
Abstract
The secondary neuroinflammatory response has come into focus of experimental stroke research. Immunological mechanisms after acute stroke are being investigated in the hope to identify novel and druggable pathways that contribute to secondary infarct growth after stroke. Among a variety of neuroimmunological events after acute brain ischemia, including microglial activation, brain leukocyte invasion, and secretion of pro-inflammatory factors, lymphocytes have been identified as the key leukocyte subpopulation driving the neuroinflammatory response and contributing to stroke outcome. Several studies have shown that pro-inflammatory lymphocyte subpopulations worsen stroke outcome and that inhibiting their invasion to the injured brain is neuroprotective. In contrast to the effector functions of pro-inflammatory lymphocytes, regulatory T cells (Treg) are critically involved in maintaining immune homeostasis and have been characterized as disease-limiting protective cells in several inflammatory conditions, particularly in primary inflammatory diseases of the central nervous system (CNS). However, due to the complex function of regulatory cells in immune homeostasis and disease, divergent findings have been described for the role of Treg in stroke models. Emerging evidence suggests that this discrepancy arises from potentially differing functions of Treg depending on the predominant site of action within the neurovascular unit and the surrounding inflammatory milieu. This article will provide a comprehensive review of current findings on Treg in brain ischemia models and discuss potential reasons for the observed discrepancies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Regulatory T Cells as Master Switches in Immune Homeostasis
The immune system has evolved several regulatory mechanisms to prevent autoreactivity to self-antigens and control excessive immune reaction after tissue injury. The presence of regulatory T cells (Treg) suppressing actively (self-reactive) immunity is one of the key mechanisms of preserving immune homeostasis and limiting inflammatory collateral damage [1]. Depletion of CD25 + CD4+ Treg naturally arising in the immune system induces autoimmune diseases, and reconstitution of this cell population prevents disease development [2]. A lack of Treg has been shown to be a primary cause of autoimmune diseases in humans [3]. In addition to maintaining autotolerance, Treg are also involved in control of immune homeostasis in a broad range of immunological conditions, including those against autologous tumor cells [4], allergens [5], and organ transplantation [6]. Although Treg might be simplistically described as immunosuppressive T cells, several phenotypically and functionally distinct Treg subpopulations have been defined [7]. Emerging evidence arises for substantial phenotypic differences between circulating and tissue-infiltrated Treg cells as well as for functional specialization of tissue-localized Treg cells in mice [8], for example, the Treg cells in muscle that promote muscle regeneration [9]. Therefore, it is essential to develop a better understanding of the biology of the site-specific function of Treg cells for control of tissue and immune homeostasis [10]. Despite the large variability of Treg function and phenotype, the best investigated population of Treg are CD4 + CD25 + Foxp3+ naturally occurring Treg [11]. This is neither a homogenous nor static cell population but consists of several specific subsets determined by their origin (thymic or peripherally induced), is resident in most non-lymphoid tissue as site-specific Treg subsets, and is determined by their recent antigen-encounter or activation status [12]. However, virtually all suppressive Treg subsets express the common transcription factor Foxp3 which is directly linked to their immunosuppressive function. Foxp3, an X-chromosome-linked factor that controls Treg cell development and function, is generally thought to positively control Treg cells, as Foxp3 expression is sufficient to determine the immunosuppressive function of conventional T cells [13]. However, at the same time, Foxp3 expression levels—in addition to other factors and effector functions of Treg—determine the “quality” of Foxp3+ cells with regard to the immunosuppressive properties [14]. However, virtually all previous analyses leading to the concept of phenotypic and qualitative differences in the Treg population have been performed by investigating secondary immunological organs such as lymph nodes or spleens. Therefore, this important information about the detailed phenotypic differences of Treg subpopulations—i.e., thymic or peripherally induced Treg, tissue-specific Treg subsets, and subsets characterized by expression of transcription factors and epitope markers [12]—and their specific functional role is missing in the context of acute brain injury.
