
Abstract
Tumor necrosis factor-α (TNF-α), a pro-apoptotic cytokine, is involved in vascular hyperpermeability, tissue edema, and inflammation. We hypothesized that TNF-α induces microvascular hyperpermeability through the mitochondria-mediated intrinsic apoptotic signaling pathway. Rat lung microvascular endothelial cells grown on Transwell inserts, chamber slides, or dishes were treated with recombinant TNF-α (10 ng/ml) in the presence or absence of a caspase-3 inhibitor, Z-DEVD-FMK (100 μM). Fluorescein isothiocyanate (FITC)-albumin (5 mg/ml) was used as a marker of monolayer permeability. Mitochondrial reactive oxygen species (ROS) was determined using dihydrorhodamine 123 and mitochondrial transmembrane potential using JC-1. The adherens junction integrity and actin cytoskeletal organization were studied using β-catenin immunofluorescence and rhodamine phalloidin, respectively. Caspase-3 activity was measured fluorometrically. The pretreatment with Z-DEVD-FMK (100 μM) attenuated TNF-α-induced (10 ng/ml) disruption of the adherens junctions, actin stress fiber formation, increased caspase-3 activity, and monolayer hyperpermeability (p < 0.05). TNF-α (10 ng/ml) treatment resulted in increased mitochondrial ROS formation and decreased mitochondrial transmembrane potential. Intrinsic apoptotic signaling-mediated caspase-3 activation plays an important role in regulating TNF-α-induced endothelial cell hyperpermeability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vascular hyperpermeability due to ischemia-reperfusion injury can give rise to serious complications leading to multiple organ dysfunction syndrome, which correlate to a worse prognosis in patients with traumatic injury-induced hemorrhagic shock (HS), burn trauma, and septic shock [5, 8, 11, 30, 35]. Furthermore, irrespective of etiology, these shock conditions lead to the release of various extracellular pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) [5, 8, 11, 30, 35].
TNF-α is also a cytotoxic cytokine known to induce cellular apoptosis by the extrinsic or receptor-mediated apoptotic pathway [2]. TNF-α, upon binding to its cell surface receptor TNF-R1, initiates intracellular signal transduction to form a death-inducing signaling complex (DISC) [2, 9]. The DISC initiates apoptosis by triggering the caspase cascade-activating downstream effector caspase-3 [2, 9]. Apoptosis can also occur when cells are subjected to apoptotic stimuli such as free radicals, ionizing radiation, or growth factor deprivation through the intrinsic or mitochondria-mediated apoptotic pathway [9, 15]. These apoptotic stimuli cause mitochondria to release apoptogenic factors such as cytochrome c. The release of cytochrome c into the cytosol initiates apoptosis by triggering the caspase cascade-activating downstream effector caspase-3 [9, 15]. The extrinsic and the intrinsic pathways are interlinked through a BH3 domain containing pro-apoptotic protein of the Bcl-2 family, BID. The receptor-mediated or extrinsic apoptotic signal translocates BID from cytosol to the mitochondria to release cytochrome c to initiate apoptosis through caspase-3 activation. Therefore, caspase-3 activation can occur through extrinsic or receptor as well as intrinsic or mitochondria-mediated apoptotic pathway [9, 15].
Apart from inducing apoptosis in different cell types, TNF-α has also been implicated in endothelial cell activation, increased endothelial cell permeability, and pulmonary edema formation [12, 23, 24]. Microvascular permeability is primarily regulated by adherens junctions between the endothelial cells [34]. Any structural change within adherens junctional proteins such as VE-cadherin, or change in the intracellular levels or dynamics of these proteins such as β-catenin, can alter the organization of the adherens junctional protein complex leading to microvascular hyperpermeability [27, 28, 31]. Intracellularly, each adherens junction is anchored to the actin cytoskeleton of the cell, and the physical integrity of this junctional protein complex is maintained through actin remodeling [13, 25, 34]. Disruption of the adherens junctional protein complex can augment actin stress fiber formation [25, 27].
