
Abstract
To investigate whether uric acid could regulate urate transporter 1 (URAT1) protein and activity level, we established uric acid nephropathy (UAN) rat model and detected their serum uric acid and URAT1 level with or without the treatment of allopurinol. Results here showed that allopurinol could reduce serum uric acid level in UAN rat model. We further found that in UAN rats, the total and surface URAT1 expression level were both increased while this increase could be blocked by allopurinol treatment. By treating URAT1 stable expressed HEK cell with monosodium urate (MSU) crystals, we found that URAT1 level showed an increase in both total and cell surface level, and it would colocalize more with Rab11 instead of Rab7. Consistently, we also found that the total URAT1 protein level will show an increase in the presence of lysosome inhibitors but not ubiquitin-proteasome inhibitors. Furthermore, we also found that MSU crystal could drive Numb, a clathrin-coated adaptor protein which performs a key function in cell division, out of cell surface and disassociated it from URAT1. Finally, we found that Numb short hairpin RNA (shRNA)-transfected showed a phenocopy as MSU treatment, while Numb-2A mutation over-expression could resist crystal-induced phenotypes. These findings indicated that uric acid crystal could increase URAT1 membrane distribution through inhibiting Numb-induced URAT1 lysosome degradation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperuricemia is one of the most common and extensive form of metabolic diseases in populations, characterized by high serum uric acid level, causing accumulation of urate crystals in the joints and kidneys [1, 2]. Hyperuricemia is also well-known as severe risk factor for gouty arthritis, uric acid nephrolithiasis, and cardiovascular and renal disease, especially hypertension [3–5]. In human, the level of serum uric acid is determined primarily by the ratio of urate production as an end product of purine metabolism in the liver (for which the liver enzyme xanthine oxidase is necessary) versus the biliary and the urinary tract elimination [6]. In the urate elimination process, the human renal urate transporter-1 (URAT1) is thought to be an essential molecule that mediates the reabsorption of urate on the apical side of the proximal tubules [7].
The complementary DNA (cDNA) of urate/anion exchanger URAT1 (SLC22A12 gene) was first identified in the human kidney, in searching for organic an anion transporter-like (OAT-like) molecules in gene databases [8]. Expression/functional studies in Xenopus oocytes have found that the properties of urate transport by URAT1 are comparable to those in human renal brush border membrane vesicles (BBMV) [9, 10]. URAT1 is a 12-transmembrane domain-containing protein found in the apical membrane of proximal tubule epithelial cells and transports urate in exchange for Cl− or organic anions [13]. The antiuricosuric agents such as lactate, pyrazinoate, and nicotinate can serve as substrate for the antiporter activity of URAT1 to increase urate reabsorption, while URAT1 is also inhibited by the classical uricosuric agents such as benzbromarone, probenecid, and losartan [14]. Furthermore, URAT1 defects by gene mutation could lead to renal hypouricemia; in those patients, the urate fractional excretion rates could reach almost 100 % [15]. All the above evidences indicated that URAT1 plays an important role in the regulation of urate metabolism homeostasis.
As previously reported, we already established a high purine diet induced uric acid nephropathy (UAN) rat model which showed an elevated serum uric acid level as well as kidney damages [21]. Considering that hyperuricemia is a major cause of uric acid nephropathy, and URAT1 is important for regulating urate metabolism, here we wanted to investigate whether and how high serum uric acid appeared in hyperuricemia and nephropathy could regulate URAT1 protein and activity level.
Material and methods
Animals
Thirty male Sprague–Dawley rats (250.2 ± 1.9 g) were obtained from the Experimental Animal Center of the Guangzhou University of Chinese Medicine (ID: 00400856). The rats were randomly divided into four experimental groups. All rats were housed in plastic cages at a room temperature of 25 °C under a 12-h light–dark cycle and provided with rodent chow and water. They were housed for 1 week to adapt to their environment before experiments. The studies were carried out in accordance with the Guidelines for Human Treatment of Animals set by the Association of Laboratory Animal Sciences and the Center for Laboratory Animal Sciences at Guangzhou University of Chinese Medicine. Our research was approved by the Committee of Biomedical Ethics of Guangzhou University of Chinese Medicine [SYXK (yue): 2013–0024].
