
Abstract
Dyslipidemia is a common risk factor of chronic kidney disease (CKD). Current notion suggests that insufficient intracellular fatty acid oxidation (FAO) and subsequently enhanced fatty acid esterification within renal resident cells, a process termed as renal lipotoxicity, is the key pathogenic event responsible for dyslipidemia-induced kidney injury. However, the detailed mechanism is not fully elucidated. Recently, accumulating data indicated that acetylation modification is an important regulating manner for both mitochondrial function and energy metabolism, while whether acetylation modification is involved in renal lipotoxicity is of little known. In the present study, the expression level of global lysine acetylation was detected by immunohistochemistry in high-fat diet mice and western blot in palmitic acid (PA) stimulated HK-2 cells. The acetylation levels of long-chain acyl-CoA dehydrogenases (LCAD) and β-hydroxyacyl-CoA dehydrogenase (β-HAD) were measured by immunoprecipitation. And a multifunction microplate reader was applied to detect FAO rate, triglyceride and acyl-CoA contents, and the enzyme activities, with cellular lipid accumulation identified by Oil Red O staining. We evidenced the acetylation levels of LCAD and β-HAD that were enhanced, which led to decreased enzymatic activities and impaired FAO rate. Furthermore, renal protein hyperacetylation induced by lipid overload was associated with increased expression of GCN5L1. And the silence of GCN5L1 in tubular epithelial cells resulted in deacetylation and activation of LCAD and β-HAD. Finally, excess lipids induced lipotoxicity and epithelial-mesenchymal transition (EMT) were ameliorated by GCN5L1 suppression, suggesting GCN5L1-mediated mitochondrial LCAD and β-HAD acetylation might be a key pathogenic event underlying excess lipids induced FAO impairment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Keypoints
-
1.
GCN5L1 is increased both in vivo and in vitro under excess lipids conditions.
-
2.
The acetylation of LCAD and β-HAD are enhanced, along with impaired FAO rate.
-
3.
GCN5L1 promotes the acetylation but decreased the activities of LCAD and β-HAD.
-
4.
GCN5L1 increases lipotoxicity and EMT by promoting LCAD and β-HAD acetylation.
Introduction
As the common clinic complication of CKD such as nephrotic syndrome and diabetic nephropathy, dyslipidemia also act as a pathogenic factor in propelling progressive kidney injury [6]. Current notion suggest that long-term systemic lipid metabolism disturbance might induce the ectopic accumulation of intracellular lipid within kidney resident cells, accordingly impairing their morphology and function and ultimately leading to kidney fibrosis, a process termed as lipotoxicity [17, 25]. During this pathogenesis, insufficient mitochondrial FAO was proposed as the key event. Ample studies, as well as our previous, have clearly demonstrated that inhibiting mitochondrial FAO could induce morphological and functional injuries on several types of kidney resident cells, while enhancing FAO has protective effects [8, 12, 24]. However, the detailed mechanisms responsible for dyslipidemia induced impairment of FAO are not fully understood, and efficient intervening method aiming at renal lipotoxicity is lacking yet.
In recent years, acetylation modification has emerged as a crucial regulating manner for mitochondrial function [8, 26]. Over 60% of mitochondrial proteins have been demonstrated to be acetylated, including those involved in energy metabolism and mitochondrial turnover [7, 16]. Furthermore, acetylation has also been proposed as one major adapting mechanism for mitochondria in sensing nutrients challenges, as nutrients oversupply leads to mitochondrial hyperacetylation, while insufficient nutrient availability results in decreased mitochondrial acetylation level [13, 18]. To some extent, renal lipotoxicity is also a status of nutrient oversupply, while the role of mitochondrial acetylation in the pathogenesis of renal lipotoxicity and potential interventional target need to be elucidated.
