig. 3; TreeBASE: M30976). We thus assume that reproductive isolation prevents inter-group intron homing among three groups of T. varia.
Summarizing, the patterns of partial SSU and COI genotypes, heterogeneities and recombination signals in complete SSU sequences, and group I intron distribution provide strong evidence that the three groups (1, 2a, 2b) of T. varia represent three biospecies which reproduce predominantly sexual. Similarly, a rapid decay of linkage disequilibrium was found for single-nucleotide polymorphisms from 137 gene fragments of wild North American strains of the cellular slime mold Dictyostelium discoideum, pointing to frequent sexual reproduction (Flowers et al. 2010). These results fit into the pattern of postulated widespread sexuality in Amoebozoa (Lahr et al. 2011b).
Our study demonstrates that the biospecies concept derived from case studies with cultivable myxomycetes is applicable in nature. Crossing experiments with several dark-spored myxomycetes (Clark and Haskins 2013) indicated the existence of several biospecies within a single morphospecies, as it was the single crossing experiment targeting a bright-spored species (Arcyria cinerea; Clark et al. 2002).
Asexual strains in T. varia?
A recent study investigating partial SSU, ITS1, and partial EF1A sequences in natural populations of the dark-spored myxomycetes Lamproderma columbinum and L. puncticulatum (Fiore-Donno et al. 2011) found groups showing identical sequences in all three markers which can be interpreted as evidence for missing or rare sexual reproduction. However, only exon sequences of SSU were considered for this study, and SSU and ITS1 markers are not independently inherited since both are linked together in one extrachromosomal molecule (Torres-Machorro et al. 2010). In addition, genotypic diversity for partial SSU sequences in Lamproderma (with a relation of specimens to genotypes of 52:11) was lower than for complete SSU in this study (66:54). When the resolution of markers is low, the observation of identical sequences in independently inherited markers is conformable with, but does not prove complete absence of recombination. Due to the lower genotypic diversity of the three (effectively two) independent markers in Lamproderma, this study may have failed to detect recombination; thus, the observed identical sequences in these markers evidence but do not prove asexual reproduction.
In the first data set composed of partial SSU, COI, and EF1A sequences of T. varia, we found among 198 specimens only 21 multilocus genotypes. With 66 specimens and 54 genotypes, our second data set of complete SSU sequences is much more polymorphic. Adding non-overlapping sequences from the first data set would only marginally enhance resolution: two specimens with the same complete SSU genotype displayed different COI sequences. Similar to the study of Fiore-Donno et al. (2011), we found as well seven cases with multiple specimens identical in all investigated markers (Supplementary Table S1; Supplementary Database S1). Two of these cases involved two identical specimens collected at distances exceeding 600 km. Therefore, the question remains if asexual strains, completing their life cycle without syngamy, occur in natural myxomycete populations of T. varia.
We must assume that amoebae divide by fission and can form large clonal populations. According to the sexual life cycle depicted in most textbooks, amoebal syngamy is a prerequisite for entering the next life stage, the diploid plasmodium, and finally the formation of fruit bodies. If this holds true, haploid ameobal clones cannot be detected by a study based on sequences from fruit bodies, since plasmodia and fruit bodies require prior syngamy of amobae. With this approach, including whole cohorts of spores, all paternal alleles will be resampled. In theory, heterogeneous sequences can occur if different alleles coexist in one fruit body. However, extrachromosomal SSU and mitochondrial COI will quickly become uniform, since one genotype will be eliminated (Ferris et al. 1983; Moriyama and Kawano 2010), and EF1A alone was found to be monomorphic in each of the three groups found within T. varia. This explains why we found only exceptional heterogeneous (SSU) or heterozygous (EF1A) sequences in T. varia.
Nevertheless, panmictic sexual reproduction should result in different genotypic combinations of multiple marker genes if they are (i) sufficiently polymorphic and (ii) independently inherited. Even then, repeated pairings within two widespread amoebal clones will produce the same multilocus genotype. The same holds true for the separation of a plasmodium into multiple parts, which migrate and fruit at different places. The five cases involving distances below 6 km with identical multiple markers may be explained by migrating amoebae or microcysts (mating repeatedly and thus producing the same genotype) and/or plasmodia (which may segregate and migrate independently). As such, this may as well be explained without assuming the existence of a second, asexual life cycle proposed in Clark and Haskins (2010) to explain the occurrence of non-heterothallic isolates.
The two cases of identical multilocus genotypes in T. varia occurring more than 600 km apart are difficult to explain by assuming migration of amoebae, microcysts, or plasmodia. But, we only found one extremely polymorphic marker, complete SSU sequences with their introns. In this marker, one of two different SSU genotypes which may be combined via amoebal syngamy is subsequenctly eliminated. Spores, which may bridge large distances, usually transport only one SSU genotype. Given the much lower variation in the other two markers (COI and EF1A), this increases the probability that identical multilocus genotypes occur by chance at two distant locations. We thus hesitate to take the two cases of identical multilocus genotypes involving specimens from distant locations as proof for the existence of asexual strains in nature. Viewing these results from the other side (different genotypic combinations of multiple markers, signs of recombination, intron patterns), we can conclude that sexual reproduction is at least the prevailing mode of reproduction in natural populations of T. varia.
