
Abstract
Middle cerebral artery steno-occlusive disease (MCAD) has been recognized as a different clinical entity from moyamoya disease (MMD). Although MCAD can progress to MMD, the extent to which patients actually progress and the risk factors for this progression have not been fully elucidated. We retrospectively reviewed patients with MCAD who underwent RNF213 genotyping. Demographic features, RNF213 p.R4810K mutation, medical history, and longitudinal changes in angiography were analyzed. Sixty patients with 81 affected hemispheres were enrolled. During the follow-up period, 17 patients developed MMD, and the RNF213 p.R4810K mutation was the only factor significantly associated with progression to MMD (odds ratio, 16.1; 95% CI, 2.13–731; P = 0.001). The log-rank test demonstrated that patients with the mutation had a higher risk of progression to MMD (P = 0.007), stenosis progression (P = 0.010), and symptomatic cerebral infarction or hemorrhage (P = 0.026). In Cox regression analysis the p.R4810K mutation remained a significant factor after adjusting for age group (childhood or adult onset) at diagnosis (hazard ratio, 8.42; 95% CI, 1.10–64.4). Hemisphere-based analysis also showed that the mutation was associated with a higher risk of progression to the MMD hemisphere (P = 0.002), stenosis progression (P = 0.005), and cerebral infarction or hemorrhage (P = 0.012). The RNF213 p.R4810K mutation was identified as a risk factor for progression from MCAD to MMD. Genotyping for this mutation may contribute to risk stratification in MCAD.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Middle cerebral artery steno-occlusive disease (MCAD) is a vascular condition that causes ischemic stroke [1]. Although atherosclerosis is recognized as the most frequent cause of MCAD, it can also be an early manifestation of moyamoya disease (MMD) [2,3,4]. Some family members display MCAD in familial MMD, suggesting that MCAD in these cases may represent incomplete expression or an early phase of MMD [5, 6].
MMD is characterized by progressive stenosis at the terminal portion of the internal cerebral artery (ICA) and dilated perforating arteries, forming various fragile collaterals known as moyamoya vessels. Although the involvement of the terminal portion of the ICA is a distinct characteristic of MMD, current insights have indicated that the distinctive angiographic characteristics of MMD may not be present in its initial stages [7]. In MMD, cerebral hemodynamics are impaired, causing transient ischemic attack (TIA) and cerebral infarction, whereas the dilated, fragile moyamoya vessels sometimes rupture and cause hemorrhage [8, 9]. RNF213 p.R4810K mutation is the most common genetic risk factor for MMD in the East Asian populations [10, 11]. A recent study demonstrated that the mutation was also associated with quasi-MMD [12].
The Asymptomatic Moyamoya Registry study (AMORE study) designated a hemisphere with stenosis in the proximal portion of the middle cerebral artery (MCA) or anterior cerebral artery (ACA), whereas the ICA terminal portion remains intact, as a “questionable (Q)” hemisphere [13]. Compared with Q hemispheres, definitive moyamoya hemispheres are at a higher risk for cerebrovascular events. Moreover, revascularization surgery is more effective in patients with MMD than in those with other steno-occlusive diseases [14]. Therefore, it is beneficial to identify patients with early manifestations of MMD among those with MCAD. However, few studies have addressed the risk factors for MCAD development. We hypothesized that the p.R4810K mutation is associated with progression from MCAD (“Q” hemisphere) to definitive MMD. To test this hypothesis, we aimed to extensively investigate the risk factors for the development of MMD, including MMD and quasi-MMD, in patients with MCAD. This study will contribute to improving the management of patients with MCAD through risk stratification and to a better understanding of the progression patterns of MMD from MCAD.
Materials and Methods
Patient Population and Imaging Study Assessment
We retrospectively reviewed consecutive patients with intracranial artery stenosis at our institution, all of whom had been genotyped for the p.R4810K mutation in RNF213. The patients were initially diagnosed between December 2000 and December 2023.
We reviewed the clinical data and radiological examinations from the patient’s medical records, including 1.5- or 3.0-T magnetic resonance (MR) images and conventional angiography, if available. Radiological evaluation included the development of MMD, stenosis progression, and grade 2 periventricular anastomosis.