GCN5L1 has recently been identified as an essential component of the mitochondrial acetyltransferase machinery, which has been shown to counter the activity of the mitochondrial deacetylase protein SIRT3 [20]. In the current study, we aimed to explore the impacts of GCN5L1 on lipid nephrotoxicity by regulating acetylation of FAO enzymes. Our research found that GCN5L1 could attenuate FAO by acetylating the key enzymes of FAO under high-fat conditions in tubular cells, thus may not only shed new light on the understanding for CKD caused by over nutrients but also hint GCN5L1 as a key intervention targets in CKD.
Materials and methods
Cell culture and treatment
Human renal tubular epithelial cells (HK-2) were purchased from American Type Culture Collection (Rockville, MD, USA) and cultured in low glucose DMEM medium (Gibco, USA) with 10% fetal bovine serum (Gibco, USA) in a humidified atmosphere of 5% CO2 at 37 °C. Two groups of culture conditions were investigated, which were treated with 200 μM PA (Sigma, coupled to BSA) for 72 h and BSA (Solarbio, Beijing) alone.
Animals
Male C57BL/6 mice, 8 weeks old, were obtained from the Experimental Animal Center of Shandong University. The mice were randomized divided into two groups (n = 8 per group) received either normal chow diet (D12450B, 20% protein, 70% carbohydrate, 10% fat, Research Diets, New Brunswick, NJ, USA) or high-fat diet (D12492, 20% protein, 20% carbohydrate, 60% fat, Research Diets, New Brunswick, NJ, USA). All animal experiments were carried out strictly according to the guidelines of the Animal Center of Shandong University, and the experimental procedures were approved by the Ethical Committee of Shandong Provincial Hospital Affiliated to Shandong University (No. S077).
General condition
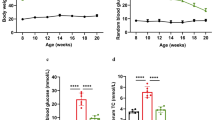
After 18 weeks, all the mice were sacrificed, and the body weights were measured at the beginning and end of the experiment. Kidney tissues were quickly dissected and blood samples were collected from the inferior vena cava and centrifuged at 3,000×g for 5 min at 4 °C to collect serum. Triglyceride and cholesterol levels were determined by ROCHE module COBAS 8000 analyzer (Roche Group, Switzerland).
Western blot analysis
Cell lysates were extracted with RIPA buffer containing protease and phosphatase inhibitor cocktail. Equal amounts of proteins were separated in a 10% Tris-Tricine SDS gel and then transferred into a 0.45 μm PVDF membrane (Millipore, USA). After incubated for 1 h with 5% non-fat milk, the membranes were incubated overnight at 4 °C with primary antibodies. The following primary antibodies were used: rabbit acetylated-lysine antibody (CST, 9441), rabbit ACADL-specific antibody (Proteintech, 17526-1-AP), rabbit HADHB antibody (Sigma, SAB2108608), rabbit COXIV antibody (Proteintech, 11242-1-AP), rabbit GCN5L1 antibody (Proteintech, 19687-1-AP). Then, all membranes were incubated with corresponding secondary antibodies. Finally, protein bands were detected by enhanced chemiluminescence (Millipore, USA).
Immunohistochemistry
Sections of freshly excised kidney tissue were fixed in 4% neutral buffered formalin, embedded in paraffin, and sectioned at 5-μm intervals. To reveal antigens, sections were then placed in sodium citrate buffer and heated in a pressure cooker for 3 min. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 10 min at room temperature. Sections were incubated with Acetylated-Lysine Antibody (rabbit pAb, 1:200, CST,) and GCN5L1 Antibody (rabbit pAb, 1:200, Proteintech) diluted in PBS at 4 °C overnight and then with the HRP-conjugated secondary antibody at 37 °C for 30 min. Slides were visualized by using 3,3′-diaminobezidine, counterstained with light hematoxylin, dehydrated, and coverslipped.