Reagents and plasmids
Allopurinol tablets were purchased from KangHe (Lot No: 111201) and diluted with distilled water to a final concentration of 5 mg/mL. Adenine tablets were purchased from Amresco Ltd. and diluted with 0.15 % CMC-Na to a final concentration of 3 %. Hematoxylin (DF001) was purchased from Dingguo. Trizol, reverse transcription buffer, SYBR green I PCR buffer, dNTPs, MMLV, and Taq were purchased from TaKaRa. Rabbit polyclonal URAT1 antibody (14937-1-AP) was purchased from Proteintech. URAT1 construct was purchased from ORIGENE in a C-terminal GFP fusion form and replaced the GFP by mRFP or Myc by routinely subclone. GFP-Numb, Numb-shRNA, GFP-Rab5, GFP-Rab7, and GFP-Rab11 were all purchased from ORIGENE. Numb-2A mutation construct (S265A and S284A) was made by quick change kit (Invitrogen). Surface protein extraction kit was purchased from BioVersion, sulfo-NHS-SS-biotin, and Avidin Agarose beads were purchased from Thermo Scientific. Ubiquintin inhibitor MG132, lysosome inhibitor chloroquine, and ammonium chloride were purchased from Sigma. Bortezomib was purchased from Santa Cruz.
Animal model and experimental procedures
Thirty Sprague–Dawley (SD) rats were randomly assigned into three groups. All rats were treated by intragastric administration. Group 1 was allocated as the normal control and fed with the same amount of CMC-Na (adenine dissolve buffer), while in the remaining two groups, rats were fed with adenine (30 mg/kg body weight) for 18 days to establish the UAN rat model. In the 19th day, we randomly selected three rats to sacrifice and collected the kidney sample. H&E staining of the kidney tissue was then performed to confirm whether the UAN model was successfully established. Then, the three groups of rats were treated in different procedures as follows: Group 1 was treated as the blank control. Rats in this group were fed with the same amount of saline buffer. Rats in group 2 were treated as the disease model control and were fed with the same amount of saline buffer as in group 1. Group 3 was treated as the positive medicine control. In this group, rats were treated with allopurinol (5 mg/kg body weight /day). In all groups, rats underwent daily drug or saline treatment for 23 days. During the process, no rats died. During sample collection, 18 days after model establishment, 5 mL of peripheral blood (fasting for 12 h) was collected for detection of serum uric acid (UA). At the end of the 42-day experiment procedure, rats were kept fasting for 12 h, and then sacrificed after 20 % urethane anesthesia. Peripheral blood was collected to detect UA, and the left kidney was resected for further research.
Tissue surface protein extraction/cell surface biotinylation and Western blot
For kidney tissues, kidney tissues from rats in different groups were frozen and stored at −20 °C immediately after dissection. It was mechanically dissociated and homogenized following manufactures instruction to get surface protein.
For HEK cells surface biotinylation, HEK cell with or without monosodium urate (MSU) treatment was washed twice with cold phosphate-buffered saline (PBS), and then incubated with PBS buffer supplemented with 0.5 mg/mL sulfo-NHS-SS-biotin for 1 h with gentle shaking, and excess biotin was quench with 50 mM Tris-PBS buffer for 20 min. Cells were lysate in RIPA buffer and then subjected to streptavidin-agarose beads in 4 °C for another 3 h.
For HEK cells internalization assay, surface biotinylated cells after quenching was put into 37 °C incubator for another 30 min to internalize, and then washed in cold cleavage buffer (50 mM glutathione, 90 mM NaCl, 1.25 mM CaCl2 dihydrate, 1.25 mM MgSO4, 0.2 % endotoxin-free BSA, pH 8.6) for 20 min with gentle shaking. Cells were lysate in RIPA buffer and then subjected to streptavidin-agarose beads in 4 °C for another 3 h.