This study enrolled patients with MCAD who, at initial presentation, had stenosis in the M1 segment of the MCA, without any stenosis in the terminal portion of the ICA [15]. Stenosis in the terminal portion of the ICA was rigorously evaluated, adhering to established diagnostic criteria [16]. Patients presenting with a unilateral MCAD hemisphere and a contralateral moyamoya hemisphere were excluded from the study. For the included patients, each hemisphere was further categorized as “Q” or unaffected. According to the AMORE study, a “Q” hemisphere is defined as one exhibiting stenosis in the proximal portion of the MCA or ACA but not in the terminal of the ICA [13].
Two experienced neurosurgeons (TS and TF) independently evaluated the radiological examinations. The κ statistic was calculated between the two readers to assess the inter-rater reliability regarding the diagnosis of MMD development from MCAD [17]. Furthermore, a judgment committee comprising three neurosurgeons (TS, TF, and YM) reviewed and discussed the radiological examinations to confirm the final diagnoses, adhering to the official diagnostic criteria of the Research Committee on the Spontaneous Occlusion of the Circle of Willis from the Ministry of Health and Welfare, Japan [16]. In the evaluation of stenosis, the cerebral artery was categorized into nine predefined segments (basilar artery, right and left intracranial ICA, right and left A1–A2 segment of the ACA, right and left M1–M2 segment of the MCA, and right and left posterior cerebral arteries) [18]. Arterial stenosis was classified based on the validated grading scale as follows: grade 1, normal or mild stenosis (0–29% diameter stenosis); grade 2, moderate stenosis (30–69% diameter stenosis); grade 3, severe stenosis (70–99% diameter stenosis); and grade 4, occlusion (100% diameter stenosis) [19]. Progression was defined by an increase in stenosis degree by at least one grade or the development of de novo stenosis in a new segment [20].
The definition and classification of periventricular anastomosis were based on previous studies [21, 22]. Grade 2 periventricular anastomosis was characterized by a clear connection in the periventricular region between the perforating or choroidal arteries and the medullary or insular arteries. Anastomoses were classified into three subtypes: (1) lenticulostriate, beginning at the lenticulostriate artery; (2) choroidal, beginning at the anterior or posterior choroidal artery; and (3) thalamic, beginning at the thalamotuberal, thalamogeniculate, or thalamoperforating artery. In this study, unless specifically stated otherwise, “periventricular anastomosis” was classified as grade 2. For patients undergoing revascularization surgery, images acquired only before surgery were used for assessment because the surgery itself can potentially reduce antegrade blood flow and moyamoya vessels [23,24,25,26]. Patients who had undergone revascularization surgery before referral to our department were excluded.
Clinical Variables
We measured the following clinical variables as potential confounders: age at initial diagnosis, sex, antiplatelet use, family history of MMD, medical history (hypertension, diabetes mellitus, autoimmune diseases, and other associated diseases), smoking habits (current or past smoker vs. nonsmoker), drinking habits (everyday alcohol drinker vs. occasional or nondrinker), and first sign at onset (ischemic stroke, including TIA, intracranial hemorrhage, or asymptomatic) for all participants. Patients aged < 17 years were considered to have childhood-onset MMD. MMD, a disease of unknown etiology, requires differentiation from similar cerebrovascular lesions associated with underlying autoimmune diseases, referred to as quasi-MMD. However, in this study, to elucidate the role of RNF213 p.R4810K mutation in patients with MCAD, we included patients with underlying diseases that cause quasi-MMD. Outcome variables in this study were defined as follows: (1) development of radiological MMD, (2) stenosis progression, (3) incidence of symptomatic ischemic infarction or hemorrhage during the follow-up period, (4) revascularization surgery, and (5) detection of grade 2 PVAs at final imaging. Neither TIA nor perioperative events were included as outcomes. In patient-based analyses, if either hemisphere reaches a clinical outcome, that patient is considered to have experienced the outcome. This prevents the redundant counting of the same outcome for one patient.
Genotyping
Genomic DNA was obtained from peripheral blood samples using a DNA Blood Mini Kit (Qiagen). Genotyping of the RNF213 p.R4810K mutation was performed using TaqMan SNP Genotyping Assays (Applied Biosystems), as previously described [12].