Regulatory T Cells in Experimental Stroke
Cerebral Treg Invasion
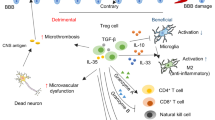
While it is still uncertain at which specific site Treg act as immunomodulators after stroke, several interfaces of Treg-brain interaction can be assumed, such as the endothelium, impact on microglial activation, or direct cell-cell interaction with neurons—which will be discussed later in more detail. In contrast, the kinetics and magnitude of Treg infiltration into the ischemic brain has been characterized in detail in several studies using models of experimental middle cerebral artery occlusion (MCAO), with the limitation that none of the previous studies has systematically characterized Treg subpopulations but focused mainly on natural CD25 + Foxp3+ Treg. One of the first studies systematically analyzing brain leukocyte invasion by flow cytometry of brain homogenates in a model of transient mechanical vascular occlusion (TMVO) using intravascular filaments was performed by Gelderblom et al. [15]. In this study, only a very low number of CD25 + Foxp3+ Treg were observed within the first week after transient ischemia. In contrast, using the distal, permanent MCAO occlusion model [16], we detected substantial T cell and Treg counts in the ischemic hemisphere, with Foxp3+ Treg constituting approx. 20 % of all CD4+ T-helper cells [17]. These findings are consistent with several reports demonstrating that distal permanent occlusion induces a significantly stronger neuroinflammatory reaction with manifold higher T cell counts in the ischemic hemisphere than in proximal transient occlusion models [18, 19]. Stubbe et al. detected after large hemispheric lesions only negligible amounts of Foxp3+ Treg early after stroke, but substantial Treg invasion occurred 14 and 30 days after MCAO, indicating probably delayed kinetics for the adaptive immune response in this TMVO model [20].
Treg Effects on Peripheral Immunity After Brain Injury
Despite their delayed recruitment into the ischemic brain, Treg have been found to influence stroke outcomes already within the first days after ischemia, arguing that effector sites outside the brain parenchyma might be critically involved. In accordance with this assumption, delayed deletion of Treg by pharmacological or genetic depletion at 3 days after MCAO did not affect stroke outcome [20, 21]. A potential extra-cerebral target of Treg in post-stroke immunity might be leukocytes in the peripheral immune system. A consistent finding in the subacute phase after extensive experimental stroke is that cellular immunosuppression and splenic atrophy are accompanied by a relative expansion of Treg in the spleen and blood [22, 23]. Interestingly, a recent report suggested that the adoptive transfer of Treg reduced the systemic inflammatory reaction in the early phase after stroke. On the other hand, Treg transfer also ameliorated the extent of immunosuppression (i.e., lymphopenia) in the subacute phase after brain injury [24]. This finding of a dualistic role of Treg in the peripheral immune system after stroke—abrogating early immune (over-)activation and subacute immunosuppression—is in accordance with the concept that the initial immune over-activation might result in an exhausted immune phenotype and even induce subsequent leukocyte cell death by currently still unclear mechanisms [25]. The potent homeostatic function of Treg might help to suppress the initial systemic inflammation, thereby attenuating the subsequent immune disturbances, such as activation-induced lymphopenia and exhaustion of antigen-presenting cells.
An additional—probably non-immunological—function has been proposed for Treg within the brain vasculature to contribute to coagulation and thrombosis of the microvasculature. It has been demonstrated that Treg might play an important role in the development of secondary microthrombosis TMVO models, which was termed thromboinflammation [26]. This concept of thromboinflammatory infarct progression proposes an interaction of Treg with platelets and the activated endothelium as an independent mechanism of Treg function [27, 28]. This is of prime clinical importance, taking into account the rapidly increasing number of mechanical recanalizations—modeled by the TMVO animal models—and the large number of insufficient reperfusions despite successful recanalization in stroke patients [28, 29].
Cerebral Targets of Treg-Dependent Mechanisms After Stroke
The most prominent immune mechanisms of Treg function in vivo are the secretion of anti-inflammatory cytokines (IL-10, tumor growth factor β (TGF-β), expression of transmembrane immunosuppressant molecules (CTLA-4, CD39, PD), consumption of vital cytokines (IL-2), and the secretion of cytolytic molecules such as granzymes and perforin [30]. Several reports have demonstrated that IL-10 is a critical cytokine-modulating post-stroke neuroinflammation in experimental brain ischemia [31–34]. The main sources of cerebral IL-10 are regulatory lymphocytes as well as microglia/macrophages. Therapeutic approaches to increase lymphocyte-derived IL-10 production [21, 33, 35, 36] or by therapeutic IL-10 administration [31, 34, 37] have resulted in improved stroke outcome. In addition, alternative Treg-related mechanisms have been found to act in ischemic brains, such as the expression of TGF-β and IL-35 [38] as well as a role for Treg in the co-inhibitory PD-1/PD-L1 pathway. It has been shown that the immune regulatory function of PD-1 limits ischemic brain lesions and blood–brain barrier damage [39]. In contrast, PD-L1 appears to have a detrimental effect in stroke [40]. This at first sight contradictory function of receptor and ligand has been attributed to two potential mechanisms: First, PD-Ls might “inhibit-the inhibitors” as PD-1 is also expressed on immunosuppressive cells; secondly, alternative receptors for PD-Ls such as CD80 might have under conditions of acute inflammation after brain ischemia an activatory net effect on T cell activation. This can be explained by the bidirectional PD-L—CD80 interaction and interference with other suppressive signals such as CD80/CD28 on effector T cells [40, 41]. The beneficial effect of Treg at later stages of ischemic stroke was frequently related to the reduced invasion of pro-inflammatory T cells [17, 42], suppression of effector T cell proliferation [21], and reduced cytokine production [38, 42, 43]. Interestingly, others and we have also detected reduced microglia/monocyte activation in the presence of Treg [17], or priming toward an M2-like microglial phenotype [38] and reduced microglial TNF-α production under the influence of Treg [17]. These findings suggested that Treg cells have an impact on the resident immune cells, most likely via soluble mediators such as IL-10 because the effect on microglia could be detected at early time points after stroke even before invasion of substantial numbers of Treg into the injured brain. In contrast, studies reporting an exacerbation of stroke by Treg-targeted therapies during the very early stage [26, 44] found an association of increased lesion volumes with amplified cerebral immune cell accumulation [44]. These effects were most likely independent of the immunological function of Treg but rather attributable to the extracerebral function of Treg in secondary microthrombosis as discussed above [26].
