
Abstract
Peroxynitrite (ONOO−) and high mobility group box 1 protein (HMGB1) are important cytotoxic factors contributing to cerebral ischemia-reperfusion injury. However, the roles of ONOO− in mediating HMGB1 expression and its impacts on hemorrhagic transformation (HT) in ischemic brain injury with delayed t-PA treatment remain unclear. In the present study, we tested the hypothesis that ONOO− could directly mediate the activation and release of HMGB1 in ischemic brains with delayed t-PA treatment. With clinical studies, we found that plasma nitrotyrosine (NT, a surrogate marker of ONOO−) was positively correlated with HMGB1 level in acute ischemic stroke patients. Hemorrhagic transformation and t-PA-treated ischemic stroke patients had increased levels of nitrotyrosine and HMGB1 in plasma. In animal experiments, we found that FeTmPyP, a representative ONOO− decomposition catalyst (PDC), significantly reduced the expression of HMGB1 and its receptor TLR2, and inhibited MMP-9 activation, preserved collagen IV and tight junction claudin-5 in ischemic rat brains with delayed t-PA treatment. ONOO− donor SIN-1 directly induced expression of HMGB1 and its receptor TLR2 in naive rat brains in vivo and induced HMGB1 in brain microvascular endothelial b.End3 cells in vitro. Those results suggest that ONOO− could activate HMGB1/TLR2/MMP-9 signaling. We then addressed whether glycyrrhizin, a natural HMGB1 inhibitor, could inhibit ONOO− production and the antioxidant properties of glycyrrhizin contribute to the inhibition of HMGB1 and the neuroprotective effects on attenuating hemorrhagic transformation in ischemic stroke with delayed t-PA treatment. Glycyrrhizin treatment downregulated the expressions of NADPH oxidase p47 phox and p67 phox and iNOS, inhibited superoxide and ONOO− production, reduced the expression of HMGB1, TLR2, MMP-9, preserved type IV collagen and claudin-5 in ischemic brains. Furthermore, glycyrrhizin significantly decreased the mortality rate, attenuated hemorrhagic transformation, brain swelling, blood-brain barrier damage, neuronal apoptosis, and improved neurological outcomes in the ischemic stroke rat model with delayed t-PA treatment. In conclusion, peroxynitrite-mediated HMGB1/TLR2 signaling contributes to hemorrhagic transformation, and glycyrrhizin could be a potential adjuvant therapy to attenuate hemorrhagic transformation, possibly through inhibiting the ONOO−/HMGB1/TLR2 signaling cascades.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tissue plasminogen activator (t-PA) remains the only FDA approved thrombolytic drug for ischemic stroke but carries a restrictive therapeutic time window of 4.5 h [1, 2]. Treatment beyond this time window increases the risk of hemorrhagic transformation (HT) [3,4,5,6]. During this process, activation of matrix metalloproteinases (MMPs) is a crucial pathological process to degrade tight junction proteins and mediate the blood-brain barrier (BBB) damage and hemorrhagic transformation [7, 8]. Independent investigators reported that inhibition of MMP-9 attenuated the incidence and severity of hemorrhagic transformation and reduced the mortality rates in rodent focal cerebral ischemia models with delayed t-PA treatment [7, 9, 10]. How MMP-9 activation is induced in ischemic stroke remains largely unknown.
High mobility group box protein 1 (HMGB1), a non-histone DNA-binding protein, could be a key player in activating MMPs and cytokines and inducing the BBB disruption and hemorrhagic transformation in cerebral ischemia-reperfusion injury with delayed thrombolytic treatment. HMGB1 can be released from necrotic neurons and promoted MMP-9 activation via binding to its receptors including RAGE, TLR2, and TLR4 [11,12,13]. Anti-HMGB1 antibody suppressed the expressions and activities of MMP-9, TNF-α, and iNOS; protected the BBB integrity; attenuated infarct volume; and improved neurological outcomes in rodent ischemic stroke models [14, 15]. We recently reported that HMGB1-binding heptamer peptide significantly inhibited HMGB1 and attenuated hemorrhagic transformation in ischemic brains with delayed t-PA treatment [16]. However, the underlying mechanisms of HMGB1 induction and release in cerebral ischemia-reperfusion injury under delayed t-PA treatment remain unclear yet.
Recanalization with delayed t-PA infusion leads to produce large amounts of reactive oxygen and nitrogen species including superoxide (O2·−), nitric oxide (NO), and peroxynitrite (ONOO−), subsequently mediating MMP activation and aggravating the BBB damage and hemorrhagic transformation in cerebral ischemia-reperfusion injury [17,18,19,20,21]. ONOO− is derived from O2·− and NO and has much higher cytotoxicity than those precursors [22]. The formation of ONOO− was concurrent with the expression of inducible nitric oxide synthase (iNOS), induced tyrosine nitration, activated MMPs, and disrupted tight junctions and neurovascular units in ischemic brains, aggravating cerebral ischemia-reperfusion injury [22,23,24]. In our recent studies, we found that peroxynitrite decomposition catalyst (PDC) could prevent hemorrhagic transformation and improve the neurological outcome of the ischemic rats with delayed t-PA treatment via inhibiting ONOO−-mediated MMP activation [25, 26]. It is of interest to address whether ONOO− could mediate the expression and release of HMGB1 and exacerbate neurological damage during cerebral ischemia-reperfusion injury.
ONOO− appears to be a mediator to induce expression and release of HMGB1. ONOO− induced overexpression of myocardial HMGB1, whereas FeTPPS, a representative PDC, reduced nitrotyrosine formation, suppressed HMGB1 overexpression, and decreased infarct size in myocardial ischemia-reperfusion injury [27]. The HMGB1-mediated inflammatory pathway was RAGE and redox signaling dependent, promoting ectopic intestinal inflammation [28]. Glycyrrhizin (GL), a triterpene isolated from the root of Glycyrrhiza glabra, is considered as BBB permeable HMGB1 inhibitor [29,30,31]. Glycyrrhizin had anti-inflammation, antioxidant, and antiexcitotoxic effects [31, 32]. Previous studies revealed that glycyrrhizin attenuated infarction volume and improved neurological outcomes in middle cerebral artery occlusion (MCAO) ischemic stroke models [31,32,33]. Therefore, in the present study, we tested the hypothesis that (1) ONOO− could directly mediate the activation and release of HMGB1 in ischemic brains with delayed t-PA treatment; (2) the neuroprotective effects of glycyrrhizin could be related to reducing the production of ONOO− and inhibiting the ONOO− induced HMGB1 expression and subsequently attenuate hemorrhagic transformation in ischemic stroke with delayed t-PA treatment.
Material and Methods
Ischemic Stroke Patients and Clinical Investigation
We conducted two independent clinical investigations to verify the roles of ONOO− and HMGB1 in ischemic stroke patients. In the first clinical investigation, we studied the correlation of nitrotyrosine (NT, a surrogate marker of ONOO−) and HMGB1 release in ischemic stroke patients. A total of 72 acute ischemic stroke patients were recruited from Huizhou First People’s Hospital, Guangdong Province, China. Both male and female stroke patients aged 40–80 years old were included in this study. Inclusion criteria of the ischemic stroke patients were as follows: (a) primary confirmed diagnosis of ischemic stroke (NIHSS > 0 or symptoms or CT/MRI detected infarction area and (b) those who are competent to give written informed consent. Exclusion criteria include (a) patients with cancer; (b) patients with transient ischemic attack (TIA) or intracranial hemorrhage or subarachnoid hemorrhage (SAH) within 6 months; (c) patients with hepatic, renal and hematologic diseases; (d) patients with mental disorders or serious dementia; (e) major surgery operation within 6 months; (f) pregnancy; and (g) epilepsy. Blood samples were collected within 24 h after ischemia onset. Baseline information of patients was described in Supplementary 1. In the second clinical study, we investigated the expression of HMGB1 in ischemic stroke patients with or without t-PA treatment. To confirm the roles of ONOO− and HMGB1 in mediating hemorrhagic transformation, we also investigated the expression of NT and HMGB1 in ischemic stroke patients with or without hemorrhagic transformation. The second clinical study was conducted at Xuzhou Medical University with the same inclusion criteria and exclusion criteria as the first clinical study for stroke patients. For the healthy control group, the inclusion criteria are as follows: (a) age older than 55, (b) no cardiovascular and cerebrovascular diseases; (c) normal liver and kidney function; (d) normal blood glucose and lipid level; (e) no major surgery within 6 months; and (f) no other basal metabolic diseases. We recruited a total of 13 control and 56 stroke patients in the second clinical study and analyzed the level of HMGB1 and NT in patients with or without t-PA treatment, with or without hemorrhagic transformation. Hemorrhagic transformation was determined by CT/MRI detection in ischemic stroke patients. Baseline characteristics of ischemic stroke patients and the healthy control group of this study are attached in Supplementary 2. In this study, the admission time point to the hospital is within 10 h from ischemic onset. Patients who received t-PA were treated within t-PA infusion within 4.5 h after ischemia onset. Detail information about the sample collection was attached in Supplementary 3.