Statistical Analyses
All statistical analyses were performed using the EZR software [27]. Log-rank tests were performed to estimate the cumulative rates and the risk of progression to MMD, stenosis progression, and development of symptomatic ischemic infarction or hemorrhage during the follow-up period. In patient-based analyses, Cox and multivariate logistic regressions were performed to adjust for covariates, such as age at diagnosis. Continuous variables were compared using the Mann–Whitney U test, and categorical variables were analyzed using Fisher’s exact test, as appropriate. Continuous data are expressed as medians (interquartile ranges [IQRs]). P values < 0.05 were considered statistically significant.
Results
Patient Demographics
A flow diagram of the patient inclusion process is shown in Fig. 1. In total, 250 patients with intracranial artery stenosis underwent RNF213 genotyping. Of these, 158 patients diagnosed with MMD were excluded from this study. Additionally, 32 patients were excluded because of stenosis in arteries other than the MCA or stenosis in the terminal portion of the ICA that was not diagnosed as MMD because moyamoya vessels were not observed. This resulted in 60 patients with MCAD being included in this study, comprising 81 with Q hemispheres and 38 with unaffected hemispheres. Among the Q hemispheres, 78 had stenosis or occlusion in the M1 segment, whereas three hemispheres exhibited isolated stenosis in the A1 segment with contralateral M1 involvement. One hemisphere was excluded from the evaluation because the intracranial arteries could not be assessed owing to occlusion of the ICA in the cervical segment. No patients underwent revascularization surgery before being referred to our hospital.

Selection process for patients with MCAD. MCAD, middle cerebral artery steno-occlusive disease
Clinical Outcomes of Patients with MCAD
Among the 60 patients with MCAD, 17 (28.3%) exhibited progression to MMD during a median follow-up period of 74 (IQR, 38–152) months. Concerning inter-rater agreement on MMD development, the κ statistic was 0.879 (95% confidence interval [CI], 0.746–1.00), demonstrating excellent reliability. Sixteen of the 17 patients who developed MMD (94.1%) had the RNF213 p.R4810K mutation, showing that the mutation was significantly associated with the progression from MCAD to MMD (odds ratio [OR] = 16.1; 95% confidence interval [CI], 2.13–731; P = 0.001) (Table 1). Of these patients, 38 (63.3%) were female, and the median follow-up duration was 74 (IQR, 38–152) months. Patients with the mutation tended to be younger at diagnosis than those with wild-type alleles, although this association was not statistically significant (P = 0.053). Thus, logistic regression analysis was performed to adjust for age at diagnosis, and the p.R4810K mutation was independently associated with the risk of progression to MMD after that adjustment (OR, 14.3; 95%CI, 1.72–120; P = 0.014).
The median follow-up periods were 68 (IQR, 37–140) months for the RNF213 wild-type group and 78 (IQR, 38–161) months for the mutation group (P = 0.506). In the mutation group, 36 patients were heterozygous, whereas one was homozygous for the mutation. Patients with the p.R4810K mutation had a significantly higher likelihood of stenosis progression (P = 0.002), required revascularization surgery (P = 0.008), and experienced cerebral infarction or hemorrhage (P = 0.011). In addition, the log-rank test indicated a significant difference in the development of MMD (P = 0.007), progression of stenosis (P = 0.010), and occurrence of cerebral infarction or hemorrhage (P = 0.026) during the follow-up (Fig. 2A–C). To determine whether the p.R4810K mutation was a significant risk factor after adjusting for childhood- or adult-onset, Cox proportional hazard analysis was performed after adjustment for age group at diagnosis. The p.R4810K mutation remained a significant predictor of progression to MMD, with a hazard ratio of 8.42 (95% CI, 1.10–64.4; P = 0.040, Table S1). In stratification analysis, in the adult-onset group (n = 51) there was a significant association between the mutation and disease progression to MMD (P = 0.021). Figure 3 presents three illustrative cases of patients with the p.R4810K heterozygous mutation and those with the wild-type alleles, in terms of disease progression to MMD.