Protein samples were boiled in Laemmli buffer, separated through sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to PVDF membranes (Hybond-C-extra; Amersham Biosciences). Membranes were blocked by 5 % BSA in TBST and then incubated overnight at 4 °C with rabbit anti-URAT1 (1:300) or mouse anti-GAPDH (1:8000) as control. After having been washed in TBST, membranes were incubated with peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies (Jackson ImmunoResearch, 1/4000) for 1 h. Protein bands were developed using ECL plus reagent (Western Lightening; Perkin Elmer).
Detection of URAT1 mRNA expression by real-time PCR
Total RNA was extracted with TRIzol (Invitrogen) and converted to cDNA by reverse kit (Takara) according to the manufacturer’s instructions. Real-time PCR was carried out according to the following procedures: 94 °C for 3 min, followed by 94 °C for 30 s, 55 °C for 45 s, and 72 °C for 1 min; 40 cycles.
URAT1:
-
Forward primer: 5′-TTG ATT GGC AGG AGG TGA CC-3′
-
Reverse primer: 5′-GGT TAA GTG GAG TCG GTC AG-3′
GAPDH:
-
Forward primer: 5′-TGG TCT ACA TGT TCC AGT ATG ACT-3′
-
Reverse primer: 5′-CCA TTT GAT GTT AGC GGG ATC TC-3′
Data were presented as normalized URAT1 messenger RNA (mRNA) level/GAPDH mRNA level.
Statistical analysis
Quantitative data was expressed as mean ± SEM. Comparisons of the means among multiple groups were performed using one-way ANOVA followed by Dunnett’s or Tukey–Kramer’s post hoc tests using a statistical software package (GraphPad Prism, version 4.0; GraphPad Software). Asterisks indicated statistically significant differences as follows: * p < 0.05, ** p < 0.01, and *** p < 0.001.
Results
UAN model rats showed an increase of both surface and total URAT1 expression level and allopurinol could repress the increasing of serum UA as well as surface/total URAT1 level
Compared with the normal control, serum UA level in adenine-treated rats (UAN model rats) were significantly higher, suggesting that UAN model has been established successfully. Allopurinol could decrease serum UA level compared with model group (Fig. 1a). To further investigate URAT1 role in our UAN model, we detected URAT1 protein expression level. As shown in Fig. 1b, total URAT1 level showed a strong increase in UAN model rats kidney tissue but can be rescued back to normal level by allopurinol. As URAT1 is a membrane protein, we next extracted total plasma membrane protein and detected surface URAT1 level. Results here showed alone with total URAT1; surface URAT1 level also showed an increase in UAN model but not in allopurinol group. Above results showed that allopurinol, which could alleviate uric acid generation, could totally block the increase of URAT1 (Fig. 1b, c). These results indicated that serum uric acid stimulation could increase the total and especially the functioning URAT1 portion (surface URAT1), and this kind of URAT1 increasing may act as the second step to aggravate uric acid-induced kidney impairments. Finally, by RT-PCR, we found that the mRNA level of URAT1 did not show any changes between all four groups (Fig. 1d).

UAN model rats showed an increase of total and surface URAT-1 in kidney tissues. UAN rats serum UA (a) was detected. b Representative Western blots of surface URAT-1 and total URAT-1 from different group rats kidney tissues. GAPDH was used as a loading control. c Quantification of surface URAT-1 expression level, normalized by GAPDH. d Real-time PCR results for URAT-1 mRNA expression levels from different groups. Data were represent here as means ± SEM (**p < 0.01, ***p < 0.001)
Uric acid crystal could increase cell surface/total URAT1 level but did not inhibit its endocytosis
To further investigate the detailed mechanism on how uric acid would regulate URAT1 expression, we employed URAT1 stable expression HEK cell here. We treated HEK cells by monosodium urate crystal (1 μM) for 24 h and found that HEK cell showed an increase of both total URAT1 and surface URAT1 but not its mRNA level (Fig. 2a–d). We then detected internalized URAT1 level but to our surprise, internalized URAT1 level did not show any significant changes with or without crystal stimulation (Fig. 2a, c), which indicated that the increase of surface URAT1 was not conducted by reduced internalization. Next, we also carried out a time lapse experiment and found that uric acid could increase the total URAT1 in a time-dependent manner (Fig. 2e, f).

