
Abstract
The epithelial-to-mesenchymal transition (EMT)-based tubulointerstitial fibrosis is the major pathological feature of diabetic kidney disease (DKD). While several studies have linked cell cycle dysregulation to various kidney injuries in recent years, its involvement in fibrosis of DKD is far from being clarified. ING2 (inhibitor of growth 2) is the second member of the inhibitor of growth family and participates in the regulation of many cellular processes. So far the role of ING2 in DKD remains largely unknown. In the present study, ING2 expression was detected by western blotting and immunofluorescent staining both in vitro high glucose-stimulated human proximal tubular epithelial cells (HK-2) and in vivo streptozotocin-induced diabetic mice. Cell proliferation was analyzed by CCK-8 and EdU assay, and cell cycle arrest was measured by flow cytometry. Quantitative polymerase chain reaction (qPCR) and western blotting were used to detect the EMT markers, and the p53 signaling activation was evaluated by chromatin immunoprecipitation (ChIP), qPCR, and western blotting. We found that the proliferation of the cells was reduced upon high glucose stimulation, which was accompanied by cell cycle arrest. The expression of ING2 was increased in hyperglycemia conditions both in vivo and in vitro. ING2 suppression ameliorated the reduced proliferation and cell cycle arrest induced by high glucose in HK-2 cells. Moreover, ING2 knockdown suppressed p21 expression by reducing p53 acetylation and finally alleviated the EMT progress in the high glucose-stimulated HK-2 cells. Our study demonstrated that cell cycle regulation is bound up with the kidney fibrosis in DKD, suggesting a novel function of ING2 as a potential therapeutic strategy targeting cell cycle arrest for DKD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic kidney disease (DKD) is one of the most common microvascular complications of diabetes and has been the leading cause of end-stage renal disease (ESRD). Besides the glomerular dysfunction, the epithelial-mesenchymal transition (EMT)-based tubulointerstitial fibrosis is the major pathological feature of DKD, which can be detected in the very early stage of DKD and contributes directly to the decline of renal function independent of glomerular dysfunction [5, 18, 32]. However, there is no effective clinical intervention to recover the tubulointerstitial fibrosis, and the molecular mechanism of this process as well as EMT of renal tubular epithelial cells is far from being clarified.
Cell cycle regulation has been linked to many cellular processes such as proliferation, apoptosis, and cell death in various cell types including renal cells such as podocytes and mesangial cells [34]. Cell cycle is controlled by cyclin/cyclin-dependent kinase (CDK) complexes, and the activity of the kinase is negatively regulated by CDK-inhibitors (CKI) including p21Cip1 and p27Kip1 [28, 29, 31]. Previous studies showed that the activation of PKC and JAK/STAT pathway and inhibition of AMPK were involved in the expression of CKIs in mesangial cells and induced glomerular hypertrophy in experimental diabetic nephropathy [1, 4, 12, 13, 33, 35]. However, there is so far no therapeutically effective treatment for the cell cycle arrest in DKD, and the mechanisms accounting for the perturbations of cell cycle progression induced by hyperglycemia in the kidney need to be fully clarified.
ING2 (inhibitor of growth 2) is the second member of the ING family, which is known as a candidate tumor suppressive protein. ING2 is also implicated in the regulation of diverse cellular processes including cell cycle, proliferation, senescence, and DNA damage repair [15, 41]. It is especially critical to control the G1 to S-phase transition. Besides, ING2 may be a key mediator of EMT-associated migration in breast cancer [6]. However, its role in the tubular injury and fibrosis process during DKD is still unknown.
In the present study, we aimed to investigate the role of ING2 in the process of high glucose-induced cell cycle arrest and EMT process and the possible mechanism underlying it. We showed that the expression of ING2 was upregulated under hyperglycemia conditions both in vitro and in vivo. And the knockdown of ING2 in proximal tubular epithelial cells abrogated the high glucose-induced G0/G1 arrest and increased cell proliferation, which was associated with an alleviated EMT progress in tubular cells. In mechanism, ING2 suppression reduced the expression of acetylated p53 and its target gene p21 in hyperglycemia condition, indicating the involvement of p53-p21 axis in the above process.