Patient-based analyses of cumulative incidence: progression from MCAD to MMD, angiographic stenosis progression, and stroke associated with the p.R4810K mutation. Cumulative incidence for (A) the development of moyamoya disease, (B) progression of stenosis, and (C) cerebral infarction or hemorrhage during the follow-up period were compared between patients with the RNF213 p.R4810K mutation and those without it

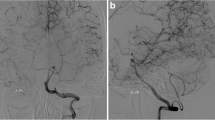
Illustrative cases demonstrating the effect of RNF213 p.R4810K mutation on progression from MCAD to MMD. A A 39-year-old man who do not have the RNF213 p.R4810K mutation presented with cerebral infarction and right MCA occlusion. No further cerebrovascular events occurred after the initiation of antiplatelet therapy. The MR angiography findings remained unchanged throughout a follow-up period of 12 years. B A 37-year-old woman carrying the heterozygous RNF213 p.R4810K mutation experienced a transient ischemic attack and was diagnosed with right MCA occlusion. Over a 5-year follow-up period, MCA stenosis progressed to moyamoya disease. Despite medication, the patient experienced frequent transient ischemic attacks and subsequently underwent STA-MCA anastomosis. C A 52-year-old man carrying the heterozygous RNF213 p.R4810K mutation experienced left cerebral infarction and was diagnosed with bilateral MCA stenosis. Over a 10-year follow-up period, left MCA stenosis progressed to moyamoya disease, and a recurrent cerebral infarction ultimately led to bypass surgery
We further compared patient characteristics and clinical outcomes between patients with the p.R4810K mutation (n = 37) and those with the wild-type alleles (n = 23, Table 2). Patients with the p.R4810K mutation were diagnosed at a median age of 37 (IQR, 26–47) years, which was significantly lower than those with the wild-type alleles, who had a median age of 47 (IQR, 30–69) years (P = 0.044).
Hemisphere-Based Analysis of Clinical Outcomes in Q and Unaffected Hemispheres
Of the 81 Q hemispheres, 34% (18/53) carrying the p.R4810K mutation progressed to moyamoya hemispheres, compared to only 3.6% (1/28) of those with wild-type alleles (P = 0.002, Table 3). Hemispheres carrying the mutation demonstrated stenosis progression (P = 0.002). Among Q hemispheres with the variant, the most frequent progression patterns were de novo stenosis in the ICA (26.4%), followed by increased stenosis severity in baseline MCA lesions (15.1%) (Table S2). Additionally, hemispheres with the mutation underwent revascularization surgery more frequently than those with wild-type alleles (P = 0.007; Table 3). Furthermore, grade 2 periventricular anastomoses were observed in 13 (24.5%) hemispheres with the p.R4810K mutation, which was significantly higher than that in one hemisphere (3.6%) with the RNF213 wild-type alleles (P = 0.028). Log-rank test also showed a significant difference in the development of MMD (P = 0.002), progression of stenosis (P = 0.005), and cerebral infarction or hemorrhage in each hemisphere (P = 0.012) between those with the wild-type alleles and those with the p.R4810K mutation (Fig. S1A–C).
Among the unaffected hemispheres, 21 out of 38 hemispheres harbored the p.R4810K mutation. In the p.R4810K mutation group, 2 (9.5%) hemispheres developed MMD, and 6 (28.6%) hemispheres exhibited progression of stenosis. In contrast, in the RNF213 wild-type group, no hemispheres developed MMD, and 1 (5.9%) showed stenosis progression (Table S3). The incidence rate of stenosis progression was higher in the p.R4810K mutation group than in the RNF213 wild-type alleles group, although this was not statistically significant in the log-rank test (P = 0.151) (Fig. S2).
Discussion
Our study revealed that the RNF213 p.R4810K mutation was significantly associated with MMD development in patients with MCAD (Q case). Actually, sixteen of 37 patients carrying the mutation developed MMD, whereas only 1 of 23 patients without the mutation developed it. We also showed that a substantial proportion of patients with MCAD develop MMD, although selection bias cannot be excluded. The results of the log-rank test further support an association between the p.R4810K variant and progression to MMD (P = 0.007). It had been reported that the p.R4810K variant is a risk factor for MCAD, but this study newly reveals its association with the progression from MCAD to MMD. Progression from MCAD to MMD had been described in case reports [3, 4], but we demonstrated that it is not an uncommon phenomenon. Recently, retrospective studies have shown that the RNF213 p.R4810K mutation is associated with contralateral progression in unilateral MMD [28, 29]. Analyzing 93 patients with unilateral MMD, Mineharu et al. revealed that the RNF213 p.R4810K mutation and contralateral abnormalities on the ACA or MCA were significantly correlated with the contralateral development of unilateral MMD [28]. Although the pathophysiological functions of the p.R4810K mutation remain unclear, these lines of evidence suggest that the mutation seems to be a clinically significant indicator for developing MMD in patients with cerebral artery stenosis [7, 30].