Uric acid crystal could increase URAT-1 expression level in HEK cells. URAT-1 stable HEK cell were treated with or without monosodium urate crystal; 24 h later, cells were harvested. a Representative Western blots of surface, internalized, and total URAT-1 level by biotin assay. b Quantification of surface URAT-1 expression level, normalized by GAPDH. c Quantification of internalized URAT-1 expression level, normalized by GAPDH. d Real-time PCR results for URAT-1 mRNA expression levels with or without monosodium urate crystal treatment. e Representative Western blots showed HEK cell treated with uric acid crystal for different time. Total URAT1 and GAPDH were detected. f Quantification of total URAT1 level in (e), normalized by GAPDH. Data were represent here as means ± SEM (*p < 0.05)
Uric acid crystal reduced the colocalization of URAT1 with lysosome and thus increased the URAT1 level
To investigate the URAT1 trafficking process, we created a URAT1-RFP fusion construct. By co-transfecting this construct with GFP-Rab5/Rab7 and Rab11 and Confocal observation for live cells, we found that without crystal stimulus, there were almost 70 % of RFP vesicles colocalized with GFP-Rab7, while after crystal stimulation this rate got decreased to 20 % (Fig. 3); instead, colocalization of RFP vesicles with GFP-Rab11 was increased to nearly 60 % after crystal stimulation. Results here indicated that uric acid crystal treatment could change URAT-1 sub-cellular trafficking process (rates of colocalization (percent) of RFP vesicles with Rab5, 7, and 11 were 19.34 ± 4.53, 66.45 ± 1.24, and 15.37 ± 7.33 before stimulation, respectively, and 18.55 ± 6.33, 21.12 ± 4.22, and 59.42 ± 8.44 after stimulation, respectively). Finally, we directly added ubiquitin-proteasome inhibitor: MG132 (500 mM) and bortezomib (Bort.) 100 nM; or lysosome inhibitor: ammonium chloride (NH4Cl, 20 mM) and chloroquine (CLQ 50 mM). While ubiquitin inhibitor did not influence a lot for total URAT1 level, results here showed that lysosome inhibitors could both dramatically increase total URAT1 protein level (Fig. 3d). The present results indicated that URAT1 would regularly get through a lysosome-dependent degradation, and uric acid stimulation may block this process.

Uric acid crystal could change URAT-1 trafficking process. URAT1-RFP and GFP-Rab5, 7, or 11 were co-transfected into HEK cell and confocol images were captured. a Representative images and colocalization ratio analysis of URAT1-RFP and GFP-Rab5. b Representative images and colocalization ratio analysis of URAT1-RFP and GFP-Rab7. c Representative images and colocalization ratio analysis of URAT1-RFP and GFP-Rab11. d HEK cell treated with ammonium chloride 20 mM; CLQ 50 mM; bortezomib (Bort.) 100 nM or MG132 500 mM. Representative Western blots showed changes of total URAT1, normalized with GAPDH. Data were represent here as means ± SEM (*p < 0.05, **p < 0.01); scale bar is 10 μm
Uric acid crystal could drive Numb out of cell surface and disassociate binding of URAT1 and Numb
Considering that Numb is an important clathrin-coated pit adapter protein, it controlled various proteins’ clathrin-conducted endocytosis process. By over-expressing an GFP fusion Numb construct and directly observing in live cells, we found that without crystal stimulation, Numb was strongly distributed on the cell membrane, while crystal treatment would drive Numb out of cell membrane and redistributed evenly in the cytoplasma (Fig. 4a). We next employed Co-IP experiment and found that crystal stimulation could disassociate binding of URAT1 and Numb, which indicated the binding of Numb and URAT1 may regulate the URAT1 trafficking destination (Fig. 4b). We then transfected Numb short hairpin RNA (shRNA) construct and found that the URAT-1 distribution in Numb knock down condition showed a pheno-mimic with crystal treatment (Fig. 4c), which indicated that monosodium urate crystal treatment may regulate URAT1 distribution through regulation of Numb location. To make sure that URAT1 lysosome degradation was actually block in the absence of Numb, we treated the Numb knock down cell with or without chloroquine, and found that combining the treatments of Numb shRNA and chloroquine could not further increase URAT1 protein level (Fig. 4d), which indicated that Numb and chloroquine would regulate URAT1 protein through some common pathways. Finally, we made Numb-2A mutation (S265A and S284A); this construct could resist phosphorylation-related Numb redistribution from membrane to cytosome. Results here showed that Numb-2A could resist crystal-induced Numb relocation (Fig. 5a), and Western blot also showed that crystal could up-regulate surface URAT1 in Numb group but not Numb-2A group (Fig. 5b). This result indicated that uric crystal may regulate Numb distribution through phosphorylation, and Numb membrane distribution is important for URAT1 membrane location.