Intron evolution in T. varia
Why do species of group 2b lack any group I introns but assume an intermediate position in all intraspecific phylogenies constructed for T. varia? Similar to the “intron early” and “intron late” scenarios proposed for red and brown algae (Bhattacharya et al. 2001), group 2b may represent either a lineage with empty insertion sites after precise intron loss or it has yet not encountered any intron-homing event. Changes in reproductive strategy (conversion from sexual to asexual reproduction) would fit with the “intron early” scenario: group 2a and 2b share an ancestor being sister to group 1, and both groups 1 and 2a still maintain HEGs and their host introns in various states. In this case, intron sequence variation between groups 1 and 2a should be interpreted as divergence after speciation. In non-heterothallic isolates, selfing (theoretically possible if the mating system collapses) or automixis can occur; such an asexual cycle was proposed by Clark and Haskins (2010). This should disfavor the preservation of HEGs and host introns, in contrast to outcrossing that increases frequency of HEGs in populations (Goddard et al. 2001). However, the observation of heterogeneous SSU sequences in three specimens of group 2b points toward outcrossing (intact sexual cycle controlled by mating types). The alternative scenario (intron late, no intron homing has yet occurred) is as well supported by S956 intron classification.
Our phylogeny of HE proteins known from myxomycetes (Fig. 5) displays two separate clades for S956 HEGs hosted in antisense strand of IC1 introns and HEGs hosted in sense strand of IE introns (the HEG at S956 from group 2a locates in the latter clade). HEGs in myxomycetes display a conserved His-Cys box, whereas free-standing HEGs (capable of moving without their host introns) from bacteria and phages show the motifs GIY-YIG or H-N-H (Hafez and Hausner 2012). We thus assume that myxomycete HEGs move together with their host introns, although exceptions were discussed (Haugen et al. 2005a, b). Joint evolution of HEGs and intronic hosts is as well suggested by the HEG phylogeny: S956 IE introns with HEG in sense strand of group 2a, and HEG-lacking S956 IC1 introns of group 1 were likely acquired from different sources, even if we did not found an example for an S956 HEG in group 1. Thus, the intron late scenario appears to be most likely: At least for insertion position 956, HEGs and host introns were not vertically inherited from the common ancestor of T. varia to groups 1 and 2a, but independently acquired by these groups after the divergence into groups 1, 2a, and 2b, and intron homing was not yet successful in group 2b.
HEG and intron transmission in T. varia
Group I introns in T. varia belong to three categories: introns with full-length HEGs (with putatively functional HEGs at positions 911, 956, and 1199, or with putatively non-functional HEGs at positions 516 and 1389), introns with partial HEGs (with putative HEG remnants at positions 516 and 956), and introns without HEGs (Fig. 3). For several organisms, coexisting introns of several categories have been found, while additional specimens lack introns (Goddard et al. 2006; Haugen et al. 2005a; Koufopanou et al. 2002; Nielsen and Johansen 2009), which corroborates the intron life cycle model (Goddard and Burt 1999; Gogarten and Hilario 2006). All five intron positions found to be occupied by HEGs in T. varia groups 1 and 2a would fit into this model.
Except for their HEG sequences in different stages of degeneration, all introns occurring in group 1 and 2a specimens can be unambiguously aligned for each group and insertion position, since only point mutations generate differences in intron sequences. This suggests that introns were acquired independently for each group and position from one common ancestor by lateral transfer from lineages outside the population of T. varia investigated herein.
If two amoebae mate, transmission of introns may be accomplished via sexual events by intron homing at the DNA level mediated by HE or at the RNA level through reverse splicing (Haugen et al. 2005a; Lambowitz and Belfort 1993). In accordance with published hypotheses about group I intron transmission routes (Bhattacharya et al. 1996; Hibbett 1996; Johansen et al. 1992; Milstein et al. 2008; Nikoh and Fukatsu 2001), HE-mediated intron homing via sexual events may have led to the current distribution of group I introns within natural populations for each of the groups 1 and 2a of T. varia. Identical or near-identical introns carrying HEGs at positions 516 and 1389 in multiple specimens of group 1 point toward HE-mediated transmission via sexual events.