All of the research protocols and procedures were approved by the Ethics Committee of Hong Kong (UW14-319). Informed consent was obtained from the patients or the legal guardian before enrollment. Blood samples were centrifuged at 1000g for 10 min, and the plasma samples were stored at − 80 °C. Plasma nitrotyrosine (NT, a surrogate biomarker of ONOO−) and HMGB1 levels were determined by commercially available ELISA kit (Uscn, Wuhan, China). Diluent standards/blank/plasma samples (50 μl) were added into the ELISA plate and incubated at 37 °C for 30 min. The remaining liquid from all wells was removed completely, and the wells were washed 5 times by auto-washer (Langpu, DNX-9620, China). Then, HRP conjugated anti-HMGB1 or anti-nitrotyrosine antibody (50 μl) was added into each well (except the blank) and incubated at 37 °C for 30 min with sealer. After washing 5 times, TMB substrate A (50 μl) and TMB substrate B (50 μl) were added into each well, followed by incubation at 37 °C for 15 min. Lastly, stop solution (50 μl) was added into each well and mixed thoroughly. The OD value of each sample was detected by the microplate reader (Tecan Infinite F50, Switzerland) with the measurement at 450 nm immediately.
MCAO Model
Male Sprague-Dawley rats weighing from 290 to 310 g were obtained from the Laboratory Animal Unit, The University of Hong Kong. The animal experiment was conducted in accordance with the national and institutional guidelines on ethics and biosafety, and the protocol was approved and regulated by the Committee on the Use of Live Animals in Teaching and Research (CULATR), The University of Hong Kong. In total, 110 rats were used in this study. Animals were housed in a temperature and humidity-controlled environment under a 12-h light/dark cycle, with free access to food and water.
We adopted the experimental stroke model with 5 h of MCAO plus reperfusion 19 h as we previously described [26]. Briefly, rats were anesthetized with 4% isofluorane and maintained at 2% isofluorane via inhalation. The left common carotid artery (CCA) was isolated from the surrounding nerve and clipped transiently using a microvascular clip. Silicon-coated suture (Doccol, Redlands, CA, USA) with a tip diameter of 0.38 mm was inserted via the lumen of the left external carotid artery (ECA) to the internal carotid artery (ICA), until its tip occlude the origin of the left middle cerebral artery (MCA). Sham control rats underwent the same anesthesia and surgical procedure without MCA occlusion. Rats were provided with a warm pad during the procedure, and the body temperature was monitored at 37 °C. After 5-h occlusion, the suture was removed and the CCA was released to allow reperfusion. The success of MCAO was confirmed by using 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) staining method as reported before [34]. In detail, we used TTC staining on a 1-mm-thick coronal slice at 5-mm away from the frontal tip, an area considered as the most injured brain region. The inclusion criteria for the success of the MCAO model was defined as the infarction in both cortex and striatum at this slice. The exclusion criteria of the MCAO model included (1) subarachnoid hemorrhage and (2) no infarction at the cortex. We got a 100% success rate in the MCAO model.
Experimental Designs and Drug Treatment
We randomly divided the rats into following seven groups: Sham, MCAO 5 h plus reperfusion 19 h (M5/R19, labeled as vehicle), M5/R19 plus t-PA (labeled as t-PA), M5/R19 plus t-PA and glycyrrhizin (15, 30, 60 mg/kg) (labeled as t-PA + GL), M5/R19 plus t-PA, and peroxynitrite decomposition catalyst (PDC) (3 mg/kg) (labeled as t-PA + PDC). Based on the hemorrhagic transformation results, we chose the dosage of glycyrrhizin at 30 mg/kg for further investigation. From our previous study and our pilot study, the number of rats per group was determined to be at least 6 to reject the null hypothesis (H0) at the 0.05 level at a power of 0.8. Given that the mortality rate could fluctuate among studies, we used at least ten animals for each group in this study. According to our previous protocol, t-PA (10 mg/kg dissolved in 1 ml saline, Actilyse, Boehringer Ingelheim) was administrated at 4.5 h of MCAO ischemia with 10% bolus, following 90% continuous infusion for 0.5 h via the femoral vein, thus defined t-PA infusion time as 5 h after MCAO [26]. Glycyrrhizin (Sigma, St. Louis, MO, USA) was dissolved in 0.5 ml saline, and ammonium hydroxide was added to achieve the pH value of 7.4 for assisting dissolution. Glycyrrhizin was administrated via the femoral vein at MCAO 4.5 h just before t-PA treatment. In a parallel experiment, the same volume of saline was used as a control. FeTmPyP (Cayman) was dissolved in saline and administrated at a dosage of 3 mg/kg via a femoral vein just before t-PA treatment. The suture was removed to allow reperfusion after finishing t-PA injection. We conducted the animal surgery, grouping, and outcome measurements in a blinded way in this study. The blinding procedure was as follows: one investigator performed the MCAO surgery, another investigator gave treatments, and a third one evaluated the neurological outcomes and hemorrhagic scores, in the absence of previous investigators.
Mortality Rates and Neurological Severity Scores
Mortality rates were calculated at 24 h after MCAO as a parameter of t-PA study. Rats died at the reperfusion stage within 24 h were excluded for other outcome parameter analysis. The neurological score was evaluated by the modified neurological severity score (mNSS) scale as previously described [35]. The mNSS scale is a composite test on motor, reflex, and balance. Neurological functions were graded on a series of scales from 0 to 18. Details of the scoring could be found in Supplementary 4. A higher score represents more severe neurological deficits. An investigator blinded to the experimental designs performed the mNSS test.
Brain Edema
After transcardial perfusion with PBS, brains were cut into 2-mm-thick coronal brain slices. Digital pictures of slices were captured and brain edema was analyzed with ImageJ software (National Institutes of Health) as a relative increase of the ischemic side volume against the non-ischemic side, as described in the previous study [36].
BBB Permeability
The BBB permeability was detected with Evans blue (EB) leakage assay [26]. Briefly, rats were anesthetized with 4% isoflurane and injected via tail vein with 2% EB (3 ml/kg, Sigma) at 1 h before sacrifice. Rats were perfused transcardially with 250 ml PBS to remove the circulating EB. Brain tissue was harvested and cut into serial 2-mm-thick coronal sections for digital capture of EB extravasation. Tissues were then divided into ischemic and non-ischemic sides, weighed and stored at − 80 °C. We homogenized the brain tissues with cold PBS and further homogenized it with the same volume of 50% trichloroacetic acid (Sigma). Supernatants were collected after centrifuging at 15,000 rpm for 20 min. OD values of supernatants were measured by a microplate reader with a wavelength of 620 nm (Bio-Rad, Hercules, CA, USA). Gradient concentrations of EB standard curve were used to calculate the quantity of EB. The amount of extravagated EB dye was quantified as μg/g of brain tissue.
Hemorrhagic Transformation
To assess hemorrhagic transformation, we detected both hemoglobin content and the hemorrhagic scores in the ischemic hemispheres [37]. For hemoglobin assay, each hemisphere was collected and stored at − 80 °C. Tissue was homogenized with 1 ml cold PBS and sonicated on ice for 30 s, followed by centrifuging at 15,000g for 15 min. The supernatant was collected to detect the hemoglobin level by using the hemoglobin assay kit (BioAssay Systems, Hayward, CA, USA) according to manufacturer’s instruction. The OD value of each sample was determined with a microplate reader at a wavelength of 400 nm (Bio-Rad). Concentration of hemoglobin in each sample was calculated as (ODsample − ODblank)/(ODstandard − ODblank) × 200 (mg/dL).