Studies from other research groups have shown involvement of TNF-α in inducing organizational change in actin cytoskeleton of endothelial cells [23, 24]. TNF-α has shown to be involved in disruption of adherens junctions by its action on VE-cadherin leading to an increase in paracellular endothelial cell permeability [12, 23, 24]. However, it is not clearly understood which signaling pathways are responsible for these changes in the adherens junctional protein complex and the actin cytoskeleton of endothelial cells leading to the microvascular barrier dysfunction. In this study, we have tried to elucidate the possible molecular mechanisms by which TNF-α triggers microvascular endothelial cell hyperpermeability.
Previous results from our laboratory have demonstrated the involvement of intrinsic or mitochondria-mediated apoptotic signaling in hemorrhagic shock-induced disruption of endothelial cell adherens junction protein complex leading to microvascular hyperpermeability [4]. The results from our earlier laboratory work have demonstrated that during hemorrhagic shock, there is an increase in formation of mitochondrial reactive oxygen species (ROS) leading to decrease in mitochondrial transmembrane potential and subsequent release of pro-apoptotic cytochrome c resulting in effector caspase-3 activation and microvascular barrier dysfunction [4, 6]. In the current study, we have hypothesized that TNF-α induces microvascular endothelial cell hyperpermeability through the mitochondria-mediated intrinsic apoptotic signaling. Furthermore, we have also used a naturally occurring endogenous antioxidant α-lipoic acid as an ameliorating agent to attenuate TNF-α-induced microvascular endothelial cell hyperpermeability.
Materials and methods
Cell culture and reagents
Rat lung microvascular endothelial cells (RLMECs) from Vec Technologies (Rensselaer, NY) were grown on cell culture dishes coated with fibronectin (0.1 % solution) from bovine plasma (Sigma-Aldrich, St. Louis, MO) using MCDB-131 complete media (Rensselaer, NY). Trypsin-EDTA solution (0.25 %; Invitrogen-Gibco, Grand Island, NY) was used to detach the cells from fibronectin-coated cell culture dishes. Fluorescein isothiocyanate-bovine albumin (FITC-albumin, 5 mg/ml) was obtained from Sigma (St. Louis, MO). β-Catenin primary and anti-rabbit secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) was obtained from Vector Laboratories (Burlingame, CA). Dihydrorhodamine 123 (DHR 123) was obtained from Invitrogen (Carlsbad, CA). JC-1 (5,5′,6,6′ tetrachoro-1,1′,3,3′ tetraethylbenzimidazolyl carbocyanine iodide) was obtained from Cell Technology, Inc. (Mountain View, CA). Caspase-3 inhibitor, Z-DEVD-FMK, was obtained from R&D Systems (Minneapolis, MN). Recombinant TNF-α and α-lipoic acid were obtained from Sigma-Aldrich (St. Louis, MO).
Effect of TNF-α on endothelial cell monolayer permeability
RLMECs were grown as monolayers on fibronectin-coated Transwell plates using MCDB-131 complete media for 72 to 96 h. The following groups were studied: an untreated control group and TNF-α treatment groups at increasing concentrations of 1, 5, 10, and 20 ng/ml. FITC-albumin (5 mg/ml) was added to the luminal (upper) chamber of the Transwell and was allowed to equilibrate for 30 min. The samples (100 μl) collected from the abluminal (lower) chambers were analyzed for FITC fluorescent intensity using a fluorometric plate reader at excitation/emission 494/520 nm, and the data were calculated as percentage of the control values.
Effect of TNF-α on mitochondrial ROS production
RLMECs were grown as monolayers on chamber slides as described above. The following groups were studied: a control or untreated group and TNF-α-treated (10 ng/ml) group for 1 h. The cells were exposed to DHR 123 for 30 min at 37 °C. DHR 123 is a cell-permeable fluorescent dye, which emits red fluorescence in the presence of mitochondrial ROS. The cells were then washed twice in phosphate-buffered saline (PBS) and observed under ×40 fluorescent microscope. This is a reliable qualitative assay to demonstrate increase in mitochondrial ROS formation [19, 32].