Immunoprecipitation assay
Immunoprecipitation assay was conducted using a Crosslink IP kit (Thermo Scientific Pierce, 26147). In brief, 10 μg acetyl-lysine antibodies were immobilized with 20 μl coupling resin slurry. A total of 250 ug of lysates was precleared with 20 μl of control agarose resin slurry. The precleared lysates were incubated with antibody cross-linked resin on a rotator overnight at 4 °C, then resin was washed, and the protein complexes bound to the antibody were eluted with 50 μl of elution buffer. The immune complexes were then subjected to immunoblot analysis as described above. As a negative control, lysates were immunoprecipitated with normal rabbit IgG coupled to agarose A/G beads. The extent of acetylation protein was quantitated by calculating the ratio of acetylated protein/total protein band intensities.
Quantitative real-time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen, USA) and reversed transcribed into cDNA using reverse transcriptase kits (Takara, RR047A). Then, real-time PCR was carried out by using Ultra SYBR Mixture kit (Cwbio, CW2601M) according to the manufacturer’s instructions. Sequence-specific primers used were as follow: GCN5L1 Fwd: 5′-TGG GGA TGT GGA GAA CTGG-3′, Rev: 5′-AGG GAA CAG GGG CTA GGAAG-3′, human β-actin Fwd: 5′-GAA GTG TGA CGT GGA CATCC-3′, Rev: 5′-CCG ATC CAC ACG GAG TACTT-3′.
Fatty acid β-oxidation rate assay
Cells were cultured in the 100 mm Petri dish and were stimulated as requested. Mitochondria isolation kit (Abcam, ab110170) was used to isolate mitochondria from cultured HK-2 cells. Then the fatty acid β-oxidation rate was measured by using mitochondria according to the Fatty Acid β-Oxidation Kit (Genmed Scientifics, USA).
LCAD and β-HAD activity assay
HK-2 cells were cultured in 100 mm Petri dishes and collected with trypsin. LCAD and β-HAD were extracted according to the long-chain acyl CoA dehydrogenase Kit (Genmed Scientifics, USA) and beta-hydroxyzine CoA dehydrogenase Kit (Genmed Scientifics, USA). Then the activity of LCAD and β-HAD was determined by spectrophotometry at 340 nm and 600 wavelength for 5 min.
Measurement of acetyl-CoA and triglyceride content
HK-2 cells were cultured in six-well plates and were stimulated as requested. Acetyl-CoA content was tested according to the manufacturer’s protocols (Sigma, MAK039). Triglyceride content was extracted according to the Tissue triglyceride assay kit (Pplygen company of Beijing). The concentration of triglyceride was measured by SpectraMax M2 microplate reader and normalized by protein concentration per mg.
Oil-Red O staining
HK-2 cells were grown on a cover slide in 24-well plates then treated with PA or BSA and transfected with GCN5L1 siRNA (KD) or scramble control siRNA (CTR). After fixed in 4% paraformaldehyde for 10 min, cells were washed thoroughly with distilled water. Then, cells were incubated with a working solution of Oil-Red O (Solarbio, Beijing) for 10 min at 37 °C. To remove the background staining, cells were incubated in 60% isopropanol for 5 s. Finally, sections were counterstained with hematoxylin and fixed in glycerol jelly-mounting medium.
Statistical analysis
Results are presented as means ± SD unless stated otherwise. The statistical analysis was carried out using SPSS 17.0 software. Graphs were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA). Data were analyzed using one-way ANOVA or Student’s t test. P < 0.05 was considered to be significant.