We adopted hemorrhagic scores as another parameter of hemorrhagic transformation. Rat brains were cut into 2-mm-thick coronal slices and digital pictures were captured. The severity of hemorrhages was macroscopically classified as follows: (1) non-hemorrhage, score 0; (2) hemorrhagic infarction type 1 (HI-1), defined as small petechiae, score 1; (3) hemorrhagic infarction type 2 (HI-2), with more confluent petechiae within the damaged area, score 2; (4) parenchymal hemorrhage type 1 (PH-1), characterized by blood occupied less than 30% of the injured tissue, score 3; and (5) parenchymal hemorrhage type 2 (PH-2) characterized by blood occupied in more than 30% of the infarct, score 4. Scores of each slice were added up as the total hemorrhagic score of each rat.
TUNEL Assay
Brain apoptotic cell death was detected by using a TUNEL staining assay. Brain tissues at 6 mm away from the frontal tips were collected at 24 h after MCAO, fixed with 4% paraformaldehyde (PFA) and then immersed in 30% sucrose. Samples were then embedded with tissue freezing medium (Leica) and made into the frozen sections. Cell apoptosis of those sections was detected by using a TUNEL assay kit (Shanghai YEASEN Biotechnology Co) according to the manufactures’ instruction. DAPI (4′,6-diamidino-2-phenylindole) stain was applied for visualizing the cell nucleus. Fluorescence images were obtained by using a fluorescence microscope (Carl Zeiss) with Axio Vision digital imaging system. Images were taken from the cortex and striatum.
Detection of Superoxide (O2·−) and Peroxynitrite (ONOO−) in Ischemic Brains
We investigated the effects of glycyrrhizin on scavenging O2·− and ONOO− in the ischemic rat brains. The brain tissues at about 6 mm away from the frontal tips of each group were collected at 24 h after ischemia onset and immediately subjected to the frozen section, and stained with our newly developed ONOO− specific probe HKYellow-AM (20 μM) [38] or superoxide probe HEt (20 μM) (Thermo Fisher Scientific) for 30 min at room temperature. The samples were then fixed with 4% PFA, and the fluorescent images were obtained by using confocal microscope Carl Zeiss LSM 780. Fluorescent images were obtained from both the cortex and striatum of the brain tissue.
Immunofluorescent Image
Immunofluorescence study was conducted to detect the expression of 3-nitrotyrosine (3-NT), HMGB1, MMP-9, claudin-5, and collagen IV. At 24 h after ischemia onset, rats were sacrificed and brain tissues at about 6 mm away from the frontal tips were collected for frozen section. After blocking the frozen sections with 5% goat serum in PBS, we incubated sections with different primary antibodies, including 3-NT (Abcam, dilution 1:50), claudin-5 (Abcam, dilution 1:200), collagen IV (Santa Cruz, dilution 1:800), HMGB1 (Santa Cruz, dilution 1:50), and MMP-9 (Santa Cruz, dilution 1:50) overnight at 4 °C. Following the washing with PBS, incubating with secondary antibody Alexa Fluor 568 Goat anti-mouse, Alexa Fluor 488 Goat anti-rabbit and washing with PBS three times, the fluorescence images were obtained by using confocal microscope Carl Zeiss LSM 780.
Gelatin Zymography
MMP-9 activity was determined by gelatin zymography. Proteins were extracted from brain tissue with RIPA buffer (Sigma) containing 1% protease/phosphatase inhibitor cocktail (Cell signaling). The same amount of proteins from each group was used for SDS-PAGE electrophoresis. After electrophoresis, gels were washed two times with renaturing buffer for 1 h (2.5% Triton in distilled water) and then incubated with fresh developing buffer at 37 °C for 24 h. Gels were then washed with ddH2O and stained with Coomassie brilliant blue (Sigma) at room temperature for 1 h, followed by washing with the disdain buffer (methanol 10 ml; acetic acid 5 ml, mini Q water 35 ml) till clear bands appear. Digital pictures of gels were captured and the bands’ intensity on gels was analyzed with the ImageJ software (National Institutes of Health, USA).
Peroxynitrite Donor SIN-1 Injection
To study the roles of ONOO− in the expressions of HMGB1 and TLR2 in rat brains, we intraventricularly injected the ONOO− donor 3-morpholino-sydnonimine (SIN-1, from Cayman) into naive rat brains. A small burr hole on the skull was drilled at 1.2 mm lateral to the left and 1.0 mm posterior to the bregma. A needle (31G) attached to a syringe (Hamilton) was inserted into the left lateral ventricle (3.5 mm in depth) and the SIN-1 solution (100 mM, 5 μl) was injected slowly and evenly within 2 min. After the injection, the needle was kept in place for 5 min and then slowly removed. After 4 h, rats were transcardially perfused with PBS and the left side brain tissues around the injection area were harvested for western blot analysis.
Cell Culture and SIN-1 Treatment
Mouse brain microvascular endothelial b.End3 cells were adopted from the American Type Culture Collection (ATCC). Cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM)-high glucose (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Life Technologies). Cells were cultured in a humidified atmosphere with 5% CO2 and 95% air at 37 °C. To investigate the role of ONOO− in mediating HMGB1 signaling, we treated the cells with SIN-1 (ONOO− donor, 500 μM) or vehicles for 4 h under normoxia condition, with or without co-treatment of FeTmPyP (10 μM).
Oxygen and Glucose Deprivation/Reoxygenation(OGD/R)
Brain microvascular endothelial b.End3 cells were subjected to OGD followed by reoxygenation treatment. In brief, cells were incubated with DMEM medium without glucose and placed in a humidified airtight chamber in which the atmosphere was saturated with 95% N2/5% CO2 at 37 °C. Oxygen concentration was less than 1% as monitored by an oxygen analyzer (Sable Systems, Las Vegas, NV, USA). For the control group, cells were incubated with fresh DMEM medium with glucose at 37 °C in a humidified incubator with 5% CO2 and 95% room air. After OGD treatment for 5 h, cells were subjected to reoxygenation and incubated with fresh DMEM medium with glucose for 5 h. During reoxygenation, cells were treated with t-PA (20 μg/ml), with or without co-treatment of glycyrrhizin (10 μM) or PDC (10 μM). The same volume of vehicle was used as a control.
Detection of O2·− and ONOO− Production in b.End3 Cells
The productions of O2·− and ONOO− in b.End3 cells were detected by using our newly developed fluorescent probes HKSOX-1 [39] and HKYellow-AM [38], respectively. HKSOX-1 and HKYellow-AM are highly specific and sensitive probes for superoxide and peroxynitrite detection, respectively. After exposed to 5 h of OGD plus 5 h of reoxygenation, with or without t-PA (20 μg/ml) treatment plus co-treatment of glycyrrhizin (10 μM) or PDC (10 μM), the b.End3 cells were incubated with HKSOX-1 and HKYellow-AM (10 μM) separately at 37 °C for 30 min and then replaced with fresh medium. The fluorescent images were then obtained by using a fluorescence microscope (Carl Zeiss) with Axio Vision digital imaging system.
Western Blot Analysis
Brain tissues or cultured cells were homogenized and lysed by RIPA buffer (Sigma) contained with 1% protease and phosphorylate inhibitor cocktail (Cell Signaling). The same amount of total protein was loaded for SDS-PAGE electrophoresis. After electrophoresis, proteins were then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% bovine serum albumin (BSA), The membranes were incubated with primary antibodies including MMP-9 (Millipore, dilution 1:1000), HMGB1 (Santa Cruz, dilution 1:200), TLR2 (Santa Cruz, dilution 1:200), TLR4 (Santa Cruz, dilution 1:200), 3-NT antibody (Abcam, dilution 1:200), p47phox (Santa Cruz, dilution 1:200), p67phox (Santa Cruz, dilution 1:200), gp91phox (Santa Cruz, dilution 1:200), p22phox (Santa Cruz, dilution 1:200), iNOS (Abcam, dilution 1:500), β-actin (Immunoway, dilution 1:4000) for overnight at 4 °C. After washing with TBST 3 times, the membrane was then incubated with goat-anti-mouse or goat-anti-rabbit HRP-conjugated secondary antibody (Cell Signaling Technology) with the standard protocol. Protein bands were visualized by using ECL Advance (GE Healthcare Bio-Sciences, USA). Band intensity was quantified with Image Lab software.