Effect of TNF-α on mitochondrial transmembrane potential
RLMECs were grown on fibronectin-coated chamber slides as described above. The following groups were studied: a control or untreated group and TNF-α-treated (10 ng/ml) group for 1 h. The cells were exposed to JC-1 fluorescent dye for 15 min at 37 °C. JC-1 is mitochondrial transmembrane potential-dependent dual fluorescent dye. JC-1 on administering to the cell has a tendency to accumulate in the normal functioning mitochondria as J-aggregates which emits red fluorescence [31]. However, decrease in mitochondrial transmembrane potential results in failure of JC-1 dye to accumulate in the mitochondria and remain in the cytosol as monomer emitting green fluorescence. The cells were then washed twice in PBS and observed under ×40 fluorescent microscope.
Effect of TNF-α on caspase-3 activity in endothelial cell
RLMECs were grown on fibronectin-coated cell culture dishes in complete MCDB-131 media. The following groups were studied: untreated or control group, TNF-α-treated (10 ng/ml) group, TNF-α (10 ng/ml) group pretreated with Z-DEVD-FMK (100 μM), and Z-DEVD-FMK (100 μM) alone group. The RLMECs were lysed by adding sample lysis buffer from the assay kit. The homogenates were used for protein estimation, followed by treatment with the substrate conjugate labeled with a fluorescent probe 7-amino-4-trifluoromethyl coumarin provided in the assay kit. The resulting fluorescent intensity was measured in a fluorescent plate reader using excitation and emission wavelength at 400 and 505 nm, respectively, for caspase-3 assay.
Effect of caspase-3 inhibitor (Z-DEVD-FMK) on TNF-α-induced disruption of adherens junctions
RLMEC monolayers were grown on fibronectin-coated chamber slides using MCDB-131 complete media. The following groups were studied: a control or untreated group, recombinant TNF-α-treated (10 ng/ml) group, recombinant TNF-α (10 ng/ml) group pretreated with Z-DEVD-FMK (100 μM), and Z-DEVD-FMK (100 μM) alone group. All the treatment and pretreatment with TNF-α and Z-DEVD-FMK, respectively, were carried out for 1 h each. The cells were processed for β-catenin immunofluorescence by fixing the cells in 4 % paraformaldehyde, followed by permeabilizing with Triton X-100 and exposing to polyclonal antibody against β-catenin for overnight at 4 °C. Following this, the cells were then washed twice with PBS and exposed to the FITC-tagged secondary antibodies. The cells were then visualized using a DAPI containing mounting medium under ×60 confocal laser scanning fluorescent Olympus Fluoview microscope.
Effect of caspase-3 inhibitor (Z-DEVD-FMK) on TNF-α-induced disruption of cytoskeletal assembly
RLMECs were grown on chamber slides as described above. The following groups were studied: a control or untreated group, recombinant TNF-α-treated (10 ng/ml) group, recombinant TNF-α (10 ng/ml) group pretreated with Z-DEVD-FMK (100 μM), and Z-DEVD-FMK (100 μM) alone group. All the treatment and pretreatment with TNF-α and Z-DEVD-FMK, respectively, were carried out for 1 h each. The cells were processed for visualization of f-actin using rhodamine phalloidin dye as described above using a DAPI-containing mounting medium under ×60 confocal laser scanning fluorescent Olympus Fluoview microscope.
Effect of caspase-3 inhibitor (Z-DEVD-FMK) on TNF-α-induced endothelial cell monolayer hyperpermeability
RLMEC monolayers grown on Transwell plates were used. Sixty minutes prior to the experiments, the monolayers were exposed to fresh media without phenol-red. The following groups were studied: an untreated control group, TNF-α-treated (10 ng/ml) group, TNF-α (10 ng/ml) group pretreated with Z-DEVD-FMK (100 μM), and Z-DEVD-FMK (100 μM) alone group. All the treatment and pretreatment with TNF-α and Z-DEVD-FMK, respectively, were carried out for 1 h each. The monolayer permeability was determined as described above using FITC-albumin (5 mg/ml) flux as a measure of endothelial cell permeability.