In addition to its association with the development of MMD, the RNF213 p.R4810K mutation is also associated with stenosis progression. A recent study reviewed 52 patients with cerebral artery stenosis without MMD and found that patients carrying the RNF213 p.R4810K mutation had a higher risk of stenosis progression [20]. Consistent with this report, our data showed that the p.R4810K mutation provided a significantly higher risk of stenosis progression in patients with MCAD compared with the RNF213 wild type (P = 0.010). The most common progression pattern was de novo stenosis in the ICA, indicating that lesions in patients with MCAD typically progress proximally, ultimately leading to the development of MMD. Furthermore, the unaffected hemispheres on initial imaging exhibited progression to the moyamoya hemisphere or de novo stenosis. Among patients with the p.R4810K mutation, 2 of 21 unaffected hemispheres (9.5%) developed MMD and 6 showed de novo stenosis (28.6%) during the median follow-up period of 71 months. An observational longitudinal study involving healthy family members of patients with MMD reported that 3 of 11 individuals (27.3%) experienced de novo stenosis during a median follow-up period of 7 years [5]. This indicates that even the unaffected hemisphere possesses a relatively high risk of cerebral artery steno-occlusive lesions in patients with the RNF213 R4810K mutation.
MCAD is a well-established vascular lesion causing ischemic stroke; thus, evaluating underlying pathophysiology would be beneficial to determine the clinical management of patients [1, 2]. Patients with low atherosclerosis burden also experience MCAD, but their pathogenesis has long been unknown [31]. However, recent studies using high-resolution MRI have shown that non-atherosclerotic MCA stenosis occurs frequently in individuals heterozygous for the RNF213 p.R4810K mutation [30,31,32]. Moreover, patients with the p.R4810K mutation in our MCAD cohort were diagnosed at a younger age compared with those without the mutation. Our results are consistent with the result of a recent study demonstrating that the RNF213 p.R4810K mutation is common in early-onset ischemic stroke with M1 or A1 stenosis [33]. The RNF213 p.R4810K mutation is considered a risk factor for anterior circulation stenosis, especially in younger patients with a lower burden of atherosclerosis [32].
In the current study, the RNF213 p.R4810K mutation significantly increased the risk of cerebrovascular events (symptomatic infarction or hemorrhage) in MCAD in both patient-based and hemisphere-based analyses. Revascularization surgery was required in 19 (35.8%) hemispheres carrying the mutation, compared to only 2 (7.1%) hemispheres with wild-type alleles. Consistently, in a recent retrospective study, MMD patients with the p.R4810K mutation often exhibited symptoms in both hemispheres, while those with wild-type alleles were more likely to only exhibit symptoms in one hemisphere [34]. Although different from MCAD cases, the AMORE study prospectively followed 109 patients with asymptomatic MMD and reported that symptomatic infarction or hemorrhage occurred in 4.9% (7/143) of moyamoya hemispheres compared to 0% (0/39) of Q hemispheres during a 5-year observation period. This is consistent with our data showing that the RNF213 mutation increased the risk of progression to moyamoya hemispheres, as well as symptomatic stroke. Although the AMORE study did not investigate RNF213 mutations, the potential association is an interesting topic for further research.
Our data demonstrate that the RNF213 p.R4810 mutation is significantly associated with periventricular anastomosis. Grade 2 periventricular anastomoses were detected in 13 of 53 affected hemispheres in patients carrying the mutation, compared with only 1 of 28 hemispheres in patients without the mutation (P = 0.028). Periventricular anastomosis is classified into three subtypes—lenticulostriate, choroidal, and thalamic—and is a substantial risk factor for cerebrovascular events in MMD [35,36,37]. Previously, Funaki et al. demonstrated that the marked development of choroidal anastomosis was closely associated with posterior hemorrhage in the territory of the choroidal artery [35,36,37]. Additionally, other types of anastomoses have also frequently been considered the vessels responsible for bleeding [25, 38]. Among patients with MMD, a recent study demonstrated that the p.R4810K mutation harbored a significantly higher risk of periventricular anastomosis development than RNF213 wild type [39].