Detection of T-PA Activity
We examined the t-PA activity by using a commercially available assay kit (Abcam). This kit measures the ability of t-PA to activate the plasminogen to plasmin. The amount of plasmin produced is quantitated using a highly specific plasmin substrate, releasing a yellow para-nitroaniline (pNA) chromophore. The change in absorbance of the pNA in the reaction solution is directly proportional to the t-PA enzymatic activity. The protocol was conducted according to the manufacturer’s instructions. Glycyrrhizin was added to the reaction system at a final concentration of 10 μM. The O.D. value was detected with a wavelength of 405 nm every hour for 10 h and at 24 h after the reaction by using a microplate reader (BioRad).
Statistical Analysis
Statistical analysis was conducted using SPSS 18.0 Software (SPSS, Chicago, IL, USA). Data were expressed as mean ± SD. χ2 test was used for the mortality rate test; non-parametric Kruskal-Wallis test was used for the studies of neurological severity scores and hemorrhagic scores, followed by Dunnett’s multiple comparison tests; one-way ANOVA followed by Dunnett tests were used in multiple groups statistical analysis. Spearman correlation coefficient was applied to assess the association between plasma HMGB1 and NT levels. P < 0.05 was set as statistically significant.
Results
Plasma Nitrotyrosine Was Positively Correlated with HMGB1 Level in Acute Ischemic Stroke Patients and Plasma Nitrotyrosine and HMGB1 Levels Were Increased in Ischemic Stroke Patients with or without Hemorrhagic Transformation
We conducted two separate clinical studies to explore the relationship between ONOO− production and HMGB1 release in ischemic stroke patients in two clinical sites. In the first clinical study, we investigated the relationship between nitrotyrosine production (NT, a surrogate marker for ONOO−) and HMGB1 level in the plasma samples of acute ischemic stroke patients within 24 h from ischemic onset. As shown in Fig. 1, the plasma nitrotyrosine level was positively correlated with the HMGB1 level in ischemic stroke patients. The results imply that the increased plasma HMGB1 level could be associated with the production of ONOO− in ischemic stroke patients. In the second clinical study, we then compared the levels of HMGB1 and NT in plasma of the ischemic stroke patients with hemorrhagic transformation and with t-PA treatment. The results showed that plasma NT level was significantly increased in ischemic stroke patients with or without hemorrhagic transformation when compared with the healthy control group at the admission time. The patients with hemorrhagic transformation (HT) had a significantly higher plasma NT level than the patients without hemorrhagic transformation (non-HT) (Fig. 2a). Meanwhile, both HT and non-HT ischemic stroke patients had significantly higher plasma HMGB1 levels than the control group (Fig. 2b, p < 0.01). The HT group had a slightly increased level of HMGB1 in the plasma than the non-HT group, but without statistic difference due to the small sample sizes (Fig. 2b). Moreover, we investigated the effects of t-PA on the level of HMGB1 in ischemic stroke patients. Plasma samples were collected at 24 h after admission to hospital in the patients with or without t-PA treatment. The results showed that ischemic stroke patients with or without t-PA treatment had a significantly higher HMGB1 level than healthy control subjects. The ischemic stroke patients who received t-PA treatment had a higher plasma HMGB1 level than the patients without t-PA treatment (Fig. 2c). Those results suggest the ONOO− production could contribute to the release of HMGB1 and hemorrhagic transformation in ischemic stroke patients, and t-PA treatment could increase the release of HMGB1 in ischemic stroke patients.

Correlation of nitrotyrosine (NT) and HMGB1 in plasma of acute ischemic stroke patients. A total of 72 both male and female acute ischemic stroke patients aged 40–80 years old were recruited in the study. Blood samples were collected within 24 h after ischemic stroke onset. Plasma NT and HMGB1 were analyzed by using ELISA kits according to the product manuals. Spearman correlation coefficient was used to assess the correlations between plasma NT and HMGB1 levels in ischemic stroke patients (n = 72, p = 0.022)

Association of plasma nitrotyrosine and HMGB1 with hemorrhagic transformation in acute ischemic stroke patients. We recruited 13 control and 56 ischemic stroke patients with or without t-PA treatment. By using ELISA kits, we analyzed the levels of HMGB1 and NT in plasma of the ischemic stroke patients with or without hemorrhagic transformation (HT). HT was determined by an CT/MRI scan. Patients who received t-PA were treated within t-PA infusion within 4.5 h after ischemia onset. a Plasma NT level at admission to hospital in healthy control (Con), and ischemic stroke patients who subsequently developed HT or not. For control group, n = 13, for non-HT group, n = 26, for HT group, n = 11, *P < 0.05, ***P < 0.001, ****P < 0.0001. b Plasma HMGB1 level at admission to hospital in healthy control (Con), and ischemic stroke patients who subsequently developed HT or not. For control group, n = 13, for non-HT group, n = 26, for HT group, n = 11, **P < 0.01. c Plasma HMGB1 level in stroke patients at 24 h with or without t-PA treatment. For control group, n = 13, for non-t-PA treatment group, n = 14, for t-PA treatment group, n = 19, *P < 0.05, **P < 0.01, ****P < 0.0001
Peroxynitrite-Mediated HMGB1 Activation in Ischemic Rat Brains with Delayed T-PA Treatment
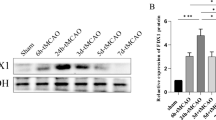
We next explored the roles of ONOO− in mediating HMGB1 activation in the rat MCAO model with delayed t-PA treatment. The rats were subjected to 5 h of MCAO cerebral ischemia following 19 h of reperfusion, in which t-PA infusion was conducted at 5 h of ischemia to mimic the t-PA treatment beyond its golden therapeutic window. We found that the increased expression of HMGB1 was co-localized with 3-nitrotyrosine (3-NT) in the ischemic brains (Fig. 3a, Supplementary 5). Treatment of FeTmPyP significantly downregulated the expression of 3-NT, HMGB1, and its receptor TLR2 in the ischemic brains (Fig. 3a–d). Glycyrrhizin treatment also downregulated the expressions of the HMGB1 and its receptor TLR2, without affecting the expression of TLR4 (Supplementary 6A). Interestingly, we found that glycyrrhizin treatment also reduced the expression of 3-NT in the ischemic brain with t-PA treatment. These results further support the roles of ONOO− production in mediating HMGB1 expression in the MCAO ischemic stroke model with delayed t-PA treatment.

Peroxynitrite activated HMGB1 signaling in an ischemic stroke model in vivo and cultured brain microvascular endothelial b.End3 cells in vitro. a Representative immunostaining of HMGB1 and 3-nitrotyrosine (3-NT) in ischemic brains at 6 mm away from the frontal tip as shown in the figure, at 24 h after ischemia onset with or without delayed t-PA treatment (10 mg/kg), plus glycyrrhizin (GL, 30 mg/kg) or peroxynitrite decomposition catalyst (PDC, FeTmPyP 3 mg/kg) treatment. Saline was used as a vehicle. Scale bar = 100 μm; n = 5. b Representative western blot of TLR2 and HMGB1 in ischemic brains after 24 h after MCAO onset. c Statistical analysis of TLR2 expression level of each group in ischemic brains (**P < 0.01, ***P < 0.001, n = 6). d Statistical analysis of HMGB1 expression level of each group in ischemic brains (*P < 0.05, ***P < 0.001, n = 6). e Representative western blot of TLR2 and HMGB1 expression in naive rat brains after intraventricular injection of peroxynitrite donor SIN-1 (100 mM, 5 μl) for 4 h. f Representative western blot of HMGB1 expression in cultured brain microvascular endothelial b.End3 cells with peroxynitrite donor SIN-1 treatment (500 μM) and FeTmPyP (10 μM) for 4 h. g Statistical analysis of TLR2 expression in rat brains after peroxynitrite donor SIN-1 treatment (**P < 0.01, n = 3). h Statistical analysis for HMGB1 expression in rat brains after peroxynitrite donor SIN-1 treatment (*P < 0.05, n = 3). i Statistical analysis for HMGB1 expression in cultured brain microvascular endothelial cells (**P < 0.01, n = 3)
We then confirmed the roles of ONOO− in activating HMGB1 signaling by intraventricular injection of 3-morpholino-sydnonimine (SIN-1, a ONOO− donor) into naïve rat brains. SIN-1 injection significantly upregulated the expressions of HMGB1 and its receptor TLR2 instead of TLR4 in naïve rat brains (Fig. 3e, g, h, Supplementary 6B). Consistently, SIN-1 treatment significantly increased the expression of HMGB1 in brain microvascular endothelial b.End3 cells in vitro, which were abolished by FeTmPyP co-treatment (Fig. 3f, i). These results suggest that ONOO− directly mediates the activation of HMGB1/TLR2 signaling in vivo and in vitro.