Specimens of T. varia group 1 contain introns with HEGs at two positions, and for group 2a at four positions. Considering each position separately, the loss of HEG together with uniform deletion sites in the intron is unlikely to occur individually for each specimen. Most probably, HEGs were lost before introns spread vertically through inheritance within the population. Consequently, the transmission of introns was not mediated by HEGs. The same argument holds true for the lateral transfers of group I introns mediated by multiple reverse splicing instead of HEG within lineages in green algae and the fungal subphylum Pezizomycotina (Bhattacharya et al. 1996, 2005). Reverse splicing can as well be involved to explain sexually mediated transmission of introns to intron-less individuals of group 2a. However, compared to HE-mediated intron homing, reverse splicing was suggested to work less efficient (Bhattacharya et al. 2005; Haugen et al. 2005a). For introns without putatively functional HEGs of T. varia group 2a, both mutation and recombination might have led to the current distribution pattern of introns. Recombination as one of the mechanisms shaping the distribution of group I introns had been discussed for lichens (DePriest 1993). In group 2a, recombination is likely to contribute: Recombination signals were detected in introns S911 and S956, and intron sequences assume a near-random association among all five insertion positions. Introns might first have spread by vertical inheritance within individuals of group 2a; later mutations accumulated and created multiple intron genotypes at each position (Fig. 3). Subsequently, introns might be gained via reverse splicing or be lost in a precise way by resuming the original insertion recognition sequences through reverse transcription of spliced RNAs (Dujon 1989), and simultaneously, recombination events may have led to individuals with a different set of intron sequences across all five positions. The current distribution of HEG-lacking group I introns in T. varia group 2a might be a result of dynamic interactions involving mutation and recombination but involves less likely intron homing and loss through reverse splicing.
S956 introns in myxomycetes
A published phylogeny of group I introns sampled from various organisms (Haugen et al. 2005b) reveals that S956 introns in dark-spored myxomycete species belong to either the IC1 clade (Didymium iridis, Fuligo septica) or the IE clade (Didymium iridis, Diderma meyerae). S956 introns from two isolates of the myxomycete Didymium iridis (CR8-1 from Costa Rica and Pan2-16 from Panama) belong to classes IC1 and IE, respectively, but their SSU genes are identical in all exon parts. It was suggested that both introns have different evolutionary histories and were most likely acquired from different sources. Our phylogeny of all hitherto known S956 introns from myxomycetes (Fig. 4) demonstrates that distribution of IC1 and IE introns is not completely lineage-specific for both bright- and dark-spored myxomycetes, suggesting multiple acquisitions through lateral transmission during hybridization events in addition to vertical inheritance attributable to the spreading of IC1 and IE S956 introns, respectively. The HE protein phylogeny shown in Fig. 5 divided HEGs at S956 introns into two clades (inserted antisense in an IC1 intron, or sense in an IE intron). We found the division between HEGs with different reading directions (sense and antisense) to be deeper than the division between HEGs inserted into IC1 and IE introns, respectively. We thus can assume that two ancestral HEGs repeatedly and sporadically invaded different myxomycete lineages. Considering the more extensive sampling of HEG sequences done by us, the two HEGs reported for Didymium iridis, an antisense-IC1 in isolate CR8-1 and a sense-IE in isolate Pan2-16 (Haugen et al. 2005b), appear now to be distantly related.
Group I intron insertion recognition sequences in SSU are rather conserved across myxomycete lineages (see SSU alignments in Fiore-Donno et al. 2012, 2013), which may facilitate intron homing via lateral transmission between distantly related species, especially if given the tolerance of HE proteins against degeneracy within the recognition sequence (Hafez and Hausner 2012). In comparison to the relatively frequent lateral transmissions in S956 introns, as inferred from their assignment to classes IC1 and IE, introns at other positions might be as motile, but we lack a signature comparable to the IC1 and IE class assignment.
Intron homing events may result in the transfer of exon sections adjacent to the intron, as it was shown for the intron L1925 of Physarum polycephalum (Muscarella and Vogt 1989, 1993). In addition to possible homologous recombination in SSU during hybridization as inferred from the IC1/IE distribution in S956 introns of myxomycetes, the transfer of flanking exons by intron homing might be recorded as evolutionary history and introduce homoplasies into flanking exons that affect as well the resolution for SSU phylogenies of myxomycetes.
Conclusions
Perhaps for their lacking economic importance, but as well due to the high variation in usually conserved markers which is common for protists (Pawlowski et al. 2012), the molecular age for myxomycetes dawned comparatively late. This is the first population genetic study targeting a natural metapopulation of a myxomycete species with three independent markers and exploring group I intron distribution in SSU as a tool to elucidate speciation processes. For the investigated species T. varia, the most likely scenario is the existence of three reproductively isolated, yet predominantly or entirely sexual, biospecies. The comparatively low variation in EF1A sequences, and as well the absence of obvious morphological traits that distinguish these biospecies, point towards a recent speciation event. Patterns of intron distribution underline the reproductive isolation of these biospecies, and their homing endonuclease genes in various stages of decay point towards the existence of the Goddard-Burt cycle. As suggested by various authors (Hafez and Hausner 2012; Hausner et al. 2014), these genes may be universally distributed among eukaryotes.
References
Adl, S. M., Simpson, A. G., Lane, C. E., Lukeš, J., Bass, D., Bowser, S. S., et al. (2012). The revised classification of eukaryotes. Journal of Eukaryotic Microbiology, 59, 429–493.
Agapow, P. M., & Burt, A. (2001). Indices of multilocus linkage disequilibrium. Molecular Ecology Notes, 1, 101–102.