Effect of α-lipoic acid on TNF-α-induced endothelial cell monolayer hyperpermeability
RLMEC monolayers grown on Transwell plates were used. Sixty minutes prior to the experiments, the monolayers were exposed to fresh media without phenol-red. The following groups were studied: an untreated control group, TNF-α-treated (10 ng/ml) group, TNF-α (10 ng/ml) group pretreated with α-lipoic acid (100 μM), and α-lipoic acid (100 μM) alone group. All the treatment and pretreatment with TNF-α and α-lipoic acid, respectively, were carried out for 1 h each. The monolayer permeability was determined as described above using FITC-albumin (5 mg/ml) flux as a measure of endothelial cell permeability.
Statistical analysis
Statistical analysis was performed utilizing analysis of variance (ANOVA), followed by the Bonferroni’s posttest for multiple comparisons. All values are expressed as mean ± SEM. A p value of ≤0.05 was considered to indicate a statistical significant difference. In monolayer permeability studies, each experimental value was compared with the initial baseline value and expressed as percentage change.
Results
TNF-α induces an increase in endothelial cell monolayer permeability
TNF-α-treated RLMEC monolayers showed an increase in permeability compared with the untreated control monolayer. The FITC-albumin fluorescent intensity in the media from abluminal chamber was significantly higher in RLMECs treated with 10 and 20 ng/ml TNF-α compared to the control group, suggesting an increase in endothelial cell monolayer permeability (p < 0.05; Fig. 1).

TNF-α-treated (1, 5, 10, and 20 ng/ml) monolayers show significant dose-dependent increase in hyperpermeability compared with the control (*p < 0.05; n = 5)
TNF-α induces an increase in mitochondrial ROS production
TNF-α-treated RLMECs showed an increase in mitochondrial ROS formation as evidenced by the increase in red fluorescence of DHR 123 dye compared with control or untreated endothelial cells (Fig. 2a).

a TNF-α-treated RLMECs show increased mitochondrial ROS formation as evidenced by increased red fluorescence of dihydrorhodamine (DHR) 123 probe compared with untreated control cells. b Following exposure to JC-1 probe, the green fluorescence indicates cytoplasmic accumulation of the dye, whereas the red fluorescence indicates J-aggregate formation in normal and healthy mitochondria. In untreated control cells, JC-1 shows greater degree of red fluorescence, indicating intact mitochondria. TNF-α treatment leads to decrease in red (J-aggregates) fluorescence, indicating the loss of mitochondrial transmembrane potential (color figure online)
TNF-α induces a decrease in mitochondrial transmembrane potential
TNF-α-treated RLMECs showed a decrease in mitochondrial transmembrane potential as evidenced by the decrease in red fluorescence, due to less accumulation of J-aggregates in the mitochondria, compared with control or untreated cells. The control cells show more of mitochondrial red fluorescence (J-aggregates) than cytoplasmic green fluorescence of JC-1 fluorescent probe, indicating normal functioning mitochondria (Fig. 2b).
Caspase-3 inhibitor (Z-DEVD-FMK) decreases TNF-α-induced increase in caspase-3 activity
TNF-α-treated RLMECs showed a significant increase in caspase-3 activity when compared with the control or untreated cells (p < 0.05; Fig. 5). Z-DEVD-FMK pretreatment significantly reduced the TNF-α-induced increase in caspase-3 activity (p < 0.05; Fig. 3).