Glycyrrhizin Inhibited the Production of O2·− and ONOO− In Vivo and In Vitro
Since we observed the reduction of 3-NT immunofluorescence by glycyrrhizin co-treatment, we stepped forward to directly detect the in vivo inhibitory effects of glycyrrhizin on ONOO− production, using our newly developed specific and sensitive probe HKYellow-AM [38]. The results showed that the delayed t-PA treatment significantly increased ONOO− level in ischemic brains, which was inhibited by co-treatment of glycyrrhizin or PDC (Fig. 4a, Supplementary 7A). We double confirmed such results by detecting 3-NT expression with western blot analysis (Fig. 4b and c). We also detected the production of O2·− by using hydroethidine (HEt) method. The fluorescent signal of the oxidized products of HEt, reflecting O2.- production, was significantly increased in the ischemic brains with the delayed t-PA treatment, which was inhibited by co-treatment of glycyrrhizin (Fig. 4a, Supplementary 7B). We then stepped forward to investigate the expressions of NADPH oxidases and inducible nitric oxide synthase (iNOS), respectively. The expression of p47phox and p67phox subunits were significantly increased in the ischemic brains with delayed t-PA treatment (Fig. 4b), whereas the expressions of gp91phox and p22phox subunits had no significant changes (Supplementary 7C). Co-treatment of glycyrrhizin significantly downregulated the expression of iNOS, p47phox and p67phox in the ischemic brains (Fig. 4b, d–f). Those results indicate that glycyrrhizin could inhibit the expressions of NADPH oxidase subunits p47phox, p67phox, and iNOS and decrease the production of O2·− and NO, subsequently reducing ONOO− formation in the ischemic rat brains after receiving delayed t-PA treatment. We then investigated the effects of glycyrrhizin on inhibiting the productions of O2·− and ONOO− in the brain microvascular endothelial b.End3 cells in vitro. Cells were subjected to OGD 5 h plus reoxygenation 5 h with or without t-PA treatment. Consistently, t-PA treatment (20 μg/ml) at reoxygenation onset increased the fluorescence of HKSOX-1 and HKYellow-AM, indicating that t-PA itself could aggravate the production of O2·− and ONOO− in the cultured endothelial cells after exposure to oxygen and glucose deprivation and reoxygenation (OGD/R). Consistently, treatment of glycyrrhizin remarkably decreased the levels of O2·− and ONOO− in the b.End3 cells (Fig. 4g, h, Supplementary 8). Taken together, these results suggest that glycyrrhizin could inhibit O2·− and ONOO− production in vivo and in vitro.

Effects of glycyrrhizin on superoxide and peroxynitrite in ischemic brains in vivo and cultured brain microvascular endothelial b.End3 cells in vitro. a Representative HEt probe staining for superoxide, HKYellow-AM probe staining for peroxynitrite in ischemic brains at 6 mm away from the frontal tip, at 24 h after ischemia onset with or without delayed t-PA treatment plus glycyrrhizin (GL, 30 mg/kg) or PDC (3 mg/kg). Saline was used a vehicle. Scale bar = 100 μm, n = 4. b Representative western blot of 3-NT, iNOS, NADPH oxidase p47 phox and p67 phox expression in ischemic brain tissues at 24 h after ischemia onset. c Statistical analysis of 3-NT level in ischemic brain tissues (*P < 0.05, **P < 0.01). d Statistical analysis of iNOS expression in ischemic brain tissues (**P < 0.01). e Statistical analysis of NADPH oxidase p47 phox expression level in ischemic brain tissues (*P < 0.05). f Statistical analysis of NADPH oxidase p67 phox expression level in ischemic brain tissues (*P < 0.05). n = 6, data are expressed as mean ± SEM. g Representative HKYellow-AM staining for peroxynitrite level in b.End3 cells after OGD 5 h plus reoxygenation 5 h with or without t-PA treatment (20 μg/ml) for 5 h during reoxygenation plus glycyrrhizin (10 μM) or PDC (10 μM) co-treatment. Scale bar = 500 μm, n = 3. h Representative HKSOX-1 staining for superoxide level in b.End3 cells after OGD 5 h plus reoxygenation 5 h with or without t-PA treatment (20 μg/ml) for 5 h during reoxygenation plus glycyrrhizin (10 μM) or PDC (10 μM) co-treatment. Scale bar = 500 μm, n = 3
Glycyrrhizin Inhibited MMP-9 Activity, Protected Claudin-5 and Collagen IV, Attenuated Apoptotic Cell Death and BBB Leakage in MCAO Rat Brains with Delayed t-PA Treatment
Given that glycyrrhizin revealed the property of inhibiting ONOO− and HMGB1 production in ischemic brains, we logically expected that glycyrrhizin treatment could inhibit MMP-9 activity and protect the BBB integrity in cerebral ischemia-reperfusion injury with the delayed t-PA treatment. We used FeTmPyP, a representative PDC, as a positive control. By using gelatin zymography analysis, we found that the delayed t-PA treatment significantly enhanced MMP-9 activity in the ischemic brain tissues. Co-treatment of glycyrrhizin (30 mg/kg) or PDC (FeTmPyP, 3 mg/kg) significantly attenuated the activation of MMP-9 (Fig. 5a, b). By using immunofluorescence staining, we also detected the expression of major vascular basal membrane protein collagen IV, tight junction protein claudin-5 and MMP-9, in which collagen IV was co-located with MMP-9. The vehicle-treated cerebral ischemia group had increased the expression of MMP-9 and reduced the expressions of collagen IV and claudin-5 in the ischemic brains. Interestingly, t-PA treatment remarkably increased the fluorescence of MMP-9 in companied with the increased fragmentations of collagen IV fluorescence and the reduced claudin-fluorescence, indicating the damage of the BBB integrity. Glycyrrhizin co-treatment remarkably inhibited the induction of MMP-9 and reduced the fragmentation of collagen IV and the loss of claudin 5, whose effects were similar to PDC (FeTmPyP) (Fig. 5c). Furthermore, we measured BBB integrity and apoptotic cell death by using Evans blue leakage assay and TUNEL staining respectively (Fig. 5d–g). The delayed t-PA treatment significantly increased the Evan blue leakage, which was attenuated by co-treatment of glycyrrhizin or PDC (Fig. 5d and e). Meanwhile, the delayed t-PA treatment group had significantly higher rates of apoptotic cell death in cortex and striatum of the ischemic brains than the MCAO vehicle group. Glycyrrhizin co-treatment (30 mg/kg) significantly reduced apoptotic cell death whose effects were similar to PDC co-treatment (FeTmPyP, 3 mg/kg) (Fig. 5f, g). These results suggest that glycyrrhizin could effectively inhibit MMP-9 activity, protect tight junction claudin-5 and extracellular matrix collagen IV, and protect the BBB integrity in the ischemia-reperfused rat brains with the delayed t-PA treatment.