Aguilar, M., Fiore-Donno, A. M., Lado, C., & Cavalier-Smith, T. (2013). Using environmental niche models to test the ‘everything is everywhere’ hypothesis for Badhamia. The ISME Journal, 8, 737–745.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25, 3389–3402.
Betterley, D. A., & Collins, O. N. R. (1983). Reproductive systems, morphology, and genetical diversity in Didymium iridis (Myxomycetes). Mycologia, 75, 1044–1063.
Bhattacharya, D., Friedl, T., & Damberger, S. (1996). Nuclear-encoded rDNA group I introns: origin and phylogenetic relationships of insertion site lineages in the green algae. Molecular Biology and Evolution, 13, 978–989.
Bhattacharya, D., Cannone, J. J., & Gutell, R. R. (2001). Group I intron lateral transfer between red and brown algal ribosomal RNA. Current Genetics, 40, 82–90.
Bhattacharya, D., Reeb, V., Simon, D. M., & Lutzoni, F. (2005). Phylogenetic analyses suggest reverse splicing spread of group I introns in fungal ribosomal DNA. BMC Evolutionary Biology, 5, 68.
Brown, M. W., Silberman, J. D., & Spiegel, F. W. (2012). A contemporary evaluation of the acrasids (Acrasidae, Heterolobosea, Excavata). European Journal of Protistology, 48, 103–123.
Bruen, T. C., Philippe, H., & Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics, 172, 2665–2681.
Burke, J. M., Belfort, M., Cech, T. R., Davies, R. W., Schweyen, R. J., Shub, D. A., Szostak, J. W., & Tabak, H. F. (1987). Structural conventions for group I introns. Nucleic Acids Research, 15, 7217–7221.
Cannone, J. J., Subramanian, S., Schnare, M. N., Collett, J. R., D'Souza, L. M., Du, Y., et al. (2002). The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics, 3, 2.
Clark, J. (1993). Didymium iridis reproductive systems: additions and meiotic drive. Mycologia, 85, 764–768.
Clark, J. (1995). Myxomycete reproductive systems: additional information. Mycologia, 87, 779–786.
Clark, J. (2000). The species problem in the myxomycetes. Stapfia, 73, 39–53.
Clark, J., & Haskins, E. F. (2010). Reproductive systems in the myxomycetes: a review. Mycosphere, 1, 337–353.
Clark, J., & Haskins, E. F. (2011). Principles and protocols for genetical study of myxomycete reproductive systems and plasmodial coalescence. Mycosphere, 2, 487–496.
Clark, J., & Haskins, E. F. (2012). Plasmodial incompatibility in the myxomycetes: a review. Mycosphere, 3, 131–141.
Clark, J., & Haskins, E. F. (2013). The nuclear reproductive cycle in the myxomycetes: a review. Mycosphere, 4, 233–248.
Clark, J., Schnittler, M., & Stephenson, S. L. (2002). Biosystematics of the myxomycete Arcyria cinerea. Mycotaxon, 82, 343–346.
Collins, O. N. R. (1975). Mating types in five isolates of Physarum polycephalum. Mycologia, 67, 98–107.
Collins, O. N. R. (1979). Myxomycete biosystematics: some recent developments and future research opportunities. Botanical Review, 45, 145–201.
Collins, O. N. R. (1981). Myxomycete genetics, 1960–1981. Journal of the Elisha Mitchell Scientific Society, 97, 101–125.
Dalgleish, D. (2014). Contexture. Excel tips, tutorials, and videos. Excel latitude and longitude calculations. http://www.contextures.com/excellatitudelongitude.html. Accessed 2 Jan 2014.
DePriest, P. T. (1993). Small subunit rDNA variation in a population of lichen fungi due to optional group-I introns. Gene, 134, 67–74.
Dujon, B. (1989). Group I introns as mobile genetic elements: facts and mechanistic speculations—a review. Gene, 82, 91–114.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797.
Edgell, D. R., Chalamcharla, V. R., & Belfort, M. (2011). Learning to live together: mutualism between self-splicing introns and their hosts. BMC Biology, 9, 22.
Elde, M., Haugen, P., Willassen, N. P., & Johansen, S. (1999). I-NjaI, a nuclear intron-encoded homing endonuclease from Naegleria, generates a pentanucleotide 3' cleavage-overhang within a 19 base-pair partially symmetric DNA recognition site. European Journal of Biochemistry, 259, 281–288.
Fenchel, T., & Finlay, B. J. (2004). The ubiquity of small species: patterns of local and global diversity. BioScience, 54, 777–784.
Ferris, P. J., Vogt, V. M., & Truitt, C. L. (1983). Inheritance of extrachromosomal rDNA in Physarum polycephalum. Molecular and Cellular Biology, 3, 635–642.
Fiore-Donno, A. M., Berney, C., Pawlowski, J., & Baldauf, S. L. (2005). Higher-order phylogeny of plasmodial slime molds (Myxogastria) based on elongation factor 1-A and small subunit rRNA gene sequences. Journal of Eukaryotic Microbiology, 52, 201–210.