Stenosis of the terminal portion of the ICA is an essential diagnostic characteristic of MMD. MCAD with intact terminal ICA (Q hemisphere) has a better prognosis than MMD, and MCAD may represent an etiology different from MMD. In contrast, in the current study, some patients with MCAD with the RNF213 p.4810 K mutation exhibited progression to definitive MMD during the follow-up period. According to current insights, the distinctive angiographic characteristics of MMD may not be present in its initial stages, particularly in adults [7]. Based on imaging techniques or patient characteristics, distinguishing MCA steno-occlusive lesions as early manifestations of MMD from other vascular pathologies remains challenging. Genotyping of RNF213 potentially contributes to the differentiation of patients with early-stage MMD from those with MCAD. In patients with the p.R4810K mutation, close clinical follow-up is required for the early detection of stenosis progression or the development of periventricular anastomosis.
This study has some limitations. First, our findings were limited by the relatively small size of our patient cohort. Therefore, subsequent studies with larger sample sizes are required to validate our results. Second, we did not systematically analyze the RNF213 p.R4810K mutation in all patients with MCAD presenting at our institutions, potentially introducing selection bias and affecting the reported incidence rate of MMD progression. A multicenter prospective cohort study is warranted to confirm the validity of our results. Third, the prevalence of the RNF213 p.R4810K mutation in patients with intracranial artery stenosis, including MMD, varies with ethnicity [40]. Therefore, the external validity of our findings may be influenced by the patient’s ethnic background.
Conclusions
Our results suggest that RNF213 p.R4810K mutation is associated with progression from MCAD (Q hemisphere) to definitive MMD and the incidence of cerebrovascular events. Genotyping RNF213 may contribute to risk stratification in patients with MCAD and adequate management, such as close follow-up. Further studies are required to confirm the clinical significance of RNF213 genotyping in MCAD.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request from any investigator.
Abbreviations
- ACA:
-
Anterior cerebral artery
- ICA:
-
Internal cerebral artery
- MCA:
-
Middle cerebral artery
- MCAD:
-
Middle cerebral artery steno-occlusive disease
- MMD:
-
Moyamoya disease
- Q:
-
Questionable
References
Korogi Y, Takahashi M, Nakagawa T, Mabuchi N, Watabe T, Shiokawa Y, et al. Intracranial vascular stenosis and occlusion: MR angiographic findings. Am J Neuroradiol. 1997;18:135–43.
Telman G, Hurani H, Sprecher E, Kouperberg E. Middle cerebral artery stenosis in patients with acute ischemic stroke and TIA in Israel. Am J Neuroradiol. 2015;36:46–9.
Shimoda Y, Fujimura M, Inoue T, Shimizu H, Tominaga T. Temporal profile of de novo development of moyamoya vasculopathy in an adult. Neurol Med Chir (Tokyo). 2012;52:339–42.
Choi HY, Lee JE, Jung YH, Cho HJ, Kim DJ, Heo JH. Progression of isolated middle cerebral artery stenosis into moyamoya disease. Neurology. 2007;68:954.
Matsuda Y, Mineharu Y, Kimura M, Takagi Y, Kobayashi H, Hitomi T, et al. RNF213 p. R4810K Variant and intracranial arterial stenosis or ccclusion in relatives of patients with moyamoya disease. J Stroke Cerebrovasc Dis. 2017;26:1841–7. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.04.019.
Mineharu Y, Takenaka K, Yamakawa H, Inoue K, Ikeda H, Kikuta KI, et al. Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting. J Neurol Neurosurg Psychiatry. 2006;77:1025–9.
Bang OY, Chung JW, Kim DH, Won HH, Yeon JY, Ki CS, et al. Moyamoya disease and spectrums of RNF213 vasculopathy. Transl Stroke Res. 2020;11:580–9.
Suzuki JTA. Cerebrovascular “moyamoya” disease: disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20:288–99.
Takahashi JC, Miyamoto S. Moyamoya disease: recent progress and outlook. Neurol Med Chir (Tokyo). 2010;50:824–32.
Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, Kikuchi A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56:34–40.
Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One. 2011;6:e22542.