Effect of glycyrrhizin on activition of MMP-9 and expression of extracellular matrix collagen IV and tight junction protein claudin-5 blood-brain barrier damage and cell apoptosis. a Representative gelatin zymography results of MMP-9 activity of ischemic brain tissues at 24 h after ischemia onset, with or without delayed t-PA treatment plus glycyrrhizin (GL, 30 mg/kg) or PDC (3 mg/kg). Saline was used as a vehicle. b Statistical analysis of MMP-9 activity in ischemic brains (**P < 0.01, ***P < 0.001, ****P < 0.0001, n = 6). c Representative immunostaining of MMP-9, collagen IV and tight junction protein claudin-5 in the ischemic brains at 6 mm away from the frontal tip, at 24 h after ischemia, with or without delayed t-PA treatment plus glycyrrhizin (GL, 30 mg/kg) or PDC (3 mg/kg). Scale bar = 100 μm. n = 4. d Representative brain slices showing Evans blue leakage at 24 h after MCAO onset, with or without delayed t-PA treatment plus glycyrrhizin (GL, 30 mg/kg) or PDC (3 mg/kg). e Statistical analysis of Evans blue leakage in the ischemic hemispheres (*P < 0.05, ****P < 0.0001, n = 6). f Representative TUNEL staining results in cortex and striatum of brain slices as shown in the figure at 24 h after MCAO onset, with or without delayed t-PA treatment plus glycyrrhizin (GL, 30 mg/kg) or PDC (3 mg/kg). Scale bar = 500 μm. g Statistical analysis of TUNEL-positive cells percentage in cortex and striatum of brain tissue (***, ###, &&&P < 0.001, n = 6)
Glycyrrhizin Reduced Mortality Rates, Attenuated Neurological Deficit Scores, and Decreased Brain Edema and Hemorrhagic Transformation in MCAO Brains with Delayed t-PA Treatment
We finally addressed whether glycyrrhizin could improve the outcomes of ischemic stroke treatment with delayed t-PA treatment. FeTmPyP was used as a positive control. The outcomes included mortality rates, neurological deficit scores, brain edema, and hemorrhage transformation. The delayed t-PA treatment significantly increased the mortality rates (Fig. 6a), neurological deficit scores (Fig. 6b), brain edema (Fig. 6c), and hemorrhagic transformation (Fig. 6d–f) in the rats with 5 h MCAO cerebral ischemia plus 19-h reperfusion. Glycyrrhizin co-treatment (30 mg/kg) or PDC (3 mg/kg) co-treatment reduced the mortality rate, which was even lower than the MCAO vehicle group. Delayed t-PA treatment worsened the neurological functions as indicated by the increase of mNSS scores (P < 0.05) (Fig. 6b). Glycyrrhizin (30 or 60 mg/kg) or PDC co-treatment significantly decreased the mNSS scores (Fig. 6b). Thus, glycyrrhizin could improve the survival rate and the neurological outcomes in the ischemia-reperfused rats with delayed t-PA treatment.

Effect of glycyrrhizin on mortality rate, neurological score, hemorrhagic transformation and brain swelling. a The mortality rate at 24 h after MCAO-reperfusion, with or without delayed t-PA treatment (10 mg/kg) plus glycyrrhizin (GL, 30 mg/kg) or PDC (3 mg/kg). The number of rats died within 24 h after MCAO and the total number of each group was labeled in each group. b Modified neurological severity scores (mNSS) of each group at 24 h after ischemia onset. Glycyrrhizin was treated at dosage of 15 mg/kg, 30 mg/kg, and 60 mg/kg (*P < 0.05, ***P < 0.001, ****P < 0.0001, ns, no significant difference, n = 12–16). c Brain swelling as calculated by an increase of brain area of ischemic side to non-ischemic side at 24 h after ischemia onset (**P < 0.01, ***P < 0.001, n = 12–16). d Representative brain slices showing the hemorrhagic transformation of each group at 24 h after ischemia onset. e Hemorrhagic transformation (HT) scores of each group at 24 h after ischemia onset was calculated as the total scores of the five slices in each group (*P < 0.05, **P < 0.01, ns, no significant difference, n = 6–8). f Hemoglobin level in the ischemic hemisphere of each group at 24 h from ischemia onset (*P < 0.05, **P < 0.01, ***P < 0.001, n = 6–8). Data are expressed as mean ± SD
We then investigated the effects of glycyrrhizin on brain edema and hemorrhagic transformation. The delayed t-PA treatment significantly increased brain edema which was dose-dependently attenuated by glycyrrhizin co-treatment (15, 30, 60 mg/kg) (Fig. 6c). Meanwhile, delayed t-PA treatment significantly increased the hemorrhagic transformation as indicated by the increased hemorrhagic transformation scores and hemoglobin level in the ischemic brain tissues of the surviving rats (P < 0.05) (Fig. 6D-F). Glycyrrhizin (15–60 mg/kg) or PDC (FeTmPyP, 3 mg/kg) co-treatment significantly decreased hemorrhagic transformation score and hemoglobin level in the ischemic brain tissues (Fig. 6d–f). One may concern whether glycyrrhizin affected the thrombolytic activity of t-PA. We found that glycyrrhizin did not affect the t-PA fibrinolytic activity as determined by the t-PA activity assay kit, which reflects the capacity of t-PA to activate plasminogen into plasmin (Supplementary 9).
Discussion
The novelty and major discoveries of this study include the following aspects: (1) Plasma nitrotyrosine level was positively correlated with the HMGB1 level in acute ischemic stroke patients; (2)Increased levels of nitrotyrosine and HMGB1 in plasma were found in hemorrhagic transformation patients and t-PA-treated ischemic stroke patients; (3) ONOO− production mediated the activation of the HMGB1 signaling in ischemic rat brains with delayed t-PA treatment, subsequently contributing to hemorrhagic transformation; (4) Glycyrrhizin downregulated the expression of iNOS, p47phox and p67phox, and inhibited the superoxide and peroxynitrite production in ischemic rat brains with delayed t-PA treatment, subsequently decreased HMGB1, TLR2, and MMP-9 expression; attenuated hemorrhagic transformation; and improved the neurological outcome.
The underlying mechanisms of the t-PA-induced hemorrhagic transformation involve multiple signaling pathways, including oxidative stress, matrix metalloproteinase activation, and inflammatory responses, etc. [40, 41]. Our recent studies demonstrated that ONOO− production localized with the ischemic brain area showing BBB leakage. ONOO− activated MMPs, aggravated the BBB disruption, and induced hemorrhagic transformation, contributing to the poor outcome in neurological functions and survival rates in the ischemic rats with delayed t-PA treatment [25, 26, 42]. Scavenging ONOO− could be a promising adjunct therapeutic strategy with t-PA for inhibiting MMP activation, protecting the BBB integrity, reducing hemorrhagic transformation, and promoting outcomes in ischemic stroke treatment [25, 26]. Meanwhile, damage-associated molecular pattern (DAMP), such as HMGB1 released from necrotic neurons can bind to toll-like receptor TLR2/4 and RAGE, induce inflammatory responses and trigger apoptotic cell death in neighboring neurons [43, 44]. HMGB1-TLR4 signaling induced the expression of MMP-9 in cultured neuronal and glial cells in vitro and mouse brains in vivo [12]. Monoclonal TLR2 antagonist T2.5 attenuated MMP-9 activation and preserved tight junction protein occludin and extracellular matrix collagen VI [45]. Thus, HMGB1/TLRs are important for MMP-9 induction in ischemic brains. HMGB1 inhibitors, including HMGB1 neutralizing antibody, HMGB1 A-box, binding heptamer peptide, ameliorated brain infarction and neuroinflammation and protected the BBB integrity [46,47,48]. We recently reported that HMGB1-binding heptamer peptide significantly inhibited HMGB1 and protected hemorrhagic transformation with delayed thrombolytic therapy [16]. Thus, HMGB1/TLR signaling activation could be important in mediating hemorrhagic transformation during ischemic stroke. Thus, we further explored the relationship between ONOO− and HMGB1 in cerebral ischemia-reperfusion injury with delayed t-PA treatment. We found that plasma nitrotyrosine, a surrogate marker of ONOO− production, was positively correlated with HMGB1 level in acute ischemic stroke patients. The following in vivo and in vitro studies confirm the role of ONOO− in HMGB1 induction: (1) we observed the co-localization of 3-NT with HMGB1 in the ischemic brain area which showing BBB leakage and hemorrhagic transformation in vivo; (2) by using the rat MCAO ischemic stroke model, we found that scavenging ONOO− with FeTmPyP decreased the expression of HMGB1 and TLR2, and attenuated the activation of MMP-9 in the ischemic brains with delayed t-PA in vivo; (3) infusion of ONOO− donor SIN-1 into the naïve rat brains upregulated the expressions of HMGB1 and its receptor TLR2 in rat brains in vivo; (4) SIN-1 treatment increased HMGB1 expression in cultured brain microvascular endothelial b.End3 cells in vitro, which were abolished by FeTmPyP treatment. Thus, ONOO− could mediate the activation of HMGB1/TLR2 signaling in cerebral ischemia-reperfusion during delayed t-PA treatment. Notably, the expression of TLR2 in b.End3 cells were not induced by SIN-1 treatment in vitro (data not shown), indicating that the in vivo increase of TLR2 after SIN-1 treatment may be due to other cell types of the brain tissue.