Results
Enhanced global lysine hyperacetylation by excess lipids in renal in renal tubular epithelial cells both in vivo and in vitro, along with increased acetylation of LCAD and β-HAD and impaired fatty acid oxidation
To investigate the effect of lipid overload on protein lysine acetylation, we fed mice with high-fat diet (HFD) for 18 weeks, and body weights, plasma triglyceride, and cholesterol levels were shown in Supplementary Fig. 1. The expression of lysine acetylation was detected by immunohistochemistry. As shown in Fig. 1a, the lysine acetylation was highly expressed in renal tubule and was greatly increased in mice on high-fat diet than in mice on regular chow diet, which was accompanied by more severe tubulointerstitial damages revealed by periodic acid-Schiff (PAS) staining. Accordingly, the tubular epithelial cells (TECs) were treated with PA for 72 h, and the mitochondrial proteins’ acetylation level was intensely increased in PA treated cells compared with BSA treated control cells (Fig. 1b). In order to understand the effects of the enhanced mitochondrial lysine hyperacetylation on fatty acid metabolism, the FAO rate was detected. As shown in Fig. 1c, the FAO rate was dramatically decreased for more than 50% in PA stimulated TECs, followed by a decreased acetyl-CoA concentration (Fig. 1d), demonstrating an impaired FAO capacity.

The global lysine acetylation was increased under excess lipids conditions in renal TECs both in vivo and in vitro, accompanied by impaired fatty acid oxidation. a Immunohistochemistry of acetylated lysine and periodic acid–Schiff (PAS) staining in the kidney of chow and HFD-fed mice. Black asterisks show tubulointerstitial damages. n = 8, scale bar represents 200 μm. b Western blotting for total acetylated lysine in mitochondria of palmitic acid (PA) treated HK-2 cells. COX IV was used for normalization. c The decreased TECs oxidation rate in PA stimulated HK-2 cells with BSA treated cells as control group. d The declined acetyl-CoA content in PA stimulated HK-2 cells. **P < 0.01
Increased acetylation and decreased activities of LCAD and β-HAD
Furthermore, the acetylation levels of two key FAO enzymes long LCAD and β-HAD were examined by immunoprecipitation and immunoblotting. As shown in Fig. 2 a and b, the acetylation of both LCAD and β-HAD were obviously increased for about 3 folds after the cells were treated with PA compared with control. As numerous metabolic enzymes, lysine acetylation has been demonstrated to be able to modulate their activities [2], the activities of LCAD and β-HAD were then examined and were both decreased significantly in PA treated TECs (Fig. 2c).

The acetylation level of LCAD and β-HAD was increased, and the enzymatic activity was decreased in PA-treated HK-2 cells. a The acetylation of LCAD and β-HAD was detected by immunoprecipitation and western blotting in PA treated HK-2 cells. COX IV was used for normalization of mitochondrial fractions. b Relative density of A. *P < 0.05. **P < 0.01. c LCAD and β-HAD activities were measured using microplate kits. *P < 0.05
The upregulated expression of GCN5L1 under lipid overload both in vivo and in vitro
Immunohistochemical staining showed that the expression of GCN5L1 was increased in kidneys of HFD mice, especially in proximal tubular cells (Fig. 3a, b). This was further confirmed by western blotting for protein extractions from kidneys of HFD or regular chow fed mice (Fig. 3c, d). The cell lysates of PA stimulated TECs also showed dramatically increased expression of GCN5L1 compared with control (Fig. 3e, f).

The expression of GCN5L1 was upregulated both in vivo and in vitro under lipid overload. a Immunohistochemical staining of GCN5L1 in kidney tissues of chow and HFD-fed mice. Scale bar represents 200 μm. b Quantifications of IHC staining of GCN5L1 in renal tissue of the different groups. n = 8, *P < 0.05. c Western blotting of GCN5L1 in kidney tissues of chow and HFD-fed mice. d Relative density of c. n = 3, *P < 0.05. e Western blotting of GCN5L1 in HK-2 cells treated with BSA or PA. f Relative density of e. n = 3, *P < 0.05
Decreased acetylation and promoted activities of LCAD and β-HAD by the downregulation of GCN5L1
To further prove that the increased GCN5L1 is directly associated with the altered acetylation and activities of FAO enzymes, we generated siRNAs to knockdown the expression of GCN5L1 in TECs. The knockdown efficiency was evaluated by both qPCR and western blotting (Fig. 4 a, b, and c). The acetylation status analysis of LCAD and β-HAD based on immunoprecipitation showed that the suppression of GCN5L1 significantly reduced the PA induced elevation of acetylated LCAD and β-HAD (Fig. 4 d, e, f, and g). Subsequently the declined enzymic activities of LCAD and β-HAD in PA stimulated cells were totally rescued by GCN5L1 knockdown (Fig. 4h, i).