Materials and methods
Animal model
Male C57BL/6 mice, 6–8 weeks old, were purchased from the Shandong University Laboratory Animal Center. Mice were randomly separated into two groups with 12 mice in each group. To accelerate renal injuries with lower mortality and higher urine glucose excretion levels without inducing significant hypertension [37, 38], unilateral nephrectomy experiments were performed before diabetes induction by streptozotocin. Briefly, mice were anesthetized with pentobarbital (1 mg/kg body weight), and approximately 1-cm incision was made on the dorso-lumbar. Subsequently, the renal artery, the vein, and the ureter were ligated and severed to remove the kidney. One week after surgery, mice were injected low doses of streptozotocin (STZ, Solarbio, China; 60 mg/kg body weight, freshly dissolved in 0.05 mol/L sterile sodium citrate, pH 4.5) for five consecutive days, and mice with unilateral nephrectomy injected the same amount of sodium citrate were served as controls. Blood glucose levels were determined from the tail vein blood samples using blood glucose meter (ACCU-CHEK, Germany). The mice with glucose level over 16.7 mmol/L on the third consecutive day were considered diabetic. After 12 weeks, 8 diabetic mice along with 11 controls were survived. Blood and kidneys were collected and stored appropriately to carry on follow-up experiments. All experiments were approved by Institutional Animal Care and Use Committee of Shandong University.
Kidney function detection
Blood samples were centrifuged at 3000 rpm for 15 min at 4 °C. Serum creatinine and urea nitrogen were assessed by commercial kits (Nanjing Jiancheng Bioengineering Institute, CN) according to the manufacturer’s instructions.
Cell culture
The human proximal tubular epithelial cells (HK-2) were obtained from American Type Culture Collection (Rockville, MD) and were cultured as previously described [39].
ING2 knockdown by small RNA interference
Cells were seeded into 6-well plates and were transfected with either small interfering RNA targeting human ING2a isoform (5’-GCC GUG AUU UAU GUC ACA UTT-3’) or the scramble negative control (GenePharma, shanghai, China) with Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s instructions.
Cell viability
The CCK-8 assay (Dojindo, Kumamoto, Japan) was used to measure the cell viability. In brief, 10 μL CCK-8 solution was added to each well of 96-well plate, and cells were incubated for 2 h at 37 °C. Cell viability was measured at 450 nm.
Histological assessment
Formalin-fixed, paraffin-embedded kidney sections were dewaxed in xylene and were rehydrated in graded alcohols. The sections were stained with hematoxylin and eosin and then dehydrated and mounted for microscopic observation. For immunohistochemical staining, heat-activated antigen retrieval was then performed with sodium citrate buffer (0.01 M, pH 6.0). Endogenous peroxidase activity was suppressed by exposure to 3% hydrogen peroxide for 10 min; sections were incubated with primary antibody overnight at 4 °C after blocking with 5% FBS for 1 h at room temperature. Then followed by 2-step plus Poly-HRP anti-rabbit IgG Detection system (ZSGB-BIO) at 37 °C. Slides were visualized with DAB and were counter stained with hematoxylin for microscopic examination. The primary antibodies were used: rabbit anti-ING2 (Abcam, ab109504), rabbit anti-E-cadherin (CST, #3195s), rabbit anti-vimentin (CST, #5741s), rabbit anti-alpha-SMA (Abcam, ab66133), rabbit anti-p21 (Proteintech, 10355-1-AP), and rabbit anti-ki-67 (CST #12202).
Real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells using the TRIzol Reagent (Life technologies) according to manufacturer’s instructions. Purity and concentration of extracted RNA were measured by UV Spectrophotometer (Merinton SMA1000). cDNA was then synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo scientific) according to the manufacturer’s instructions. Real-time PCR was performed on the Applied Biosystems Real-Time PCR System (ABI, USA) with 2 × RealStar Green Power Mixture (GenStarBiosolutions). Relative expression levels of target mRNAs were normalized to that of the endogenous control (β-actin) and was calculated based on the 2-ΔΔCt comparative method. PCR for each sample was performed in triplicates. Primer sequences were as follows: ING2 Fwd, 5’-GCG AGA GCT GGA CAA CAAAT-3’, and Rev, 5’-GAC ACT TGG TTG CAT AAGCAG-3’; TGF-β1 Fwd, 5’-CTG CAA GTG GAC ATC AACGG-3’, and Rev, 5’-TCC GTG GAG CTG AAG CAATA-3’; CTGF Fwd, 5’-TGG AAG AGA ACA TTA AGA AGG GCA-3’, and Rev, 5’-TGC AGC CAG AAA GCT CAAAC-3’; p21 Fwd, 5’-AGT CAG TTC CTT GTG GAGCC-3’, and Rev, 5’-GCA TGG GTT CTG ACG GACAT-3’; vimentin Fwd, 5’-AGT CCA CTG AGT ACC GGA GAC-3’, and Rev, 5’-CAT TTC ACG CAT CTG GCG TTC-3’; α-SMA Fwd, 5’-GTG TTG CCC CTG AAG AGCAT-3’, and Rev 5’-GCT GGG ACA TTG AAA GTC TCA-3’; E-cadherin Fwd, 5’-CAT GAG TGT CCC CCG GTATC-3’, and Rev, 5’-CAG TAT CAG CCG CTT TCAGA-3’; and β-actin Fwd, 5’-GAA GTG TGA CGT GGA CATCC-3’, and Rev, 5’-CCG ATC CAC ACG GAG TACTT-3’.