As an HMGB1 inhibitor, glycyrrhizin (GL) showed its neuroprotective effects against cerebral ischemia-reperfusion injury [31, 49]. Here, for the first time, we report that the GL treatment reduced hemorrhagic transformation, improved the neurological outcomes, and decreased the mortality rate in the MCAO rats with delayed t-PA at 5 h of cerebral ischemia. GL had similar effects to PDC treatment. GL inhibited MMP-9 activity, protected tight junction claudin-5 and extracellular matrix collagen IV, attenuated ischemic brain damages, and protected the BBB integrity, subsequently reducing brain edema and hemorrhagic transformation. These results highlight glycyrrhizin as a potential adjuvant therapy with t-PA for ischemic stroke treatment. We then stepped forward to investigate whether glycyrrhizin has the properties of inhibiting the productions of O2·− and ONOO−. By using various fluorescent probes for O2.− and ONOO−, we found that glycyrrhizin inhibited the production of O2·− and ONOO− in ischemic brains with delayed t-PA treatment in vivo, and in cultured brain microvascular endothelial cells under oxygen and glucose deprivation condition in vitro. Glycyrrhizin treatment downregulated the expressions of NADPH oxidase p47 phox, p67 phox, and iNOS for reducing the productions of NO and O2.−, and subsequently decreasing ONOO− production in the ischemia-reperfused brain tissues with delayed t-PA treatment in vivo. Glycyrrhizin downregulated the HMGB1 and TLR2 expression in ischemic brains with delayed t-PA treatment. Given that ONOO− could mediate the expressions of HMGB1 and TLR2, we logically conclude that the antioxidant property of glycyrrhizin might contribute to the inhibitive effects on HMGB1 and TLR2 in the ischemic brains with delayed t-PA treatment. Therefore, glycyrrhizin could have direct and indirect inhibitive effects on HMGB1 signaling. However, we should keep in mind that glycyrrhizin also has pleiotropic effects on top of the HMGB1 inhibition. Thus, the downregulation of ONOO−/HMGB1/TLR2/MMP-9 signaling could be the consequence of reduced brain injury by glycyrrhizin treatment, which needs further investigation.
Since glycyrrhizin has glucuronic acid moiety which limits its BBB permeability, one may concern the bioavailability of glycyrrhizin in CNS. However, a recent UPLC/ESI-MS study revealed the capacity of glycyrrhizin in penetrating the BBB [31]. Importantly, we found that glycyrrhizin has no influence on the thrombolytic bioactivity of t-PA. Thus, glycyrrhizin could be a promising adjunct therapy candidate with t-PA for attenuating brain edema and hemorrhagic transformation, and improving the neurological outcomes in ischemic stroke treatment. In future studies, we should investigate whether the attenuation of hemorrhagic transformation by glycyrrhizin at the acute phase of stroke could bring long-term benefits for stroke recovery.
In conclusion, as shown in Fig. 7, the delayed t-PA treatment induces ONOO− production in the ischemic brains, inducing HMGB1/TLR2 expression, MMP-9 activation, leading to tight junction and extracellular matrix degradation, BBB disruption and eventually hemorrhagic transformation. Glycyrrhizin can inhibit the ONOO− production, reduce HMGB1/TLR2 expression and MMP-9 activation, subsequently protect the BBB integrity, and attenuate hemorrhagic transformation.

Schematic diagram showing the protective effect of glycyrrhizin on reducing hemorrhagic transformation in ischemic brains with delayed t-PA treatment. Delayed t-PA treatment triggered the ONOO−/HMGB1/TLR2/MMP-9 signaling cascade, subsequently inducing the hemorrhagic transformation. Glycyrrhizin or FeTmPyP inhibits this signaling cascade and reduces hemorrhagic transformation. BBB blood-brain barrier, HMGB1 high mobility group box protein 1, HT hemorrhagic transformation, MMP-9 matrix metalloproteinase-9, TJ tight junction, TLR2 Toll-like receptor 2, t-PA tissue plasminogen activator
References
Fugate JE, Giraldo EA, Rabinstein AA. Thrombolysis for cerebral ischemia. Front Neurol. 2010;1:139. https://doi.org/10.3389/fneur.2010.00139.
Emberson J, Lees KR, Lyden P, Blackwell L, Albers G, Bluhmki E, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet. 2014;384(9958):1929–35. https://doi.org/10.1016/S0140-6736(14)60584-5.
Ho WM, Reis C, Akyol O, Akyol GY, Applegate R, Stier G, et al. Pharmacological management options to prevent and reduce ischemic hemorrhagic transformation. Curr Drug Targets. 2017;18(12):1441–59. https://doi.org/10.2174/1389450117666160818115850.
Knecht T, Story J, Liu J, Davis W, Borlongan CV, Dela Pena IC. Adjunctive therapy approaches for ischemic stroke: innovations to expand time window of treatment. Int J Mol Sci. 2017;18(12). https://doi.org/10.3390/ijms18122756.
Chen HS, Qi SH, Shen JG. One-compound-multi-target: combination prospect of natural compounds with thrombolytic therapy in acute ischemic stroke. Curr Neuropharmacol. 2017;15(1):134–56.
Zhang L, Zhang ZG, Chopp M. The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol Sci. 2012;33(8):415–22. https://doi.org/10.1016/j.tips.2012.04.006.
Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002;33(3):831–6.
Tsuji K, Aoki T, Tejima E, Arai K, Lee SR, Atochin DN, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36(9):1954–9. https://doi.org/10.1161/01.STR.0000177517.01203.eb.
Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminogen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31(12):3034–40.
Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003;34(8):2025–30. https://doi.org/10.1161/01.STR.0000083051.93319.28.
Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28(5):927–38. https://doi.org/10.1038/sj.jcbfm.9600582.
Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Lo EH, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke. 2010;41(9):2077–82.
Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86(5):1125–31. https://doi.org/10.1002/jnr.21555.
Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke. 2011;42(5):1420–8. https://doi.org/10.1161/STROKEAHA.110.598334.
Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21(14):3904–16. https://doi.org/10.1096/fj.07-8770com.
Li M, Chen S, Shi X, Lyu C, Zhang Y, Tan M, et al. Cell permeable HMGB1-binding heptamer peptide ameliorates neurovascular complications associated with thrombolytic therapy in rats with transient ischemic stroke. J Neuroinflammation. 2018;15(1):237.
Sun MS, Jin H, Sun X, Huang S, Zhang FL, Guo ZN, et al. Free radical damage in ischemia-reperfusion injury: an obstacle in acute ischemic stroke after revascularization therapy. Oxidative Med Cell Longev. 2018;2018:3804979. https://doi.org/10.1155/2018/3804979.
Chen HS, Chen X, Li WT, Shen JG. Targeting RNS/caveolin-1/MMP signaling cascades to protect against cerebral ischemia-reperfusion injuries: potential application for drug discovery. Acta Pharmacol Sin. 2018;39(5):669–82. https://doi.org/10.1038/aps.2018.27.
Gu Y, Dee CM, Shen J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front Biosci (Schol Ed). 2011;3:1216–31.
Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21(12):1393–400. https://doi.org/10.1097/00004647-200112000-00003.
Jian Liu K, Rosenberg GA. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic Biol Med. 2005;39(1):71–80. https://doi.org/10.1016/j.freeradbiomed.2005.03.033.