Regulatory T Cells in Stroke Patients
Only few studies analyzed Treg cells in human stroke patients, and these studies were understandably limited to the analysis of blood samples from patients with different stroke entities. Hug et al. have found that Treg function is preserved in the subacute phase after stroke in contrast to the dysfunction of effector cell populations such as circulating monocytes or helper T cells [45] in the context of post-stroke immunosuppression. An opposing study detected an impaired Treg suppressive function in female but not male stroke patients, proposing gender-specific effects in post-stroke immunomodulation [46]. These discrepant findings can be attributed to differing patient characteristics regarding comorbidities and stroke severity. While the latter study has detected a robust increase of Treg cell counts after stroke in accordance with rodent experiments, others have shown the opposite. Li et al. detected a significant reduction of circulating Treg in stroke patients [47]. Overall, clinical data is supporting the experimental finding of substantial peripheral immunomodulation after stroke including the Treg population. However, specific changes might depend on stroke entity, severity, and patient characteristics, and further studies to better characterize the involvement of Treg cells in human stroke are urgently needed.
Treg Depletion in Experimental Brain Ischemia
Depletion of the Treg population is a common experimental paradigm to investigate the functional role of Treg in experimental stroke. Antibody-mediated cell depletion using anti-CD25 antibodies and genetic depletion using transgenic mice with a diphtheria toxin receptor (DTR) transgene under the control of the Foxp3 promoter have been used for efficient Treg depletion. Using Treg-depletion paradigms, about half of the experiments performed revealed an increase in infarct volume16, 19, 28, while the other half did not detect any effect on stroke outcome [21, 26, 48] and one study even observed a reduction of infarct size in Treg-deficient mice [21, 26, 48]. This irreproducibility of findings in-between several studies from independent laboratories led to an intense debate on the biological role of Treg in stroke [49–51]. Notably, inconsistencies cannot be attributed to the specific depletion approach, since transgenic mouse models have been used on both sides of the “efficacy spectrum.” Moreover, each of the three studies using genetic depletion of Treg used a different inducible Foxp3-KO mouse line [21, 26, 48]. Each of these transgenic Foxp3-deletion models are derived from different transgenic constructs and differ in their depletion efficacy. Moreover, previous reports have demonstrated a substantial dysregulation of peripheral immune homeostasis upon Treg depletion [52]. Treg depletion induces rapid expansion of lymphocytes and dysbalance in peripheral immune homeostasis in the first week after depletion. Hence, small methodological differences in the depletion paradigm itself or by priming of lymphocytes during this vulnerable phase by environmental factors, such as housing conditions, the specific microbiota conditions, or experimental stressors, might have an unforeseen impact on immunological outcome [53, 54]. The immunological effects of Treg depletion on the neuroinflammatory response after brain ischemia have been investigated only in a fraction of the published reports. All three studies detecting an exacerbation of stroke outcome after Treg depletion also observed an associated increase in neuroinflammatory biomarkers [17, 21, 38]. The most robust findings included an increase in cerebral leukocyte invasion and an increase in pro-inflammatory cytokine secretion. Interestingly, a previous study investigating the role of Treg in secondary microthrombosis and stroke infarct progression has detected a detrimental role of Treg in mediating post-stroke neurodegeneration which was independent of their immunological function and alterations in post-stroke neuroinflammation [26]. Accordingly, “wannabe Treg” without immunomodulatory function were still able to exert their impact on stroke progression without affecting neuroinflammatory markers, supporting the hypothesis of non-immunological functions of Treg in stroke models with potential endothelial injury, vascular inflammation, and microthrombosis.