Suppression of GCN5L1 decreased the acetylation but promoted the activities of LCAD and β-HAD. The cells were transfected by siRNA targeting GCN5L1 (KD) or scramble control (CTR), and the knockdown efficiency were detected by a RT-qPCR on mRNA level and b, c western blotting on protein level. d, e The acetylation of LCAD and β-HAD was detected by immunoprecipitation (IP) and western blotting in PA treated HK-2 cells after GCN5L1 was knocked down. f, g Relative density of acetylated LCAD and β-HAD compared to total LCAD and β-HAD in d, e. h, i LCAD and β-HAD activities were detected by microplate kits. *P < 0.05, **P < 0.01
Ameliorated FAO defects and lipids accumulation by GCN5L1 downregulation in PA treated TECs
Then we analyzed whether downregulation of GCN5L1 had an effect on FAO indeed. As shown in Fig. 5a, the diminished FAO rate by PA stimulation was attenuated after GCN5L1 was knocked down. The dramatically impaired acetyl-CoA production was partially attenuated (Fig. 5b). Consequently, the increased concentration of cellular triglyceride in PA treated cells was significantly reduced after GCN5L1 was suppressed (Fig. 5c). This was further confirmed by Oil-Red O staining, which showed that the PA induced accumulation of neutral lipid droplets in scramble control siRNA transfected cells were nearly depleted in cells transfected with siRNA targeting GCN5L1 (Fig. 5d).

The fatty acid oxidation defects were ameliorated by GCN5L1 knockdown in PA treated HK-2 cells. a, b, and c Fatty acid oxidation rate, acetyl-CoA level and triglyceride contents were detected by corresponding microplate kits in PA or BSA treated HK-2 cells transfected with GCN5L1 siRNA (KD) or scramble control siRNA (CTR). *P < 0.05. **P < 0.01. d Oil-Red O staining showed the lipid droplet deposit in PA or BSA treated HK-2 cells transfected with GCN5L1 siRNA (KD) or scramble control siRNA (CTR)
Alleviated EMT by GCN5L1 downregulation in PA treated TECs
As impaired FAO in renal tubular cells has been proved to play a key role in kidney tubulointerstitial fibrosis by numerous studies [23, 24], the expression of EMT markers was investigated in our following experiments. As shown in Fig. 6 a, b, and c, loss of epithelial marker E-cadherin and acquisition of mesenchymal marker vimentin were observed in PA treated cells, and the knockdown of GCN5L1 completely inhibited the effects of PA on E-cadherin and vimentin expression in TECs, suggesting that GCN5L1 suppression may protect TECs from PA induced fibrosis process.