Uric acid crystal could drive Numb out of cell membrane and disassociate from URAT-1. URAT-1 stable HEK cell was transfected with GFP-Numb and treated with or without monosodium urate crystal for 24 h. a Live cell imaging showed GFP-Numb distribution. b Co-IP experiment showed binding of GFP-Numb and URAT1-Myc. c Upper panel: representative Western blots of surface and total URAT-1 level with Numb knock down or luciferase knock down (control); GAPDH was used as a loading control. Lower panel: quantification of surface URAT-1 expression level, normalized by GAPDH. d Numb knock down cell treated with or without CLQ, and representative Western blots showed changes of total URAT1, normalized with GAPDH. Data were represent here as means ± SEM (*p < 0.05, **p < 0.01)

Uric acid crystal regulates Numb membrane distribution through phosphorylation on S265 and S284. URAT1 stable HEK cell was transfected with GFP, GFP-Numb, or GFP-Numb-2A and treated with or without monosodium urate crystal for 24 h. a Live cell imaging showed GFP-Numb-2A distribution. b Upper panel: representative Western blots of surface and total URAT-1 level with control of Numb construct. GAPDH was used as a loading control. Lower panel: quantification of surface URAT-1 expression level, normalized by GAPDH. Data were represent here as means ± SEM (*p < 0.05, **p < 0.01)
Discussion
It has long been suggested that except from synthesis pathways, surface proteins’ trafficking process would also control the proteins amount by determining their transport destinations. Surface protein/receptors would go through endocytosis process spontaneously under a physiological condition or after some certain stimulation [12]. Endocytosis process itself involved in all kinds of basic cellular activity, such as receptor’s desensitization and resensitization, Integrin’s endocytosis and recycling back to the rear side of cell, signal peptides release from surface precursor protein in early endosomes, as well as protein amount regulation through lysosome or proteasome degradation [11]. Under endocytosis process, adaptor protein, which would bind and connect surface protein to different subcellular cargos, is usually considered as a key molecule for the whole trafficking process as they tightly regulate the selection of cargos which surface protein would go through with. In the present study, we found that uric acid direct stimulation in vitro or hyperuricemia in vivo could both increase surface and total URAT1 level. As URAT1 biosynthesis pathway was not strengthened under uric acid treatment, we raise the hypothesis that the increased URAT1 level may be caused by the changes of internalization pathways from lysosome to recycling endosomes.
Among the different trafficking cargos, early endosome, late endosome/lysosome, and recycling endosome, which were represented by three Rab family members, namely Rab5, Rab7, and Rab11, respectively, were most researched throughout [16]. At the early stage of endocytosis, recruiting of Rab5 to the newly formed endocytosis vesicle would finally fuse it to EEA1 positive vesicles and form early endosomes. Similarly, Rab7 is involved in the selection of early endosome to late endosome while Rab11 would drive early endosome to recycling endosome. Working together with cytoskeletons, those vesicles formed a highly regulated trafficking system. In the present study, we found that after uric acid direct stimulation, URAT1 would more colocalize with Rab11 instead of Rab7 as normal. This phenotype strongly indicated and would raise a hypothesis that normally internalized URAT1 would be more contained in Rab7 positive late endosome/lysosome and get through lysosome degradation to eliminate extra URAT1, but under uric acid stimulation, URAT1 would be more contained in Rab11 positive recycling endosome and get recycled back to cell membrane, and thus rise the surface and total URAT1 level. Furthermore, we also noticed that the ratio of surface URAT1 to total URAT1 was not consistent in vivo and in vitro. It may be caused by prolonged treatment time in vivo (42 days) versus in vivo treatment time (24 h), which would facilitate an accumulation phenotype for total URAT1, or caused by the different recycling speed and mechanism in the kidney tissue cells and HEK cells.