Cell cycle analysis
A total of 4 × 105 cells were harvested and washed with PBS. Cells were fixed in 1 mL 70% ethanol and were incubated for at least 3 h at − 20 °C. After washing, cells were centrifuged at 300g for 5 min at RT. The supernatant was discarded, and cell pellets were resuspended in 200 μL Flow Cytometry Staining all-in-one Buffer. The mixed cells were incubated in the dark at room temperature for 30 min; the cell cycles were measured by flow cytometry (MuseTM, Merck Millipore).
Western blotting
Cell lysates were collected, and protein concentration was determined by BCA. Proteins were denatured for 10 min at 98 °C. After running on 10% SDS-PAGE, proteins were transferred onto PVDF membranes, which were blocked with 5% non-fat milk at room temperature (RT) for 1 h, and were incubated in primary antibodies at a proper dilution at 4 °C for overnight. After the final wash, membranes were incubated with horseradish peroxidase-conjugated secondary antibody at 1:5000 dilution for 1 h at RT. Membranes were developed using the Immobilon™ Western Chemiluminescent HRP Substrate (Millipore). The primary antibodies were used: rabbit anti-ING2 (Abcam, ab109504), rabbit anti-Vimentin (CST, #5741s), rabbit anti-α-SMA (Abcam, ab66133), rabbit anti-p21 (Proteintech, 10355-1-AP), rabbit anti-p53 (Proteintech, 10442-1-AP), and rabbit anti-Acetyl-p53 (CST, #2525 Lys382). Primary antibody against β-actin and horseradish peroxidase-conjugated secondary antibodies were from ZSGB-BIO.
Cell proliferation
HK-2 cells were seeded at a density of 3 × 104cells/well in a 24-well plate. The cell proliferation was detected by DNA incorporation using 5-ethynyl- 2’-deoxyuridine (EdU) kit (RiboBio, China) according to the manufacturer’s instructions. Images were acquired by immunofluorescence microscopy (Nikon Ti-S, Tokyo, Japan).
Chromatin immunoprecipitation
ChIP experiments were performed using the EZ-Magna ChIP kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. Immunoprecipitation analysis was carried out using anti-p53 antibody (Proteintech, 10442-1-AP). Real-time PCR was performed with primers (p21 forward, 5’-GTT CCC AGC ACT TCC TCTCC-3’; p21 reverse, 5’-GAA GCA GGC AGC ATA GGGAT-3’) that amplified the part of the human p21 promoter that contains a putative p53 binding site.
Statistical analysis
Data are presented as means ± SD and were analyzed using graph prism 6.0 software. Significance was assessed using unpaired Student’s t-test. Three independent experiments were performed and p < 0.05 was considered as significant.
Results
Reduced proliferation accompanied by cell cycle arrest in high glucose-treated proximal tubular epithelial cells
Human proximal tubular epithelial cells (HK-2) were treated with high glucose for up to 72 h. As shown in Fig. 1a, CCK-8 assay first revealed a reduced viability in high glucose-treated cells at 72 h. Then the cell proliferation was evaluated by EdU incorporation, and the EdU-positive cells were dramatically reduced by nearly 75% in high glucose-treated cells compared with low glucose control group (Fig. 1b-c). Further cell cycle was analyzed by flow cytometry. As shown in Fig. 1d, high glucose treatment arrested more cells in G0/G1 phase with the accumulation of approximate 66% cells in G0/G1, compared with 56% in the control group.