EMT was alleviated by GCN5L1 knockdown in PA treated HK-2 cells. a Western blotting for E-cadherin and vimentin in PA or BSA treated HK-2 cells transfected with GCN5L1 siRNA (KD) or scramble control siRNA (CTR). b, c, and d Relative density for E-cadherin (ECAD), vimentin, and GCN5L1 in a. Bands were normalized to GAPDH. *P < 0.05, **P < 0.01
Discussion
Renal proximal tubule epithelial cells have high energy demand and prefer FAO as the major energy source, as oxidation of fatty acids is more efficient to generate ATP compared with oxidation of glucose [22].Thus, insufficient intracellular FAO has been recognized as the key pathogenic event and potential intervention target in mediating renal lipotoxicity. In recent years, acetylation modification has emerged as an important mechanism for multiple dysfunction of mitochondrial energy metabolism, such as the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), and FAO [9, 26]. Lysine acetylation is mainly controlled via enzymatic regulation, which contains a number of acetyltransferase and deacetylase enzymes [11]. The role of histone deacetylases (HDACs), especially sirtuins, in the regulation of lysine acetylation and energy metabolism has been extensively investigated. For example, Alrob et al. found that cardiac proteins were hyperacetylated in HFD-fed mice, which was associated with a decrease in SIRT3 expression [1]. SIRT3 was also found to regulate mitochondrial fatty-acid oxidation by reversible enzyme deacetylation in fasting liver [1]. On the contrary, the role of acetyltransferase in this process has not been well-characterized. Some histone acetyltransferases (such as p300, CBP, and GCN5) were identified and found to be able to regulate chromatin dynamics and transcriptional activation [14]. GCN5L1 is a novel gene with sequence homology to the histone acetyltransferase GCN5 [19]. There are only a few studies exploring the direct function of GCN5L1 and the relative investigation in the kidney is lacking. In the present study, we first found that the expression of GCN5L1 was elevated both in the kidney of HFD mice and in PA stimulated TECs and identified GCN5L1 as a potential intervening target for renal lipotoxicity in that downregulation of GCN5L1 dramatically enhanced FAO rate by reducing the acetylation of key FAO enzymes, indicating that the active regulation of acetylation by GCN5L1 besides sirtuins has an important role in the renal lipotoxicity. In addition to the enzymatic regulation of lysine acetylation, it has recently been proposed that acetyl-CoA can promote non-enzymatic regulation of mitochondrial protein acetylation. The incubation of acetyl-CoA with isolated mitochondria in vitro can promote global protein acetylation [15]. However, in our study, the PA induced hyperacetylation was totally reduced by GCN5L1 suppression (Fig. 4), along with an elevated acetyl-CoA concentration (Fig. 5). This result indicates that the enzymatic regulation, especially GCN5L1, is likely to prevail in the regulation of renal mitochondrial protein acetylation and mitochondrial energy metabolism under excess lipids conditions, and the acetyl-CoA pool does not seem to be a major contributor in this process.
Mitochondrial protein acetylation has been proved to be one of the major sensory and adaptive mechanisms of mitochondria under different pathological conditions [4, 9]. A recent study has shown that multiple enzymes of the kidney are differentially acetylated in diabetes especially in tubular cells, suggesting acetylation might be involved in the development of CK [13].In agreement with this study, we demonstrated a significant increase of lysine acetylation levels in high-fat diet mice kidney samples (Fig. 1a), indicating that the kidney proteins are generally hyperacetylated under excess energy supply status. In addition, we provided evidence that the mitochondrial proteins, which are known to involve many metabolic enzymes, were acetylation in PA treated renal tubular epithelial cells (Fig. 1b). Particularly, we identified GCN5L1 as a novel intermediating intervening target to control the hyperacetylation of FAO enzymes in the kidney.
In recent years, lysine acetylation has emerged as a key protein posttranslational modification in metabolic regulation [26]. Our study further found the acetylation level of LCAD and β-HAD, which are mitochondrial fuel substrate oxidation key enzymes, were enhanced in TECs (Fig. 2). This is in accordance with the study in the heart of HFD mice that both the enzymes were found to be hyperacetylated [21]. In addition, we observed that the hyperacetylation of LCAD and β-HAD was along with the decrease of enzymatic activity and FAO rate in PA stimulated renal tubular epithelial cells (Fig. 2). Consistent with our results, Hirschey and colleagues also found that hyperacetylation of LCAD reduced its enzymatic activity with defective FAO in liver [10]. However, the enzymatic activity was upregulated along with the hyperacetylation status in the heart of HFD mice and with maturation [5, 21]. Reasons for these discrepancies remain enigmatic but may be explained by tissue differences in controlling acetylation status and energy balance or tissue specific acetylated sites of the enzymes. For example, there are eight acetylated lysine residues identified in LCAD, and Lys-42 was identified as a critical lysine residue for the regulation of LCAD enzymatic activity in liver during fasting [9], while two other sites, Lys-318 and Lys-322, were found to suppress LCAD activity by modulating its confirmation in vitro [3]. This difference as to what effects lysine acetylation has on fatty acid β-oxidation needs to be explored in the future study.