TNF-α induces caspase-3 activity in RLMECs. TNF-α-treated RLMECs show increase in caspase-3 activity compared with untreated control RLMECs (*p < 0.05). The Z-DEVD-FMK pretreatment decreases TNF-α-induced increase in caspase-3 activity significantly in RLMECs (a p < 0.05)
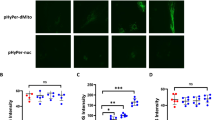
Caspase-3 inhibitor (Z-DEVD-FMK) prevents TNF-α-induced disruption of adherens junctions
TNF-α-treated RLMECs showed diffuse punctate distribution of β-catenin in the form of intercellular gaps at the cell junction which was reversed by Z-DEVD-FMK to characteristics continuous distribution of β-catenin immunofluorescence as seen in control or untreated endothelial cells (Fig. 4a).

a TNF-α-treated RLMECs show discontinuity of β-catenin at cell-cell contact demonstrating the disruption of the barrier (indicated by arrows). The cells treated with Z-DEVD-FMK prior to the treatment of TNF-α show improvement in the junctional damage as evidenced by the continuity of the β-catenin immunofluorescence. The blue color indicates DAPI staining for the nucleus. b Rhodamine phalloidin fluorescence showing f-actin cytoskeletal assembly in RLMECs. TNF-α-treated cells show increase in actin stress fiber formation intracellularly (indicated by arrows). The pretreatment with Z-DEVD-FMK prevents TNF-α-induced actin stress fiber formation. The control cells or untreated cells show no visible increase in stress fiber formation. The blue color indicates DAPI staining for the nucleus (color figure online)
Caspase-3 inhibitor (Z-DEVD-FMK) prevents TNF-α-induced disruption of cytoskeletal assembly
TNF-α-treated RLMECs showed a remodeling of the actin cytoskeleton in the form of increased actin stress fiber formation. However, Z-DEVD-FMK protected actin cytoskeleton and decreased TNF-α-induced increase in the actin stress fiber formation as evidenced by rhodamine phalloidin staining for f-actin (Fig. 4b).
Caspase-3 inhibitor (Z-DEVD-FMK) attenuates TNF-α-induced endothelial cell monolayer hyperpermeability
TNF-α-treated RLMEC monolayers showed an increase in FITC-albumin fluorescent intensity in the media from abluminal chamber compared with the control group, suggesting an increase in endothelial cell monolayer permeability (p < 0.05; Fig. 5a). However, the monolayers pretreated with Z-DEVD-FMK followed by TNF-α treatment showed significantly less fluorescent intensity of FITC-albumin in the media from the abluminal chamber (p < 0.05, Fig. 5a).

a TNF-α-treated monolayers show significant increase in permeability compared with the untreated control monolayers (*p < 0.05). TNF-α-treated monolayers when pretreated with Z-DEVD-FMK show significant decrease in permeability compared with the TNF-α-treated monolayers (a p < 0.05). b TNF-α-treated monolayers show significant increase in permeability compared with the untreated control monolayers (*p < 0.05). TNF-α-treated monolayers when pretreated with α-lipoic acid show significant decrease in permeability compared with the TNF-α-treated monolayers (a p < 0.05)
α-Lipoic acid attenuates TNF-α-induced endothelial cell monolayer hyperpermeability
The RLMEC monolayers pretreated with α-lipoic acid followed by TNF-α treatment showed significantly less fluorescent intensity of FITC-albumin in the media from the abluminal chamber (p < 0.05; Fig. 5b) compared with the cells treated with TNF-α only. Thus, α-lipoic acid attenuates TNF-α-induced increase in endothelial cell monolayer permeability.
Discussion
The results of this study have illustrated that TNF-α induces a dose-dependent increase in permeability in RLMEC monolayers. The TNF-α-treated RLMECs showed oxidative stress with increase in mitochondrial ROS in the form of increase in red fluorescence of DHR 123, a mitochondrial specific fluorescent probe, compared with the control or untreated endothelial cells under fluorescent microscopy. Furthermore, TNF-α also induced a decrease in mitochondrial transmembrane potential and was visualized by using JC-1 fluorescent probe in the form of decrease in red fluorescent of J-aggregates in TNF-α-treated RLMECs compared with the control or untreated cells under fluorescent microscopy. The caspase-3 enzyme activity was also assayed fluorometrically, which showed significant increase in caspase-3 activity in the endothelial cells treated with TNF-α. The endothelial cells with prior treatment with caspase-3 inhibitor, Z-DEVD-FMK, showed reversal in TNF-α-induced increase in caspase-3 activity.