Fiore-Donno, A. M., Meyer, M., Baldauf, S. L., & Pawlowski, J. (2008). Evolution of dark-spored Myxomycetes (slime-molds): molecules versus morphology. Molecular Phylogenetics and Evolution, 46, 878–889.
Fiore-Donno, A. M., Nikolaev, S. I., Nelson, M., Pawlowski, J., Cavalier-Smith, T., & Baldauf, S. L. (2010). Deep phylogeny and evolution of slime moulds (Mycetozoa). Protist, 161, 55–70.
Fiore-Donno, A. M., Novozhilov, Y. K., Meyer, M., & Schnittler, M. (2011). Genetic structure of two protist species (Myxogastria, Amoebozoa) suggests asexual reproduction in sexual amoebae. PLoS ONE, 6, e22872.
Fiore-Donno, A. M., Kamono, A., Meyer, M., Schnittler, M., Fukui, M., & Cavalier-Smith, T. (2012). 18S rDNA phylogeny of Lamproderma and allied genera (Stemonitales, Myxomycetes, Amoebozoa). PLoS ONE, 7, e35359.
Fiore-Donno, A. M., Clissmann, F., Meyer, M., Schnittler, M., & Cavalier-Smith, T. (2013). Two-gene phylogeny of bright-spored Myxomycetes (slime moulds, superorder Lucisporidia). PLoS ONE, 8, e62586.
Flick, K. E., Jurica, M. S., Monnat, R. J., Jr., & Stoddard, B. L. (1998). DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature, 394, 96–101.
Flowers, J. M., Li, S. I., Stathos, A., Saxer, G., Ostrowski, E. A., Queller, D. C., et al. (2010). Variation, sex, and social cooperation: molecular population genetics of the social amoeba Dictyostelium discoideum. PLoS Genetics, 6, e1001013.
Frommlet, J. C., & Iglesias-Rodríguez, M. D. (2008). Microsatellite genotyping of single cells of the dinoflagellate species Lingulodinium polyedrum (Dinophyceae): a novel approach for marine microbial population genetic studies. Journal of Phycology, 44, 1116–1125.
Goddard, M. R., & Burt, A. (1999). Recurrent invasion and extinction of a selfish gene. Proceedings of the National Academy of Sciences of the United States of America, 96, 13880–13885.
Goddard, M. R., Greig, D., & Burt, A. (2001). Outcrossed sex allows a selfish gene to invade yeast populations. Proceedings of the Royal Society of London B, 268, 2537–2542.
Goddard, M. R., Leigh, J., Roger, A. J., & Pemberton, A. J. (2006). Invasion and persistence of a selfish gene in the Cnidaria. PLoS ONE, 1, e3.
Gogarten, J. P., & Hilario, E. (2006). Inteins, introns, and homing endonucleases: recent revelations about the life cycle of parasitic genetic elements. BMC Evolutionary Biology, 6, 94.
Goodman, E. M. (1980). Physarum polycephalum: a review of a model system using a structure-function approach. International Review of Cytology, 63, 1–58.
Gott, J. M., Visomirski, L. M., & Hunter, J. L. (1993). Substitutional and insertional RNA editing of the cytochrome c oxidase subunit 1 mRNA of Physarum polycephalum. The Journal of Biological Chemistry, 268, 25483–25486.
Gray, W. D., & Alexopoulos, C. J. (1968). Biology of the Myxomycetes. New York: The Ronald Press Co.
Guindon, S., & Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52, 696–704.
Hafez, M., & Hausner, G. (2012). Homing endonucleases: DNA scissors on a mission. Genome, 55, 553–569.
Haskins, E. F., & Wrigley de Basanta, D. (2008). Methods of agar culture of Myxomycetes: an overview. Revista Mexicana de Micologia, 27, 1–7.
Haubold, B., & Hudson, R. R. (2000). LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics, 16, 847–848.
Haugen, P., Huss, V. A. R., Nielsen, H., & Johansen, S. (1999). Complex group-I introns in nuclear SSU rDNA of red and green algae: evidence of homing-endonuclease pseudogenes in the Bangiophyceae. Current Genetics, 36, 345–353.
Haugen, P., De Jonckheere, J. F., & Johansen, S. (2002). Characterization of the self-splicing products of two complex Naegleria LSU rDNA group I introns containing homing endonuclease genes. European Journal of Biochemistry, 269, 1641–1649.
Haugen, P., Coucheron, D. H., Rønning, S. B., Haugli, K., & Johansen, S. (2003). The molecular evolution and structural organization of self-splicing group I introns at position 516 in nuclear SSU rDNA of myxomycetes. Journal of Eukaryotic Microbiology, 50, 283–292.
Haugen, P., Reeb, V., Lutzoni, F., & Bhattacharya, D. (2004). The evolution of homing endonuclease genes and group I introns in nuclear rDNA. Molecular Biology and Evolution, 21, 129–140.
Haugen, P., Simon, D. M., & Bhattacharya, D. (2005a). The natural history of group I introns. Trends in Genetics, 21, 111–119.