The decreased proliferation and cell cycle arrest in high glucose-stimulated HK-2 cells. a CCK-8 assay from day 0 to day 3 after HK-2 cells were incubated with medium containing 5 mM glucose (low glucose, LG) or 30 mM glucose (high glucose, HG). b Cell proliferation was measured by EdU corporation assay. Nuclei were counterstained with DAPI. Bar = 50 um. c Quantification of EdU-positive cells. Eight images were collected for each group, and percentages of EdU-positive cells were calculated. d Cell cycle was measured by FACS after the HK-2 cells were stimulated with 30 mM glucose for 48 h. Data were shown as mean ± SD, n = 3, *p < 0.05
Aggravated EMT along with increased ING2 expression under hyperglycemia conditions
ING2 expression was then evaluated both in vivo in the diabetic mice by immunohistochemistry and in vitro in high glucose-stimulated HK-2 cells by qPCR, western blotting, and immunofluorescent staining. The body weights and the elevation of blood glucose level, serum creatine, and urea nitrogen in diabetic mice were shown in Supplementary Fig. 1. As is shown in Fig. 2a, the positive ING2 staining was significantly enhanced in the nuclear of diabetic mice compared with normal controls. The qPCR showed an obvious increase of ING2 expression in HK-2 cells treated by high glucose for 48 h (Fig. 2c), which was also proved by western blotting (Fig. 2d) and immunostaining (Fig. 2e).

Increased ING2 expression along with aggravated EMT under hyperglycemia conditions. a Immunohistochemical staining for ING2, E-cadherin, vimentin, and α-SMA of kidneys from control mice (CTR) and diabetic mice (DM). Bar = 100 um. The expression of ING2 was measured by qPCR. b Hematoxylin-eosin staining for the kidney morphologies of control mice (CTR) and diabetic mice (DM). The arrows indicated the mesangial matrix thickening, glomerular hypertrophy, and renal tubular atrophy. Scale bars = 100 μm. The expression of ING2 was measured by qPCR (c), western blotting (d), and immunofluorescent staining (e) of in HK-2 cells cultured in medium with 5.5 mM glucose (LG) or 30 mM glucose (HG). Scale bars = 50 um. The expression of E-cadherin (f), vimentin (g), and α-SMA (h) were measured by qPCR after the HK-2 cells were stimulated with 30 mM glucose for 48 h
To investigate the changes of fibrotic related proteins in hyperglycemia conditions, the expression of E-cadherin, vimentin, and α-SMA were evaluated by immunohistochemistry on serial sections with ING2 staining. As shown in Fig. 2a, E-cadherin immunohistochemistry staining in diabetic mice was reduced compared with control mice, while vimentin and α-SMA were increased, with H/E staining on serial sections showing the kidney injuries in the diabetic mice (Fig. 2b). In mRNA level, the expression of E-cadherin was also downregulated (Fig. 2f), while the expression of vimentin (Fig. 2g) and α-SMA (Fig. 2h) were upregulated in high glucose-stimulated HK-2 cells.
ING2 suppression ameliorated the proliferation defect and cell cycle arrest induced by high glucose in HK-2 cells
To investigate the role of ING2 in the regulation of cell cycle arrest and fibrosis in hyperglycemia conditions more precisely, the expression of ING2 was suppressed in HK-2 cells by small interfering RNA; the knockdown efficiency was evaluated by both qPCR at mRNA level and western blotting and immunostaining at protein level. As shown in Fig. 3, the ING2 expression was knocked down by approximately 60% in mRNA level in ING2 knockdown group (ING2kd) compared with scramble siRNA-transfected control group (CTR) (Fig. 3a), while the protein level was downregulated by more than 60% (Fig. 3b–d).

ING2 knockdown ameliorated high glucose-induced proliferation defect and cell cycle arrest in HK-2 cells. ING2 was knocked down by siRNA transfection, and the efficiency was measured in mRNA level by qPCR (a) and protein level by immunofluorescent staining (b) and western blotting (c). d Relative density of C. Data was shown as mean ± SD (n = 3); *p < 0.05, **p < 0.01. Cells transfected with scramble control siRNA (CTR) or specific siRNA targeting ING2 (ING2kd) were cultured in medium with 5.5 mM (LG) or 30 mM glucose (HG). e Cell viability was measured by CCK-8 assay. f Cell proliferation was evaluated by EdU corporation. g Quantification of EdU-positive cells. Eight images were collected for each group, and percentages of EdU-positive cells were calculated. h Cell cycle analysis by FACS. Data were shown as mean ± SD, *p < 0.05, **p < 0.01
The cell proliferation and cell cycle were further investigated after ING2 was knocked down. Interestingly, as shown in Fig. 3e, the cell viability had no significant difference in the first 2 days either in high glucose-treated group or ING2 knockdown group compared with control group. However, the cell viability was clearly suppressed by high glucose at day 3, which was significantly ameliorated by additional ING2 downregulation. Consistently, the EdU incorporation assay revealed a dramatic reduction in cell proliferation after the cells were treated with high glucose for 72 h, which was restored by ING2 suppression (Fig. 3f–g).