The cells pretreated with the Z-DEVD-FMK demonstrated a decrease in TNF-α-induced endothelial cell hyperpermeability by fluorometrically measuring FITC-albumin flux across the monolayers. Z-DEVD-FMK pretreatment also showed reversal of TNF-α-induced disruption of the adherens junctions in the form of diffused and punctate distribution on β-catenin. Similarly, caspase-3 inhibition via Z-DEVD-FMK also prevented TNF-α-induced organizational change in the actin cytoskeleton of the endothelial cells in the form of increased actin stress fiber formation as visualized on rhodamine phalloidin staining. Moreover, the role of mitochondria in TNF-α-induced endothelial cell hyperpermeability was substantiated with the help of endogenous antioxidant α-lipoic acid. The cells pretreated with α-lipoic acid showed reversal of TNF-α-induced increase in endothelial cell permeability.
In normal mitochondrial physiology, oxidative phosphorylation is carried out by the respiratory complexes of the electron transport chain situated in the inner mitochondrial membrane [10, 20]. The transport of electrons along with movement of H+ ions across the mitochondrial membrane generates a gradient, which is utilized by the ATP synthase enzyme to synthesize the ATP. This transfer of H+ ions results in a potential known as mitochondrial transmembrane potential [10, 20]. During oxidative stress, due to improper oxidative phosphorylation, ROS formation is accelerated many folds. The increase in the production of ROS alters the integrity of the mitochondrial membrane, resulting in an influx of water and electrolytes, and subsequent disruption of the proton gradient across the mitochondrial membrane [10, 20]. This disorderly process gives rise to a phenomenon known as mitochondrial membrane permeabilization, leading to the collapse of the mitochondrial transmembrane potential and loss of mitochondrial function [10, 20].
One of the possible mechanisms through which TNF-α exacerbates ROS production in mitochondria is by depleting endoplasmic reticulum (ER) Ca2+ stores and increasing cytosolic levels of Ca2+ via inositol-1,4,5-triphosphate (IP3) receptors [7, 26]. This in turn leads to an increased uptake of Ca2+ in mitochondria through mitochondrial voltage-dependent anion channels, the mitochondrial Ca2+ uniporter channels, and specifically, mitochondrial calcium uptake 1 channel [7, 22]. An increase in mitochondrial levels of Ca2+ causes rapid movement of ions through mitochondrial permeability transition pore leading to further accelerating ROS generation through complex III of electron transport chain [7, 26]. Similarly, earlier studies from our laboratory have shown involvement of mitochondrial complex III in intrinsic apoptotic signaling-mediated microvascular endothelial cell hyperpermeability [3]. Also, TNF-α is known to generate extramitochondrial or cellular ROS [1]. And so, we cannot rule out the role of extramitochondrial or cellular ROS in causing mitochondrial dysfunction and subsequent activation of caspase-3. Furthermore, TNF-α being a pro-apoptotic cytokine that mainly act through the receptor mediated pathway, there might be a component of receptor mediated pathway in the activation of caspase-3.
However, in order to demonstrate importance of mitochondrial ROS in TNF-α-induced microvascular endothelial cell hyperpermeability, we have used α-lipoic acid, an endogenous mitochondrial antioxidant that scavenges ROS and promotes other antioxidants like glutathione, vitamin C, and vitamin E [14, 16–18, 21]. It also reduces intracellular Ca2+ levels and thus inhibits mitochondrial Ca2+ surge [16]. RLMECs pretreated with α-lipoic acid showed decrease in TNF-α-induced endothelial cell hyperpermeability. Moreover, previous studies from our laboratory have demonstrated that α-lipoic acid attenuated hemorrhagic shock-induced microvascular endothelial cell hyperpermeability by decreasing mitochondrial ROS formation and preventing drop in mitochondrial transmembrane potential, thus limiting release of cytochrome c from mitochondria in to cytosol and averting an increase in effector caspase-3 activity [32, 33]. We have also demonstrated in our earlier study that even though there is an increase in endothelial cell caspase-3 activity during microvascular hyperpermeability that does not correlate to increase in apoptotic process or cell death in the endothelial cells, which in a way reaffirms that the microvascular endothelial cell hyperpermeability is a reversible process [29].