Haugen, P., Wikmark, O. G., Vader, A., Coucheron, D. H., Sjøttem, E., & Johansen, S. D. (2005b). The recent transfer of a homing endonuclease gene. Nucleic Acids Research, 33, 2734–2741.
Hausner, G., Hafez, M., & Edgell, D. R. (2014). Bacterial group I introns: mobile RNA catalysts. Mobile DNA, 5, 8.
Hedberg, A., & Johansen, S. D. (2013). Nuclear group I introns in self-splicing and beyond. Mobile DNA, 4, 17.
Hibbett, D. S. (1996). Phylogenetic evidence for horizontal transmission of group I introns in the nuclear ribosomal DNA of mushroom-forming fungi. Molecular Biology and Evolution, 13, 903–917.
Hood, G. M. (2010). PopTools version 3.2.5. http://www.poptools.org. Accessed 1 April 2014.
Horton, T. L., & Landweber, L. F. (2000). Evolution of four types of RNA editing in myxomycetes. RNA, 6, 1339–1346.
Huson, D. H., & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23, 254–267.
Johansen, S., Johansen, T., & Haugli, F. (1992). Structure and evolution of myxomycete nuclear group I introns: a model for horizontal transfer by intron homing. Current Genetics, 22, 297–304.
Johansen, S., Embley, T. M., & Willassen, N. P. (1993). A family of nuclear homing endonucleases. Nucleic Acids Research, 21, 4405.
Johansen, S., Elde, M., Vader, A., Haugen, P., Haugli, K., & Haugli, F. (1997). In vivo mobility of a group I twintron in nuclear ribosomal DNA of the myxomycete Didymium iridis. Molecular Microbiology, 24, 737–745.
Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780.
Koufopanou, V., Goddard, M. R., & Burt, A. (2002). Adaptation for horizontal transfer in a homing endonuclease. Molecular Biology and Evolution, 19, 239–246.
Lado, C. (2014). An on line nomenclatural information system of Eumycetozoa. 2005–2014. http://www.nomen.eumycetozoa.com. Accessed 8 April 2014.
Lahr, D. J. G., Grant, J., Nguyen, T., Lin, J. H., & Katz, L. A. (2011a). Comprehensive phylogenetic reconstruction of Amoebozoa based on concatenated analyses of SSU rDNA and actin genes. PLoS ONE, 6, e22780.
Lahr, D. J. G., Parfrey, L. W., Mitchell, E. A., Katz, L. A., & Lara, E. (2011b). The chastity of amoebae: re-evaluating evidence for sex in amoeboid organisms. Proceedings of the Royal Society of London B, 278, 2081–2090.
Lambowitz, A. M., & Belfort, M. (1993). Introns as mobile genetic elements. Annual Review of Biochemistry, 62, 587–622.
Lazo, W. R. (1961). Growth of green algae with myxomycete plasmodia. The American Midland Naturalist Journal, 65, 381–383.
Li, Z., & Zhang, Y. (2005). Predicting the secondary structures and tertiary interactions of 211 group I introns in IE subgroup. Nucleic Acids Research, 33, 2118–2128.
Lieb, B. (2014). PCR Additives. http://www.staff.uni-mainz.de/lieb/additiva.html. Accessed 6 March 2014.
Linne, C. (1792). Systema Naturae. Tom. II. Pars II. listed under the name Stemonitis. 1467–1470.
Lundblad, E. W., Einvik, C., Rønning, S., Haugli, K., & Johansen, S. (2004). Twelve group I introns in the same pre-rRNA transcript of the myxomycete Fuligo septica: RNA processing and evolution. Molecular Biology and Evolution, 21, 1283–1293.
Machouart, M., Lacroix, C., Bui, H., Feuilhade de Chauvin, M., Derouin, F., & Lorenzo, F. (2004). Polymorphisms and intronic structures in the 18S subunit ribosomal RNA gene of the fungi Scytalidium dimidiatum and Scytalidium hyalinum. Evidence of an IC1 intron with an His-Cys endonuclease gene. FEMS Microbiology Letters, 238, 455–467.
Marchler-Bauer, A., Zheng, C., Chitsaz, F., Derbyshire, M. K., Geer, L. Y., Geer, R. C., et al. (2013). CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Research, 41, D348–D352.
Martin, G. W., & Alexopoulos, C. J. (1969). The Myxomycetes. Iowa City: Iowa Univ. Press.
Michel, F., & Westhof, E. (1990). Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. Journal of Molecular Biology, 216, 585–610.
Milne, I., Lindner, D., Bayer, M., Husmeier, D., McGuire, G., Marshall, D. F., & Wright, F. (2009). TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics, 25, 126–127.
Milstein, D., Oliveira, M. C., Martins, F. M., & Matioli, S. R. (2008). Group I introns and associated homing endonuclease genes reveals a clinal structure for Porphyra spiralis var. amplifolia (Bangiales, Rhodophyta) along the eastern coast of South America. BMC Evolutionary Biology, 8, 308.