There are many adaptor proteins that would help to decide the certain vesicles that a surface protein would go through with, and Numb is one of them. Numb is a clathrin-coated pit adapter protein and has a key function in cell division; among its many aspects of functions, the ability to drive protein to lysosome and degradation is one of the most important function [17]. It has been found that many surface protein/receptors can bind with Numb and then start the lysosome-conducted degradation, such as glycoprotein CD8α, claudins, integrin, and amyloid precursor protein (APP) [18–20, 22]. Although the detailed mechanism is still unclear, people just found that Numb could bind to certain motif on surface protein through its PTB domain, and thus facilitate them to transport to lysosome and finally degrade there [23]. Here, we found that uric acid crystal stimulation could drive Numb out of the cell surface and disassociated it from URAT1; although the mechanism is still unclear, we would presume that it may be caused by Numb phosphorylation under crystal stimulation, since the phosphorylation resistant form of Numb-2A mutation did not show redistribution after crystal treatment.
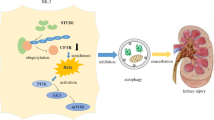
In summary, in the present study, we demonstrated for the first time that high purine diet could cause an increase of both cell surface and total URAT1 protein amount in vivo, while uric acid stimulation could mimic this phenotype in vitro. Furthermore, we also found that instead of biosynthesis pathway, the increase of URAT1 protein level induced by uric acid crystal was more likely to go through shifting URAT1 trafficking pathways from lysosome to recycling endosomes. Finally, we found that Numb, a clathrin-coated pit adapter protein, which could bind to URAT1 and targeted it into lysosome, could be driven out of cell surface and disassociated from URAT1 by uric acid crystal stimulation, and thus reduced URAT1 degradation and increased total and surface URAT1 protein level. This kind of uric acid-induced URAT1 increase may aggravate uric acid metabolism dyshomeostasis and exacerbate the kidney damages.
References
Arromdee E, Michet CJ, Crowson CS, O’Fallon WM, Gabriel SE (2002) Epidemiology of gout: is the incidence rising? J Rheumatol 29:403–406
Bogdanović O, Delfino-Machín M, Nicolás-Pérez M, Gavilán MP, Gago-Rodrigues I, Fernández-Miñán A, Lillo C, Ríos RM, Wittbrodt J, Martínez-Morales JR (2012) Numb/Numbl-Opo antagonism controls retinal epithelium morphogenesis by regulating integrinendocytosis. Dev Cell 23:782–795
D’Agostino M, Tornillo G, Caporaso MG, Barone MV, Ghigo E, Bonatti S, Mottola G (2011) Ligand of Numb proteins LNX1p80 and LNX2 interact with the human glycoprotein CD8α and promote its ubiquitylation and endocytosis. J Cell Sci 124:3545–3556
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H (2002) Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 23:447–452
Eraly SA, Hamilton BA, Nigam SK (2003) Organic anion and cation transporters occur in pairs of similar and similarly expressed genes. Biochem Biophys Res Commun 300:333–342
Johnson RJ, Kang DH, Feig D, Kivlighn S, Kanellis J, Watanabe S, Tuttle KR, Rodriguez-Iturbe B, Herrera-Acosta J, Mazzali M (2003) Is there a pathogenic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension 41:1183–1190
Kai H, Kaneyuki M, Shihara M, Toyama Y, Mitsutake Y, Umei H, Kusaba K, Ueda T, Adachi H, Imaizumi T, MAPPY Study Investigators (2013) Reduction in morning blood pressure is a key factor for ameliorating urinary albumin excretion in patients with morning hypertension irrespective of treatment regimen. Circ J 77:1551–1557