In conclusion, RLMECs exposed to TNF-α demonstrate increase in mitochondrial ROS production, decrease in mitochondrial transmembrane potential, and increase in caspase-3 activity. However, pretreatment with caspase-3 inhibitor, Z-DEVD-FMK, attenuates TNF-α-induced disruption of adherens junctional protein complex, actin stress fiber formation, and endothelial cell hyperpermeability. Furthermore, α-lipoic acid, a mitochondrial antioxidant, attenuated TNF-α-induced increase in endothelial cell barrier dysfunction. Thus, our results indicate the role of mitochondria-mediated “intrinsic” apoptotic signaling in regulating TNF-α-induced endothelial cell barrier dysfunction leading to an increase in permeability. The clinical significance of this study is that TNF-α, being a prominent player in the pathophysiology of inflammatory and ischemia-reperfusion injury, the results from this study have elucidated possible molecular mechanisms through which TNF-α induces microvascular endothelial cell hyperpermeability. The findings also provide avenues which have therapeutic potential by targeting different steps in the intrinsic apoptotic signaling pathway. Moreover, the desired result can be achieved either by blocking TNF-α-induced apoptotic signal transduction at the receptor level, or through protecting mitochondrial physiology by limiting intracellular Ca2+ surge and/or restoring mitochondrial function by preventing escalation in ROS formation from incoming apoptotic signal by using antioxidant therapy.
References
Bubici C, Papa S, Dean K, Franzoso G (2006) Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene 25:6731–6748
Campbell MT, Dagher P, Hile KL, Zhang H, Meldrum DR, Rink RC, Meldrum KK (2008) Tumor necrosis factor-α induces intrinsic apoptotic signaling during renal obstruction through truncated Bid activation. J Urol 180:2694–2700
Childs EW, Tharakan B, Hunter FA, Isong M, Liggins ND (2008) Mitochondrial complex III is involved in proapoptotic BAK-induced microvascular endothelial cell hyperpermeability. Shock 29:636–641
Childs EW, Tharakan B, Hunter FA, Tinsley JH, Cao X (2007) Apoptotic signaling induces hyperpermeability following hemorrhagic shock. Am J Physiol Heart Circ Physiol 292:H3179–3189
Childs EW, Udobi KF, Hunter FA, Dhevan V (2005) Evidence of transcellular albumin transport after hemorrhagic shock. Shock 23:565–570
Childs EW, Udobi KF, Wood JG, Hunter FA, Smalley DM, Cheung LY (2002) In vivo visualization of reactive oxidants and leukocyte-endothelial adherence following hemorrhagic shock. Shock 18:423–427
Dada LA, Sznajder JI (2011) Mitochondrial Ca2+ and ROS take center stage to orchestrate TNF-α-mediated inflammatory responses. J Clin Invest 121:1683–1685
Dewar D, Moore FA, Moore EE, Balogh Z (2009) Postinjury multiple organ failure. Injury 40:912–918
Fulda S, Debatin KM (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25:4798–4811
Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 10:481–494
Groeneveld AB (2002) Vascular pharmacology of acute lung injury and acute respiratory distress syndrome. Vasc Pharmacol 39:247–256
Goldblum SE, Hennig B, Jay M, Yoneda K, McClain CJ (1989) Tumor necrosis factor alpha-induced pulmonary vascular endothelial injury. Infect Immun 57:1218–1226
Hartsock A, Nelson WJ (2008) Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta 1778:660–669
Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER (2001) L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J Biol Chem 276:40–47
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE (2009) Cell death. N Engl J Med 361:1570–1583
Jia L, Liu Z, Sun L, Miller SS, Ames BN, Cotman CW, Liu J (2007) Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci 48:339–348