Moriyama, Y., & Kawano, S. (2010). Maternal inheritance of mitochondria: multipolarity, multiallelism and hierarchical transmission of mitochondrial DNA in the true slime mold Physarum polycephalum. Journal of Plant Research, 123, 139–148.
Müller, K. M., Cannone, J. J., Gutell, R. R., & Sheath, R. G. (2001). A structural and phylogenetic analysis of the group IC1 introns in the order Bangiales (Rhodophyta). Molecular Biology and Evolution, 18, 1654–1667.
Muscarella, D. E., & Vogt, V. M. (1989). A mobile group I intron in the nuclear rDNA of Physarum polycephalum. Cell, 56, 443–454.
Muscarella, D. E., & Vogt, V. M. (1993). A mobile group I intron from Physarum polycephalum can insert itself and induce point mutations in the nuclear ribosomal DNA of Saccharomyces cerevisiae. Molecular and Cellular Biology, 13, 1023–1033.
Muscarella, D. E., Ellison, E. L., Ruoff, B. M., & Vogt, V. M. (1990). Characterization of I-Ppo, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Molecular and Cellular Biology, 10, 3386–3396.
Nandipati, S. C., Haugli, K., Coucheron, D. H., Haskins, E. F., & Johansen, S. D. (2012). Polyphyletic origin of the genus Physarum (Physarales, Myxomycetes) revealed by nuclear rDNA mini-chromosome analysis and group I intron synapomorphy. BMC Evolutionary Biology, 12, 166.
Nannenga-Bremekamp, N. B. (1991). A guide to temperate Myxomycetes (Feest A, Burgraff E: De Nederlandse Myxomyceten, Engl. transl.). Bristol: Biopress Lim.
Neubert, H., Nowotny, W., & Baumann, K. (1993). Die Myxomyceten Deutschlands und des angrenzenden Alpenraumes unter besonderer Berücksichtigung Österreichs. Band 1. Ceratiomyxales, Echinosteliales, Liceales, Trichiales. Gomaringen: Baumann Verl.
Nielsen, H., & Johansen, S. D. (2009). Group I introns: moving in new directions. RNA Biology, 6, 375–383.
Nikoh, N., & Fukatsu, T. (2001). Evolutionary dynamics of multiple group I introns in nuclear ribosomal RNA genes of endoparasitic fungi of the genus Cordyceps. Molecular Biology and Evolution, 18, 1631–1642.
Novozhilov, Y. K., Okun, M. V., Erastova, D. A., Shchepin, O. N., Zemlyanskaya, I. V., García-Carvajal, E., & Schnittler, M. (2013a). Description, culture and phylogenetic position of a new xerotolerant species of Physarum. Mycologia, 105, 1535–1546.
Novozhilov, Y. K., Schnittler, M., Erastova, D. A., Okun, M. V., Schepin, O. N., & Heinrich, E. (2013b). Diversity of nivicolous myxomycetes of the Teberda State Biosphere Reserve (Northwestern Caucasus, Russia). Fungal Diversity, 59, 109–130.
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., Berney, C., et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the Animal, Plant, and Fungal kingdoms. PLoS Biology, 10, e1001419.
Persoon, C. H. (1794). Neuer Versuch einer systematischen Eintheilung der Schwämme. Neues Magazin für die Botanik in ihrem ganzen Umfange, 1, 63–128.
Poulain, M., Meyer, M., & Bozonnet, J. (2011). Les myxomycetes. Delémont: Féd. Mycol. Bot. Dauphiné-Savoie.
Rätzel, V., Ebeling, B., Hoffman, X. K., Tesmer, J., & Marwan, W. (2013). Physarum polycephalum mutants in the photocontrol of sporulation display altered patterns in the correlated expression of developmentally regulated genes. Development Growth and Differentiation, 55, 247–259.
Ronquist, F., & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542.
Sauer, H. W. (1982). Developmental Biology of Physarum. Cambridge: Cambridge University Press.
Schnittler, M., & Mitchell, D. W. (2000). Species diversity in Myxomycetes based on the morphological species concept—a critical examination. Stapfia, 73, 55–62.
Schnittler, M., & Tesmer, J. (2008). A habitat colonisation model for spore-dispersed organisms—does it work with eumycetozoans? Mycological Research, 112, 697–707.
Schnittler, M., Novozhilov, Y. K., Romeralo, M., Brown, M., & Spiegel, F. W. (2012). Myxomycetes and Myxomycete-like organisms. In F. W. Stuttgart (Ed.), Englers Syllabus of Plant Families. Volume 4. 13th edition (pp. 40–88). Bornträger.
Smirnov, A. V., Chao, E., Nassonova, E. S., & Cavalier-Smith, T. (2011). A revised classification of naked lobose amoebae (Amoebozoa: Lobosa). Protist, 162, 545–570.
STAR. (2014). StarORF. Software Tools for Academics and Researchers. http://star.mit.edu/orf. Accessed 25 March 2014.