Suzuki M, Tabuchi M, Ikeda M, Tomita T. Concurrent formation of peroxynitrite with the expression of inducible nitric oxide synthase in the brain during middle cerebral artery occlusion and reperfusion in rats. Brain Res. 2002;951(1):113–20.
Virag L, Szabo E, Gergely P, Szabo C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett. 2003;140–141:113–24. https://doi.org/10.1016/s0378-4274(02)00508-8.
Gursoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35(6):1449–53. https://doi.org/10.1161/01.STR.0000126044.83777.f4.
Chen HS, Chen XM, Feng JH, Liu KJ, Qi SH, Shen JG. Peroxynitrite decomposition catalyst reduces delayed thrombolysis-induced hemorrhagic transformation in ischemia-reperfused rat brains. CNS Neurosci Ther. 2015;21(7):585–90. https://doi.org/10.1111/cns.12406.
Chen H, Guan B, Chen X, Chen X, Li C, Qiu J, et al. Baicalin attenuates blood-brain barrier disruption and hemorrhagic transformation and improves neurological outcome in ischemic stroke rats with delayed t-PA treatment: involvement of ONOO(−)-MMP-9 pathway. Transl Stroke Res. 2018;9(5):515–29. https://doi.org/10.1007/s12975-017-0598-3.
Loukili N, Rosenblatt-Velin N, Li J, Clerc S, Pacher P, Feihl F, et al. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc Res. 2011;89(3):586–94. https://doi.org/10.1093/cvr/cvq373.
Chandrashekaran V, Seth RK, Dattaroy D, Alhasson F, Ziolenka J, Carson J, et al. HMGB1-RAGE pathway drives peroxynitrite signaling-induced IBD-like inflammation in murine nonalcoholic fatty liver disease. Redox Biol. 2017;13:8–19. https://doi.org/10.1016/j.redox.2017.05.005.
Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, et al. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol. 2007;14(4):431–41. https://doi.org/10.1016/j.chembiol.2007.03.007.
Girard JP. A direct inhibitor of HMGB1 cytokine. Chem Biol. 2007;14(4):345–7. https://doi.org/10.1016/j.chembiol.2007.04.001.
Kim SW, Jin Y, Shin JH, Kim ID, Lee HK, Park S, et al. Glycyrrhizic acid affords robust neuroprotection in the postischemic brain via anti-inflammatory effect by inhibiting HMGB1 phosphorylation and secretion. Neurobiol Dis. 2012;46(1):147–56. https://doi.org/10.1016/j.nbd.2011.12.056.
Gong G, Xiang L, Yuan L, Hu L, Wu W, Cai L, et al. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS One. 2014;9(3):e89450. https://doi.org/10.1371/journal.pone.0089450.
Zhang J, Wu Y, Weng Z, Zhou T, Feng T, Lin Y. Glycyrrhizin protects brain against ischemia-reperfusion injury in mice through HMGB1-TLR4-IL-17A signaling pathway. Brain Res. 2014;1582:176–86. https://doi.org/10.1016/j.brainres.2014.07.002.
Liu W, Hendren J, Qin XJ, Liu KJ. Normobaric hyperoxia reduces the neurovascular complications associated with delayed tissue plasminogen activator treatment in a rat model of focal cerebral ischemia. Stroke. 2009;40(7):2526–31. https://doi.org/10.1161/STROKEAHA.108.545483.
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32(11):2682–8.
Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, et al. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107(5):1196–205. https://doi.org/10.1111/j.1471-4159.2008.05664.x.
Fagan SC, Lapchak PA, Liebeskind DS, Ishrat T, Ergul A. Recommendations for preclinical research in hemorrhagic transformation. Transl Stroke Res. 2013;4(3):322–7. https://doi.org/10.1007/s12975-012-0222-5.
Peng T, Chen X, Gao L, Zhang T, Wang W, Shen J, et al. A rationally designed rhodamine-based fluorescent probe for molecular imaging of peroxynitrite in live cells and tissues. Chem Sci. 2016;7(8):5407–13. https://doi.org/10.1039/c6sc00012f.
Hu JJ, Wong NK, Ye S, Chen X, Lu MY, Zhao AQ, et al. Fluorescent probe HKSOX-1 for imaging and detection of endogenous superoxide in live cells and in vivo. J Am Chem Soc. 2015;137(21):6837–43. https://doi.org/10.1021/jacs.5b01881.
Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34(2):185–99. https://doi.org/10.1038/jcbfm.2013.203.
Kanazawa M, Takahashi T, Nishizawa M, Shimohata T. Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. J Atheroscler Thromb. 2017;24(3):240–53. https://doi.org/10.5551/jat.RV16006.
Chen H, Chen X, Luo Y, Shen J. Potential molecular targets of peroxynitrite in mediating blood-brain barrier damage and haemorrhagic transformation in acute ischaemic stroke with delayed tissue plasminogen activator treatment. Free Radic Res. 2018:1–20. https://doi.org/10.1080/10715762.2018.1521519.
Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, et al. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26(24):6413–21. https://doi.org/10.1523/JNEUROSCI.3815-05.2006.
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28(46):12023–31. https://doi.org/10.1523/JNEUROSCI.2435-08.2008.
Zhu H, Dai R, Fu H, Meng Q. MMP-9 Upregulation is attenuated by the monoclonal TLR2 antagonist T2. 5 after oxygen–glucose deprivation and reoxygenation in rat brain microvascular endothelial cells. J Stroke Cerebrovasc Dis. 2019;28(1):97–106.
Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther. 2014;141(3):347–57.
Wang C, Jiang J, Zhang X, Song L, Sun K, Xu R. Inhibiting HMGB1 reduces cerebral ischemia reperfusion injury in diabetic mice. Inflammation. 2016;39(6):1862–70. https://doi.org/10.1007/s10753-016-0418-z.
Kim ID, Shin JH, Lee HK, Jin YC, Lee JK. Intranasal delivery of HMGB1-binding heptamer peptide confers a robust neuroprotection in the postischemic brain. Neurosci Lett. 2012;525(2):179–83. https://doi.org/10.1016/j.neulet.2012.07.040.
Xiong X, Gu L, Wang Y, Luo Y, Zhang H, Lee J, et al. Glycyrrhizin protects against focal cerebral ischemia via inhibition of T cell activity and HMGB1-mediated mechanisms. J Neuroinflammation. 2016;13(1):241. https://doi.org/10.1186/s12974-016-0705-5.
Funding
This work is supported by Hong Kong General Research Fund (GRF No. 17102915, GRF No. 17118717), Research Grant Council, Hong Kong SAR and Health and Medical Research Fund, Hong Kong SAR (NO. 13142901), AoE/P-705/16 Areas of Excellence Scheme, RGC, Hong Kong SAR; SIRI/04/04/2015/06 Shenzhen Basic Research Plan Project. National Natural Science Foundation of China (No. 81671164).
Author information
Authors and Affiliations
Contributions
Hansen Chen performed in vivo and in vitro experiments, analyzed data, and wrote the manuscript; Binghe Guan, Bin Wang, and Haiwei Pu performed clinical investigations and analyzed the data; Caiming Li, Jihong Liu, and Jinhua Qiu recruited the ischemic stroke patients, made clinical diagnosis, collected blood samples, and performed clinical studies; Xiaoyu Bai and Xi Chen performed in vitro study and conducted data analysis; Dan Yang synthesized the probes for ONOO− detection and data analysis; Kejian Liu and Qi Wang contributed to the study design and discussion; Suhua Qi contributed to the study design in vivo study, clinical investigation and manuscript preparation; Jiangang Shen received the funding, guided and designed all experiments, interpreted the results, and prepared and revised the manuscripts.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 1125 kb).
Rights and permissions
About this article

Cite this article
Chen, H., Guan, B., Wang, B. et al. Glycyrrhizin Prevents Hemorrhagic Transformation and Improves Neurological Outcome in Ischemic Stroke with Delayed Thrombolysis Through Targeting Peroxynitrite-Mediated HMGB1 Signaling. Transl. Stroke Res. 11, 967–982 (2020). https://doi.org/10.1007/s12975-019-00772-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-019-00772-1