Morimoto T, Mineharu Y, Kobayashi H, Harada KH, Funaki T, Takagi Y, et al. Significant association of the RNF213 p. R4810K polymorphism with quasi-moyamoya disease. J Stroke Cerebrovasc Dis. 2016;25:2632–6. https://doi.org/10.1016/j.jstrokecerebrovasdis.2016.07.004.
Kuroda S, Yamamoto S, Funaki T, Fujimura M, Kataoka H, Hishikawa T, et al. Five-year stroke risk and its predictors in asymptomatic moyamoya disease: asymptomatic moyamoya registry (AMORE). Stroke. 2023;54:1494–504.
Fujimura M, Tominaga T, Kuroda S, Takahashi JC, Endo H, Ogasawara K, et al. 2021 Japanese Guidelines for the Management of Moyamoya Disease: Guidelines from the Research Committee on Moyamoya Disease and Japan Stroke Society. Neurol Med Chir (Tokyo). 2022;62:165–70.
Gibo H, Carver CC, Rhoton AL Jr, Lenkey CMR. Microsurgical anatomy of the middle cerebral artery. J Neurosurg. 1981;54:151–69.
Kuroda S, Fujimura M, Takahashi J, Kataoka H, Ogasawara K, Iwama T, et al. Diagnostic criteria for moyamoya disease-2021 revised version. Neurol Med Chir. 2022;62:307–12.
McHugh ML. Interrater reliability: the kappa statistic. Biochem Medica. 2012;22:276–82.
Samuels OB, Joseph GJ, Lynn MJ, Smith HA, Chimowitz MI. A standardized method for measuring intracranial arterial stenosis. Am J Neuroradiol. 2000;21:643–6.
Lavados P, Oppenheimer S, Enger C, Wong KS, Lam WWM. Variability of magnetic resonance angiography and computed tomography angiography in grading middle cerebral artery stenosis using the unweighted kappa statistic [4]. Stroke. 1996;27:2340.
Okazaki S, Yoshimoto T, Ohara M, Takagaki M, Nakamura H, Watanabe K, et al. Effect of the RNF213 p.R4810K variant on the progression of intracranial artery stenosis. Neurol Genet. 2022;8:e200029.
Funaki AT, Takahashi JC, Yoshida K, Takagi Y, Fushimi Y, Kikuchi T, et al. Periventricular anastomosis in moyamoya disease: detecting fragile collateral vessels with MR angiography. J Neurosurg. 2016;124:1766–72.
Funaki T, Fushimi Y, Takahashi JC, Takagi Y, Araki Y, Yoshida K, et al. Visualization of periventricular collaterals in moyamoya disease with flow-sensitive black-blood magnetic resonance angiography: preliminary experience. Neurol Med Chir. 2015;55:204–9.
Funaki T, Miyakoshi A, Kataoka H, Takahashi JC, Takagi Y, Yoshida K, et al. Larger posterior revascularization associated with reduction of choroidal anastomosis in moyamoya disease: a quantitative angiographic analysis. Am J Neuroradiol. 2022;43:1279–85.
Connolly F, Alsolivany J, Czabanka M, Vajkoczy P, Valdueza JM, Röhl JE, et al. Blood volume flow in the superficial temporal artery assessed by duplex sonography: predicting extracranial-intracranial bypass patency in moyamoya disease. J Neurosurg. 2021;135:1666–73.
Sasagasako T, Funaki T, Tanji M, Arakawa Y, Suzuki H, Miyakoshi A, et al. Intractable medial anastomotic branches from the lenticulostriate artery causing recurrent hemorrhages in moyamoya disease. World Neurosurg. 2019;127:279–83. https://doi.org/10.1016/j.wneu.2019.04.066.
Funaki T, Kataoka H, Yoshida K, Kikuchi T, Mineharu Y, Okawa M, et al. The targeted bypass strategy for preventing hemorrhage in moyamoya disease: technical note. Neurol Med Chir. 2019;59:517–22.
Kanda Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 2013;48:452–8.
Mineharu Y, Takagi Y, Koizumi A, Morimoto T, Funaki T, Hishikawa T, et al. Genetic and nongenetic factors for contralateral progression of unilateral moyamoya disease: the first report from the SUPRA Japan Study Group. J Neurosurg. 2022;136:1005–14.