Furthermore, the cell cycle was addressed by flow cytometry. As shown in Fig. 3h, the cell cycle arrest in G0/G1 phase by high glucose stimulation was attenuated by additional ING2 knockdown.
ING2 knockdown alleviated the high glucose-induced profibrogenic cytokines and EMT markers expression in HK-2 cells
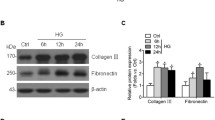
To investigate the role of ING2 in the process of kidney fibrosis, the expression of profibrogenic cytokines and EMT-related markers were detected after ING2 was knocked down under hyperglycemia conditions. As shown in Fig. 4a–b, the mRNA levels of transforming growth factor-β1 (TGF-β1) and connective tissue growth factor (CTGF) were upregulated after treated with high glucose for 72 h and were downregulated back to the level of control group after ING2 was knocked down compared with scramble siRNA-transfected cells. Correspondingly, the expression of EMT markers vimentin and α-SMA was evaluated at protein level by western blotting. The expression of vimentin and α-SMA were upregulated upon high glucose stimulation and were downregulated after ING2 was knocked down by siRNA transfection (Fig. 4c–e).

ING2 knockdown alleviated the high glucose-induced profibrogenic cytokines and EMT marker expression in HK-2 cells. Cells were transfected with scramble siRNA (CTR) or specific siRNA targeting ING2 (ING2kd) and were cultured in medium with 5.5 mM (LG) or 30 mM glucose (HG) for 72 h. a–b mRNA levels of TGF-β1 and CTGF were evaluated by qPCR. c Vimentin and α-SMA expression were analyzed by western blotting. d–e Relative density of vimentin and α-SMA. Data were shown as mean ± SD, *p < 0.05, **p < 0.01
ING2 knockdown suppressed the high glucose activated p53-p21 axis in HK-2 cells by regulating p53 acetylation
Based on previous researches, ING2 expression was related with p21 in a p53 dependent or independent way. To evaluate the mechanism in greater details, the p53-p21 axis was investigated in our following experiments. First, p21 expression was evaluated in diabetic mice by immunohistochemistry, and the positive p21 staining was significantly enhanced in diabetic mice compared with normal control (Fig. 5a). We next determined whether high glucose stimulation impaired the ability of p53 to interact with the promoter of p21 by ChIP assay. Indeed, the p53 binding to the p21 promoter was enhanced under high glucose condition (Fig. 5b). The mRNA level of p21 was further evaluated by qPCR and is shown in Fig. 5c, and the high glucose-induced expression of p21 was reduced after ING2 was knocked down, indicating that ING2 plays a role in regulating the activity of p53. While the expression of total p53 was quite constant after high glucose stimulation and ING2 siRNA transfection, the p21 and acetylated p53 were markedly enhanced upon high glucose stimulation, which was totally decreased by downregulating ING2, revealing that ING2 may regulate the activity of p53 through its acetylation (Fig. 5d–h).