Johansen JS, Harris AK, Rychly DJ, Ergul A (2005) Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol 4:5
Li AE, Ito H, Rovira II, Kim KS, Takeda K, Yu ZY, Ferrans VJ, Finkel T (1999) A role for reactive oxygen species in endothelial cell anoikis. Circ Res 85:304–310
Liu J, Ames BN (2005) Reducing mitochondrial decay with mitochondrial nutrients to delay and treat cognitive dysfunction, Alzheimer’s disease, and Parkinson’s disease. Nutr Neurosci 8:67–89
Moffitt KL, Martin SL, Walker B (2010) From sentencing to execution—the processes of apoptosis. J Pharm Pharmacol 62:547–562
Packer L, Roy S, Sen CK (1997) Alpha-lipoic acid: a metabolic antioxidant and potential redox modulator of transcription. Adv Pharmacol 38:79–101
Perocchi F, Gohil VM, Girgis HS, Huertas A, Quadri SK, Horiuchi K, Inamdar N, Emin MT, Lindert J, Ten VS, Bhattacharya S, Bhattacharya J (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467:291–296
Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD (2003) The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol 28:574–581
Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG (2001) Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 280:L1168–L1178
Prasain N, Stevens T (2009) The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res 77:53–63
Rowlands DJ, Islam MN, Das SR et al (2011) Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J Clin Invest 121:1986–1999
Sawant DA, Tharakan B, Adekanbi A, Hunter FA, Smythe WR, Childs EW (2011) Inhibition of VE-cadherin proteasomal degradation attenuates microvascular hyperpermeability. Microcirculation 18:46–55
Sawant DA, Tharakan B, Hunter FA, Smythe WR, Childs EW (2011) Role of β-catenin in regulating microvascular endothelial cell hyperpermeability. J Trauma 70:481–487
Sawant DA, Tharakan B, Tobin RP, Reilly J, Hunter FA, Newell MK, Smythe WR, Childs EW (2013) Microvascular endothelial cell hyperpermeability induced by endogenous caspase 3 activator staurosporine. J Trauma Acute Care Surg 74:516–523
Shankar R, Melstrom KA Jr, Gamelli RL (2007) Inflammation and sepsis: past, present, and the future. J Burn Care Res 28:566–571
Tharakan B, Hellman J, Sawant DA, Tinsley JH, Parrish AR, Hunter FA, Smythe WR, Childs EW (2012) β-Catenin dynamics in the regulation of microvascular endothelial cell hyperpermeability. Shock 37:306–311
Tharakan B, Holder-Haynes JG, Hunter FA, Childs EW (2008) Alpha lipoic acid attenuates microvascular endothelial cell hyperpermeability by inhibiting the intrinsic apoptotic signaling. Am J Surg 195:174–178
Tharakan B, Hunter FA, Smythe WR, Childs EW (2008) Alpha-lipoic acid attenuates hemorrhagic shock-induced apoptotic signaling and vascular hyperpermeability. Shock 30:571–577
Vandenbroucke E, Mehta D, Minshall R, Malik AB (2008) Regulation of endothelial junctional permeability. Ann NY Acad Sci 1123:134–145
van Nieuw Amerongen GP, van Hinsbergh VW (2002) Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vasc Pharmacol 39:257–272
Acknowledgments
We acknowledge the Texas A&M Health Science Center College of Medicine Integrated Microscopy and Imaging Laboratory.
Funding
This work was supported by a grant (1K01HL07815-01A1) from National Heart, Lung and Blood Institute, National Institutes of Health, USA.
Conflict of interest
The authors have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sawant, D.A., Wilson, R.L., Tharakan, B. et al. Tumor necrosis factor-α-induced microvascular endothelial cell hyperpermeability: role of intrinsic apoptotic signaling. J Physiol Biochem 70, 971–980 (2014). https://doi.org/10.1007/s13105-014-0366-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-014-0366-8