Ok T, Jung YH, Kim J, Park SK, Park G, Lee S, et al. RNF213 R4810K variant in suspected unilateral moyamoya disease predicts contralateral progression. J Am Heart Assoc. 2022;11(15):e025676.
Asselman C, Hemelsoet D, Eggermont D, Dermaut B, Impens F. Moyamoya disease emerging as an immune-related angiopathy. Trends Mol Med. 2022;28:939–50. https://doi.org/10.1016/j.molmed.2022.08.009.
Kim YJ, Lee JK, Ahn SH, Kim BJ, Kang DW, Kim JS, et al. Nonatheroscleotic isolated middle cerebral artery disease may be early manifestation of moyamoya disease. Stroke. 2016;47:2229–35.
Bang OY, Toyoda K, Arenillas JF, Liu L, Kim JS. Intracranial large artery disease of non-atherosclerotic origin: recent progress and clinical implications. J Stroke. 2018;20:208–17.
Kamimura T, Okazaki S, Morimoto T, Kobayashi H, Harada K, Tomita T, et al. Prevalence of RNF213 p. R4810K variant in early-onset stroke with intracranial arterial stenosis. Stroke. 2019;50:1561–3.
Ishigami D, Miyawaki S, Imai H, Shimizu M, Hongo H, Dofuku S, et al. RNF213 p.Arg4810Lys heterozygosity in moyamoya disease indicates early onset and bilateral cerebrovascular events. Transl Stroke Res. 2022;13:410–9.
Funaki T, Takahashi JC, Houkin K, Kuroda S, Fujimura M, Tomata Y, et al. Effect of choroidal collateral vessels on de novo hemorrhage in moyamoya disease: analysis of nonhemorrhagic hemispheres in the Japan Adult Moyamoya Trial. J Neurosurg. 2020;132:408–14.
Funaki T, Takahashi JC, Houkin K, Kuroda S, Takeuchi S, Fujimura M, et al. High rebleeding risk associated with choroidal collateral vessels in hemorrhagic moyamoya disease: analysis of a nonsurgical cohort in the Japan Adult Moyamoya Trial. J Neurosurg. 2019;130:525–30.
Funaki T, Takahashi JC, Houkin K, Kuroda S, Takeuchi S, Fujimura M, et al. Angiographic features of hemorrhagic moyamoya disease with high recurrence risk: a supplementary analysis of the Japan Adult Moyamoya Trial. J Neurosurg. 2018;128:777–84.
Miyakoshi A, Funaki T, Fushimi Y, Kikuchi T, Kataoka H, Yoshida K, et al. Identification of the bleeding point in hemorrhagic moyamoya disease using fusion images of susceptibility-weighted imaging and time-of-flight MRA. Am J Neuroradiol. 2019;40:1674–80.
Xue Y, Zeng C, Ge P, Liu C, Li J, Zhang Y, et al. Association of RNF213 variants with periventricular anastomosis in moyamoya disease. Stroke. 2022;53:2906–16.
Koizumi A, Kobayashi H, Hitomi T, Harada KH, Habu T, Youssefian S. A new horizon of moyamoya disease and associated health risks explored through RNF213. Environ Health Prev Med. 2016;21:55–70.
Funding
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (KAKENHI 17H06397, 19H03770) and from Japan Agency for Medical Research and Development (JP21bm0804032h0001, JP22bm0804032h0002, JP23bm0804032h0003).
Author information
Authors and Affiliations
Contributions
Author contributions Conception: T.F.; Study design: T.S., Y.M., and T.F.; acquisition of data: T.S., Y.M., T.F., and Y.F.; analysis and interpretation of data: all authors; drafting of the article: T.S.; critical review of the article: all authors; administrative/technical/material support: Y.F., S.P., M.O., and T.K.; and study supervision: Y.A. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
This study was approved by the Ethics Committee of Kyoto University Hospital (G1109 and R2088). All participants or their guardians provided written informed consent (and assent if possible) after receiving a detailed explanation of the study. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sasagasako, T., Mineharu, Y., Funaki, T. et al. RNF213 Mutation Associated with the Progression from Middle Cerebral Artery Steno-Occlusive Disease to Moyamoya Disease. Transl. Stroke Res. (2024). https://doi.org/10.1007/s12975-024-01293-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12975-024-01293-2