ING2 knockdown suppressed the high glucose activated p53-p21 axis in HK-2 cells by regulating p53 acetylation. a Immunohistochemical staining for p21 in kidneys from control mice (CTR) and diabetic mice (DM). b Binding of p53 to the p21-promoter analyzed by ChIP assays in HK-2 cells under high glucose condition. Normal IgG served as control. c p21 mRNA level was evaluated by qPCR. Cells were transfected with scramble siRNA (CTR) or specific siRNA targeting ING2 (ING2kd) and were cultured in medium with 5.5 mM (LG) or 30 mM glucose (HG). d ING2, p53, p21, and acetylated p53 expression was evaluated by western blotting. e–h Relative density of ING2, p53, p21, and ac-p53. Data were shown as mean ± SD, *p < 0.05, **p < 0.01
Discussion
Inhibitor of growth 2 (ING2) is known as a candidate tumor suppressor gene [14] and has been implicated in the cell proliferation and death of many cell types in recent years [27]. However, the role of ING2 in the kidney diseases remains largely unknown. Here, we first reported that the expression of ING2 was enhanced both in high glucose-stimulated renal tubular epithelial cells and in the kidney of diabetic mice (Fig. 2). We observed that long time exposure to high glucose leads to proliferation suppression in HK-2 cells, which is probably due to the cell cycle arrest at G1 phase, and the function of ING2 in this process was further investigated by small RNA interference in greater details. Interestingly, knocking down of ING2 rescued the high glucose-induced proliferation defect in HK-2 cells probably by alleviating the high glucose-induced cell cycle arrest as shown in Fig. 3. It has been reported previously that the inhibition of ING2 expression accelerated the progression of cells from G1 to S phase in U2OS osteosarcoma cells [15]. This is consistent with our results that ING2 suppression could ameliorate the high glucose-induced cell growth defect by permitting transition from G1 to S phase.
More interestingly, we identified a new potential function for ING2 in the renal tubular fibrosis process. Renal tubulointerstitial fibrosis has been established as the hallmark and major pathological feature of most progressive chronic kidney disease and ESRD [30]. Though it has been controversial if EMT program does exist in the renal fibrosis progress in vivo, more and more experiments support that EMT of TECs is central to the kidney tubulointerstitial fibrosis [7, 16, 24]. TGF-β1 has been well recognized as a multi-functional cytokine that can drive fibrosis [26, 36]. And CTGF is a matricellular protein that can exacerbate extracellular matrix production and cooperate with TGF-β1 to induce sustained fibrosis in the kidney [11, 21, 25]. As shown in Fig. 4, we could show that suppression of ING2 alleviated the EMT progress by reducing the expression of profibrogenic cytokines TGF-β1 and CTGF.
The correlation of cell cycle arrest and the development of kidney fibrosis have been established in AKI models by Yang et al. in 2010, in which they have demonstrated that the arrest of proximal tubule epithelial cells in G2/M causes a profibrotic phenotype [40]. In the present study, the rescued cell cycle arrest in G1 phase by ING2 knockdown ameliorated the EMT induced by high glucose in TECs. We did not detect any significant alteration in G2/M phase in our experiment, which could be due to the difference between AKI and diabetic models. And more strikingly, we provided evidence showing the correlation of cell cycle arrest with kidney fibrosis process in chronic kidney diseases. Furthermore, our study indicates that ING2 might serve as a potential new target for the prevention and treatment of DKD by regulating cell cycle. The role of ING2 in the kidney fibrosis in vivo needs to be addressed in the future with ING2 deficient mice.
In mechanism, we further showed that the acetylation of p53 on lysine 382 in HK-2 cells was increased under high glucose stimulation, which was decreased by ING2 knockdown (Fig. 5), while the total p53 expression was constant. It has been previously reported that p33ING1b (an alternative splice variant of ING1) could inhibit the deacetylation of p53 through binding to SIRT1 and resulting in an increase of acetylated p53 [10]. On the other hand, ING2 could enhance the interaction between p53 and p300 and acted as a cofactor for p300-mediated p53 acetylation during the onset of replicative senescence [22]. The increase of p53 acetylation on lysines 382 has been related to an augmentation of site-specific DNA binding of p53 and enhanced activity [17, 23]. Indeed, we further found that the p53 target gene p21 was transcriptionally activated in high glucose-treated HK-2 cells, which was decreased by ING2 knockdown (Fig. 5). The increased expression of p21 has been identified both in acute kidney injury (AKI) models [20] and various experimental models of diabetic nephropathy [3, 8, 12] in previous studies. However, the role of p21 in these two series of models was contradictory. In AKI models, p21-deficient mice develop more severe damage in comparison with wild-type animals, and p21 induction ameliorates AKI [19], while in diabetic nephropathy models, p21-deficient mice are protected from progression of diabetic nephropathy and glomerular hypertrophy [2, 9]. In our present study, p21 was upregulated in high glucose-stimulated renal tubular epithelial cells and downregulated by ING2 knockdown, which may play a vital role in the process of ING2 protecting the TECs from cell cycle arrest and EMT.
Collectively, our study demonstrates that the cell cycle regulation is bound up with the kidney fibrosis in DKD and ING2 may serve as a potential therapeutic strategy targeting cell cycle arrest for DKD.
Change history
19 June 2020
The above article was published online first with the following errors which have now been corrected.