Stephenson, S. L., Schnittler, M., & Novozhilov, Y. K. (2008). Myxomycete diversity and distribution from the fossil record to the present. Biodiversity and Conservation, 17, 285–301.
Tamura, K., Nei, M., & Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America, 101, 11030–11035.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Tanabe, Y., Yokota, A., & Sugiyama, J. (2002). Group I introns from Zygomycota: evolutionary implications for the fungal IC1 intron subgroup. Journal of Molecular Biology, 54, 692–702.
Torres-Machorro, A. L., Hernández, R., Cevallos, A. M., & López-Villaseñor, I. (2010). Ribosomal RNA genes in eukaryotic microorganisms: witnesses of phylogeny? FEMS Microbiology Reviews, 34, 59–86.
Traphagen, S. J., Dimarco, M. J., & Silliker, M. E. (2010). RNA editing of 10 Didymium iridis mitochondrial genes and comparison with the homologous genes in Physarum polycephalum. RNA, 16, 828–838.
Urich, T., Lanzén, A., Qi, J., Huson, D. H., Schleper, C., & Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE, 3, e2527.
Vader, A., Nielsen, H., & Johansen, S. (1999). In vivo expression of the nucleolar group I intron-encoded I-DirI homing endonuclease involves the removal of a spliceosomal intron. The EMBO Journal, 18, 1003–1013.
Walker, L. M., Dewsbury, D. R., Parks, S. S., Winsett, K. E., & Stephenson, S. L. (2011). The potential use of mitochondrial cytochrome c oxidase I for barcoding myxomycetes. In VII International Congress on Systematics and Ecology of Myxomycetes: 11–16 September 2011; Recife, Brazil (pp. 138).
Wikmark, O. G., Haugen, P., Haugli, K., & Johansen, S. D. (2007a). Obligatory group I introns with unusual features at positions 1949 and 2449 in nuclear LSU rDNA of Didymiaceae myxomycetes. Molecular Phylogenetics and Evolution, 43, 596–604.
Wikmark, O. G., Haugen, P., Lundblad, E. W., Haugli, K., & Johansen, S. D. (2007b). The molecular evolution and structural organization of group I introns at position 1389 in nuclear small subunit rDNA of myxomycetes. Journal of Eukaryotic Microbiology, 54, 49–56.
Xu, C., Wang, C., Sun, X., Zhang, R., Gleason, M. L., Eiji, T., & Sun, G. (2013). Multiple group I introns in the small-subunit rDNA of Botryosphaeria dothidea: implication for intraspecific genetic diversity. PLoS ONE, 8, e67808.
Yokoyama, E., Yamagishi, K., & Hara, A. (2002). Group-I intron containing a putative homing endonuclease gene in the small subunit ribosomal DNA of Beauveria bassiana IFO 31676. Molecular Biology and Evolution, 19, 2022–2025.
Zhou, Y., Lu, C., Wu, Q. J., Wang, Y., Sun, Z. T., Deng, J. C., & Zhang, Y. (2008). GISSD: group I intron sequence and structure database. Nucleic Acids Research, 36, D31–D37.
Acknowledgments
This research was supported by the Deutsche Forschungsgemeinschaft (DFG) to MS (SCHN 1080/2-1). The authors owe thanks for technical support to Anja Klahr, Greifswald. Fieldwork in the Bavarian Forest National Park was supported by Claus Bässler from the research unit of the park administration. We wish to thank Anna Maria Fiore-Donno for suggesting four primer sequences. For loans of specimens of T. varia, we are indebted to A.M. Fiore-Donno, Germany; Myriam de Haan, Belgium; Hans van Hooff, Netherlands; Yuri K. Novozhilov, Russia; Anna Ronikier, Poland; and Jos Van Roy, Belgium.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure S1
Position of Trichia varia in a Bayesian majority-rule consensus tree of the complete SSU gene for bright-spored myxomycetes with Ceratiomyxa fruticulosa as outgroup. Support values are shown for nodes with both Bayesian posterior probability >0.70 and bootstrap replicates >50. (PDF 343 kb)
Supplementary Figure S2
ML tree constructed from exon parts of complete SSU for 66 accessions of Trichia varia. Support values are shown for nodes with both Bayesian posterior probability >0.70 and bootstrap replicates >50. (PDF 269 kb)
Supplementary Table S1
Specimens used in this study and their assignment to genotypes with GenBank accession numbers. (DOC 563 kb)
Supplementary Table S2
Known SSU sequences of myxomycetes with group I intron S956. (DOC 92 kb)
Supplementary Database S1 (Microsoft Excel 2013)
Locality descriptions for all specimens investigated in this study. Geographic coordinates are given in the format dd°mm'ss.s", elevations in meter above sea level. (XLS 109 kb)
Rights and permissions
About this article

Cite this article
Feng, Y., Schnittler, M. Sex or no sex? Group I introns and independent marker genes reveal the existence of three sexual but reproductively isolated biospecies in Trichia varia (Myxomycetes). Org Divers Evol 15, 631–650 (2015). https://doi.org/10.1007/s13127-015-0230-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-015-0230-x