Renal TECs have high levels of baseline energy consumption and abundant mitochondria [22] and preferred FAO as major energy source [12]. When the cells were exposed to excess FAs, FAO rate was depressed (Fig. 1). It has been reported that defective FAO in renal tubular epithelial cells had a key role in kidney fibrosis development [12]. Facing the high fat metabolic constraints, TECs exhibit a phenotypic switch including the extracellular matrix proteins rearrangement and contribute to renal fibrogenesis. Our present study reveals that this process is controlled by lysine acetylation, and as shown in Fig. 6, the GCN5L1 knockdown attenuated the EMT phenotype of TECs and protected the cells from PA induced lipotoxicity. Collectively, GCN5L1 may serve as a critical component of the mitochondrial acetyltransferase machinery in the regulation of FA metabolism in kidney, and it may also be a potential therapeutic target for CKD by controlling mitochondrial enzymes acetylation (Fig. 7).

Model for the role of GCN5L1 in the regulation of fatty acid metabolism in renal tubule cells. In renal tubule cells, GCN5L1 is upregulated upon PA stimulation, which leads to the increase of acetylation levels of key FAO enzymes LCAD and β-HAD and further weakened their activities. Correspondingly, the FAO rate was decreased, resulting in the accumulation of FAO intermediates and occurrence of lipotoxicity. However, when GCN5L1 was suppressed, the enzyme activities are recovered, which accelerates the FAO rate and ameliorates lipotoxicity eventually in the cells
References
Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD (2014) Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res 103:485–497. https://doi.org/10.1093/cvr/cvu156
Amado FM, Barros A, Azevedo AL, Vitorino R, Ferreira R (2014) An integrated perspective and functional impact of the mitochondrial acetylome. Expert Rev Proteomics 11:383–394. https://doi.org/10.1586/14789450.2014.899470
Bharathi SS, Zhang Y, Mohsen AW, Uppala R, Balasubramani M, Schreiber E, Uechi G, Beck ME, Rardin MJ, Vockley J, Verdin E, Gibson BW, Hirschey MD, Goetzman ES (2013) Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J Biol Chem 288:33837–33847. https://doi.org/10.1074/jbc.M113.510354
Campbell SL, Wellen KE (2018) Metabolic signaling to the nucleus in cancer. Mol Cell 71:398–408. https://doi.org/10.1016/j.molcel.2018.07.015
Fukushima A, Alrob OA, Zhang L, Wagg CS, Altamimi T, Rawat S, Rebeyka IM, Kantor PF, Lopaschuk GD (2016) Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol 311:H347–H363. https://doi.org/10.1152/ajpheart.00900.2015
Hager MR, Narla AD, Tannock LR (2017) Dyslipidemia in patients with chronic kidney disease. Rev Endocr Metab Disord 18:29–40. https://doi.org/10.1007/s11154-016-9402-z
Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, Westphall MS, Pagliarini DJ, Prolla TA, Assadi-Porter F, Roy S, Denu JM, Coon JJ (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell 49:186–199. https://doi.org/10.1016/j.molcel.2012.10.024
Henriksen P, Wagner SA, Weinert BT, Sharma S, Bacinskaja G, Rehman M, Juffer AH, Walther TC, Lisby M, Choudhary C (2012) Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol Cell Proteomics 11:1510–1522. https://doi.org/10.1074/mcp.M112.017251
Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125. https://doi.org/10.1038/nature08778
Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV Jr, Kahn CR, Verdin E (2011) SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell 44:177–190. https://doi.org/10.1016/j.molcel.2011.07.019
Hosp F, Lassowskat I, Santoro V, De Vleesschauwer D, Fliegner D, Redestig H, Mann M, Christian S, Hannah MA, Finkemeier I (2017) Lysine acetylation in mitochondria: from inventory to function. Mitochondrion 33:58–71. https://doi.org/10.1016/j.mito.2016.07.012
Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K (2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21:37–46. https://doi.org/10.1038/nm.3762
Kosanam HT, K, Zhang Y, Advani A, Connelly KA, Diamandis EP, Gilbert RE (2014) Diabetes induces lysine acetylation of intermediary metabolism enzymes in the kidney. Diabetes 63:2432–2439. https://doi.org/10.2337/db12-1770/-/DC1
Menzies KJ, Zhang H, Katsyuba E, Auwerx J (2016) Protein acetylation in metabolism-metabolites and cofactors. Nat Rev Endocrinol 12:43–60. https://doi.org/10.1038/nrendo.2015.181
Paik WK, Pearson D, Lee HW, Kim S (1970) Nonenzymatic acetylation of histones with acetyl-CoA. Biochim Biophys Acta 213
Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A 110:6601–6606. https://doi.org/10.1073/pnas.1302961110
Ruan XZ, Varghese Z, Moorhead JF (2009) An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol 5:713–721. https://doi.org/10.1038/nrneph.2009.184
Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB (2009) Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8:604–606. https://doi.org/10.1111/j.1474-9726.2009.00503.x
Scott I, Wang L, Wu K, Thapa D, Sack MN (2018) GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell Biol 28:346–355. https://doi.org/10.1016/j.tcb.2018.01.007
Scott I, Webster BR, Li JH, Sack MN (2012) Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J 443:655–661. https://doi.org/10.1042/BJ20120118
Thapa D, Zhang M, Manning JR, Guimaraes DA, Stoner MW, O’Doherty RM, Shiva S, Scott I (2017) Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am J Physiol Heart Circ Physiol 313:H265–H274. https://doi.org/10.1152/ajpheart.00752.2016
Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, Heymsfield SB, Muller MJ (2010) Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr 92:1369–1377. https://doi.org/10.3945/ajcn.2010.29885
Xin W, Zhao X, Liu L, Xu Y, Li Z, Chen L, Wang X, Yi F, Wan Q (2015) Acetyl-CoA carboxylase 2 suppression rescues human proximal tubular cells from palmitic acid induced lipotoxicity via autophagy. Biochem Biophys Res Commun 463:364–369. https://doi.org/10.1016/j.bbrc.2015.05.070
Xu Y, Huang J, Xin W, Chen L, Zhao X, Lv Z, Liu Y, Wan Q (2014) Lipid accumulation is ahead of epithelial-to-mesenchymal transition and therapeutic intervention by acetyl-CoA carboxylase 2 silence in diabetic nephropathy. Metabolism 63:716–726. https://doi.org/10.1016/j.metabol.2014.02.010
Zager RA, Johnson AC, Hanson SY (2005) Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int 67:111–121. https://doi.org/10.1111/j.1523-1755.2005.00061.x
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004. https://doi.org/10.1126/science.1179689
Funding
This work was supported by the National Natural Science Foundation of China (Grants No. 81570654, 31600699, 81770729, 91749111, and 81471007) and Shandong Province Taishan Scholar Project (Grants No.tsqn 20161073).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All animal experiments were carried out strictly according to the guidelines of the Animal Center of Shandong University, and the experimental procedures were approved by the Ethical Committee of Shandong Provincial Hospital affiliated to Shandong University (No. S077).
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Figure 1
(DOCX 801 kb)
Rights and permissions
About this article

Cite this article
Lv, T., Hu, Y., Ma, Y. et al. GCN5L1 controls renal lipotoxicity through regulating acetylation of fatty acid oxidation enzymes. J Physiol Biochem 75, 597–606 (2019). https://doi.org/10.1007/s13105-019-00711-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-019-00711-6