References
Al-Douahji M, Brugarolas J, Brown PA, Stehman-Breen CO, Alpers CE et al (1999) The cyclin kinase inhibitor p21 (WAF1/CIP1) is required for glomerular hypertrophy in experimental diabetic nephropathy. Kidney Int 56:1691–1699
Awazu M, Omori S, Ishikura K, Hida M, Fujita H (2003) The lack of cyclin kinase inhibitor p27(Kip1) ameliorates progression of diabetic nephropathy. J Am Soc Nephrol 14:699–708
Baba M, Wada J, Eguchi J, Hashimoto I, Okada T, Yasuhara A, Shikata K, Kanwar YS, Makino H (2005) Galectin-9 inhibits glomerular hypertrophy in db/db diabetic mice via cell-cycle-dependent mechanisms. J Am Soc Nephrol 16:3222–3234
Bonventre JV (2012) Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol 32:452–462
Chevalier RL (2016) The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311(1):F145–F161
Gao Y, Ma H, Gao C, Lv Y, Chen X, Xu R, Sun M, Liu X, Lu X, Pei X, Li P (2018) Tumor-promoting properties of miR-8084 in breast cancer through enhancing proliferation, suppressing apoptosis and inducing epithelial-mesenchymal transition. J Transl Med 16:38
Hills CE, Squires PE (2010) TGF-beta 1 induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol 31:68–74
Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto KI, Nagata M (2002) Podocyte injury promotes progressive nephropathy in Zucker diabetic fatty rats. Lab Investig 82:25–35
JuditMegyesi, Adel Tarcsafalvi, Shenyang Li, Rawad Hodeify, Nang San Hti Lar Seng et al (2015) Increased expression of p21WAF1/CIP1 in kidney proximal tubules mediates fibrosis. Am J Physiol Renal Physiol 308(2):F122–F130
Kataoka H, Bonnefin P, Vieyra D, Feng X, Hara Y, Miura Y, Joh T, Nakabayashi H, Vaziri H, Harris CC, Riabowol K (2003) ING1 represses transcription by direct DNA binding and through effects on p53. Cancer Res 63:5785–5792
Kok HM, Falke LL, Goldschmeding R, Nguyen TQ (2014) Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. Nat Rev Nephrol 10:700–711
Kuan CJ, Al-Douahji M, Shankland SJ (1998) The cyclin kinase inhibitor p21 WAF1/Cip1 is increased in experimental diabetic nephropathy: potential role in glomerular hypertrophy. J Am Soc Nephrol 9:986–993
Lan R, Geng H, Polichnowski A, Singha P, Saikumar P, McEwen D, Griffin K, Koesters R, Weinberg J, Bidani A, Kriz W, Venkatachalam K (2012) PTEN loss defines a TGF-β induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol 302:F1210–F1223
Larrieu D, Ythier D, Binet R, Brambilla C, Brambilla E, Sengupta S, Pedeux R (2009) ING2 controls the progression of DNA replication forks to maintain genome stability. EMBO Rep 10:1168–1174
Larrieu D, Ythier D, Brambilla C, Pedeux R (2010) ING2 controls the G1 to S-phase transition by regulating p21 expression. Cell Cycle 9:3984–3990
Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K et al (2015) Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21:998–1009
Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A 101:2259–2264
Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC (1984) Structural-functional relationships in diabetic nephropathy. J Clin Invest 74:1143–1155
Megyesi J, Safirstein RL, Price PM (1998) Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest 101:777–782
Megyesi J, Udvarhelyi N, Safirstein RL, Price PM (1996) The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am J Phys 271:1211–1216
Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K (1999) Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: a mouse fibrosis model. J Cell Physiol 181:153–159
Pedeux R, Sengupta S, Shen JC, Demidov ON, Saito S et al (2005) ING2 regulates the onset of replicative senescence by induction of ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol Cell Biol 25:6639–6648
Prives C, Manley JL (2001) Why is p53 acetylated? Cell 107:815–818
Rastaldi MP (2006) Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J Nephrol 19:407–412
Rayego-Mateos S, Morgado-Pascual JL, Rodrigues-Diez RR, Rodrigues-Diez R, Falke LL, Mezzano S, Ortiz A, Egido J, Goldschmeding R, Ruiz-Ortega M (2018) Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J Pathol 244(2):227–241