Klemp P, Stansfield SA, Castle B, Robertson MC (1997) Gout is on the increase in New Zealand. Ann Rheum Dis 56:22–26
Koepsell H, Endou H (2004) The SLC22 drug transporter family. Pflugers Arch 447:666–676
Kyriazis GA, Wei Z, Vandermey M, Jo DG, Xin O, Mattson MP, Chan SL (2008) Numb endocytic adapter proteins regulate the transport and processing of the amyloid precursor protein in an isoform-dependent manner: implications for Alzheimer disease pathogenesis. J Biol Chem 283:25492–25502
Li Z, Ding H, Chen C, Chen Y, Wang DW, Lv Y (2013) Gene 512:97–101
Li PS, Fu ZY, Zhang YY, Zhang JH, Xu CQ, Ma YT, Li BL, Song BL (2014) The clathrin adaptor Numb regulates intestinal cholesterol absorption through dynamic interaction with NPC1L. Nat Med 20:80–86
Li Y, Stamler J, Xiao Z, Folsom A, Tao S, Zhang H (1997) Serum uric acid and its correlates in Chinese adult populations, urban and rural, of Beijing. The PRC-USA collaborative study in cardiovascular and cardiopulmonary epidemiology. Int J Epidemiol 26:88–96
Miura D, Anzai N, Jutabha P, Chanluang S, He X, Fukutomi T, Endou H (2011) Human urate transporter 1 (hURAT1) mediates the transport of orotate. J Physiol Sci 61:253–257
Nakanishi T, Ohya K, Shimada S, Anzai N, Tamai I (2013) Functional cooperation of URAT1 (SLC22A12) and URATv1 (SLC2A9) in renal reabsorption of urate. Nephrol Dial Transplant 28:603–611
Sun M, Zhou T, Zhou L, Chen Q, Yu Y, Yang H, Zhong K, Zhang X, Xu F, Cai S, Yu A, Zhang H, Xiao R, Xiao D, Chui D (2012) Formononetin protects neurons against hypoxia-induced cytotoxicity through upregulation of ADAM10 and sAβPPα. J Alzheimers Dis 28:795–808
Takahashi S, Iwamoto N, Sasaki H, Ohashi M, Oda Y, Tsukita S, Furuse M (2009) The E3 ubiquitin ligase LNX1p80 promotes the removal of claudins from tight junctions in MDCK cells. J Cell Sci 122:985–994
Tseng CH (2005) Independent association of uric acid levels with peripheral arterial disease in Taiwanese patients with type 2 diabetes. Diabet Med 21:24–29
Vázquez-Mellado J, Jiménez-Vaca AL, Cuevas-Covarrubias S, Alvarado-Romano V, Pozo-Molina G, Burgos-Vargas R (2007) Molecular analysis of the SLC22A12 (URAT1) gene in patients with primary gout. Rheumatology (Oxford) 46:215–219
Villarroel-Campos D, Gastaldi L, Conde C, Caceres A, Gonzalez-Billault C (2014) Rab-mediated trafficking role in neurite formation. J Neurochem 129:240–248
Wang H, Sun M, Yang H, Tian X, Tong Y, Zhou T, Zhang T, Fu Y, Guo X, Fan D, Yu A, Fan M, Wu X, Xiao W, Chui D (2014) Hypoxia-inducible factor-1α mediates upregulation of neprilysin by histone deacetylase-1 underhypoxia condition in neuroblastoma cells. J Neurochem 131:4–11
Wu X, Liu L, Xie H, Liao J, Zhou X, Wan J, Yu K, Li J, Zhang Y (2012) Tanshinone IIA prevents uric acid nephropathy in rats through NF-κB inhibition. Planta Med 78:866–873
Yu J, Sun M, Chen Z, Lu J, Liu Y, Zhou L, Xu X, Fan D, Chui D (2010) Magnesium modulates amyloid-beta protein precursor trafficking and processing. J Alzheimers Dis 20:1091–1106
Acknowledgments
This work was supported by the Natural Science Foundation of GuandDong provence (No. 2011B080701007).
Author information
Authors and Affiliations
Corresponding author
Additional information
Xinlin Wu and Jianqing Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Wu, X., Zhang, J., Liu, T. et al. Uric acid crystal could inhibit Numb-induced URAT1 lysosome degradation in uric acid nephropathy. J Physiol Biochem 71, 217–226 (2015). https://doi.org/10.1007/s13105-015-0399-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-015-0399-7