Salazar KD, Lankford SM, Brody AR (2009) Mesenchymal stem cells produce Wnt isoforms and TGF-beta1 that mediate proliferation and procollagen expression by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 297(5):L1002–L1011
Sarker KP, Kataoka H, Chan A, Netherton SJ, Pot I, Huynh MA, Feng X, Bonni A, Riabowol K, Bonni S (2008) ING2 as a novel mediator of transforming growth factor-beta-dependent responses in epithelial cells. J Biol Chem 283:13269–13279
Shankland SJ, Al'Douahji M (1999) Cell cycle regulatory proteins in glomerular disease. Exp Nephrol 7:207–211
Shankland SJ, Wolf G (2000) Cell cycle regulatory proteins in renal disease: role in hypertrophy, proliferation, and apoptosis. Am J Physiol Renal Physiol 278:515–529
Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin FI (1994) Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes 43:1046–1051
Terada Y, Inoshita S, Nakashima O, Kuwahara M, Sasaki S et al (1998) Cyclins and the cyclin-kinase system--their potential roles in nephrology. Nephrol Dial Transplant 13:1913–1916
Thomas MC, Burns WC, Cooper ME (2005) Tubular changes in early diabetic nephropathy. Adv Chronic Kidney Dis 12:177–186
Vallon V, Thomson SC (2012) Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol 74:351–375
Wolf G (2000) Cell cycle regulation in diabetic nephropathy. Kidney Int Suppl 77:59–66
Wolf G, Schroeder R, Ziyadeh FN, Thaiss F, Zahner G et al (1997) High glucose stimulates expression of p27 Kip1 in cultured mouse mesangial cells: relationship to hypertrophy. Am J Phys 273:348–356
Wu CF, Chiang WC, Lai CF, Chang FC, Chen YT, Chou YH, Wu TH, Linn GR, Ling H, Wu KD, Tsai TJ, Chen YM, Duffield JS, Lin SL (2013) Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol 182:118–131
ParkM HH, Nam S, Kim M, Kang M, Lee G, Han J, Woo M (2015) A novel mouse model of diabetes mellitus using unilateral nephrectomy. Lab Anim 50:88–93
Tesch H, Allen J (2007) Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology (Carlton) 12:261–266
Xin W, Zhao X, Liu L, Xu Y, Li Z, Chen L, Wang X, Yi F, Wan Q (2015) Acetyl-CoA carboxylase 2 suppression rescues human proximal tubular cells from palmitic acid induced lipotoxicity via autophagy. Biochem Biophys Res Commun 463:364–369
Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV (2010) Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16:535–543
Zhong J, Yang L, Liu N, Zheng J, Lin CY (2003) Knockdown of inhibitor of growth protein 2 inhibits cell invasion and enhances chemosensitivity to 5-fu in human gastric cancer cells. Dig Dis Sci 58:3189–3197
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 81970427; grant no. 31600699; grant no. 81770729; grant no. 91749111), Science and Technology Development Project of Shandong Province (2016GSF201023), and Shandong Province Taishan Scholar Project (grant no. tsqn 20161073).
Author information
Authors and Affiliations
Contributions
Conceptualization, W.X.; data acquisition, Y.M. R.Y. and Q.W.; methodology, W.X., Y.M., R.Y., Y.Y., and B.L; investigation, W.X., Y.M., R.Y., and Q.W.; resources, Y.M. and R.Y.; data curation, Y.M., R.Y., and B.L.; writing, review, and editing, W.X., Y.M., R.Y., and T.L.; visualization, W.X. and Q.W.; supervision, W.X.; project administration and funding, Q.W. and W.X.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All experiments were approved by Institutional Animal Care and Use Committee of Shandong University.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
•ING2 was increased both in vivo and in vitro under hyperglycemia conditions.
•ING2 knockdown ameliorated the reduced proliferation and cell cycle arrest in the high glucose-stimulated HK-2 cells.
•ING2 knockdown alleviated the EMT progress in the high glucose-stimulated HK-2 cells.
•ING2 knockdown suppressed p21 expression by reducing p53 acetylation.
Electronic supplementary material
ESM 1
(PDF 156 kb)
Rights and permissions
About this article

Cite this article
Ma, Y., Yan, R., Wan, Q. et al. Inhibitor of growth 2 regulates the high glucose-induced cell cycle arrest and epithelial-to-mesenchymal transition in renal proximal tubular cells. J Physiol Biochem 76, 373–382 (2020). https://doi.org/10.1007/s13105-020-00743-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-020-00743-3