Therapeutic Enhancement of Treg Function in Brain Ischemia
Despite the discrepancies on Treg function in Treg-depletion paradigms, several reports have tested Treg expansion approaches for stroke therapy. These studies have investigated very different methods to increase Treg numbers and/or function. The most intuitive approach might be the adoptive cell transfer of purified Treg to recipient animals to increase circulating Treg counts. The second most widely used approach was the administration of a CD28 superagonist (CD28SA), which induces in vivo expansion of Treg and amplification of their suppressive function. A pathomechanistically distinctive approach used in previous studies was the paradigm of “mucosal immunization” by administration of cerebrovascular antigens, such as myelin oligodendrocyte glycoprotein, or selectins, which should result in the expansion of autoantigen-specific Treg [43, 55]. Out of 16 experiments reported until December 2015 in 14 independent studies, 13 found an improvement in stroke outcome [21, 24, 35, 38, 39, 42, 43, 55–57], while two described an increase in infarct volume when using two independent Treg-targeted therapies [26, 44] and one study did not detect an effect at all [58] (Table 1). This discrepancy cannot be easily explained by the therapeutic paradigm used, since the methods of adoptive Treg transfer into wild-type mice and the CD28SA treatment utilized in the two studies showing an exacerbation of lesion volume were also tested in three or more other experiments that showed an improved outcome (Table 1). It appears that stroke severity or other factors directly eminent to the specific stroke model might predict the net biologic effect of Treg: The majority of the 13 experiments detecting an overall benefit of Treg boosting was performed in stroke models with small- to moderate-sized lesion volumes. Surprisingly, however, the two studies [35, 44] in which the identical CD28SA has been used in very similar stroke models reported opposing results. Na et al. described improved stroke outcome following treatment with the CD28SA [35], while Schuhmann et al. observed a significant deterioration of stroke outcome [44].
We have performed a meta-analysis to better estimate the efficacy of Treg-targeted therapeutic approaches in all studies published until December 2015 using therapeutic paradigms of Treg targeting in experimental stroke models. All studies with the aim to therapeutically modulate Treg (defined at least by CD25 or Foxp3 expression in CD4+ T-helper cells) numbers and/or Treg function in models of experimental brain ischemia were included in the analysis. The studies were clustered by the respective treatment paradigm: mucosal immunization [43, 57], adoptive Treg transfer [24, 26, 39, 42, 56], including intracerebroventricular Treg injection [58], CD28SA therapy [35, 44, 56], and others (i.e., HDAC inhibition [21] and mTOR inhibition [38]). Odds ratios for effect size estimation were calculated and the forest plot illustrated using RevMan software (version 5.3). The overall effect size estimation revealed an odds ratio favoring each subgroup of interventions and for all studies in total (Fig. 1a). However, these results have to be interpreted with caution due to several critical caveats: Funnel plot analysis displayed substantial asymmetry, suggesting publication bias and lack of reporting neutral or negative results (Fig. 1b). Moreover, a large heterogeneity between the included studies has to be taken into account regarding study design, experimental model, and species (rat and mice). In addition, most studies included in this meta-analysis did not adhere to the guidelines on data reporting according to the Stroke Therapy Academic Industry Roundtable (STAIR) recommendations. Therefore, further studies investigating Treg function in acute brain injury are urgently needed to increase the robustness of effect size estimation. Also, the publication of negative results should be encouraged to avoid a publication bias in this emerging research field. We have recently reported the feasibility and value of performing multicenter preclinical studies (pRCTs) adopting key elements of clinical randomized controlled trials (RCT) in such situations where reports from individual laboratories are discrepant, irreproducible, or simply not comparable due to their vast heterogeneity [19, 59]. Performing such a multicenter pRCT using a unified and highly standardized study protocol across laboratories might resolve the uncertainty about the role of Treg-targeted therapies in experimental stroke and guide further translational research in this field.

Meta-analysis for Treg-targeted therapies in experimental brain ischemia. a The meta-analysis, depicted as a forest plot, includes all published studies until 30th of November 2015 investigating Treg (at least characterized by CD25− and/or Foxp3-expressing CD4+ cells) in rodent stroke models. Odds ratios were calculated using inverse variance in a fixed effects model. CI confidence interval. b Publication bias was estimated by illustration of the odds ratio (OR, x-axis) and the logOR standard error (SE, y-axis) in a funnel plot
Reasons for Discrepant Treg Function on Stroke Outcome
The Role of Infarct Size in Post-stroke Neuroinflammation
Evaluating all published reports on Treg function in experimental stroke suggests that lesion size might be a systematic confounder independent of reperfusion. It has been consistently reported that post-stroke peripheral immunosuppression occurs in stroke patients and animal stroke models only after extensive brain tissue injury [23, 60–62]. While mice and humans with substantial brain lesions develop lymphopenia and changes in monocyte subpopulation, small brain lesions induce only a minor immunomodulation, but no immunosuppressive syndrome [23, 63]. Surprisingly, the impact of peripheral immunosuppression after substantial brain injuries on the neuroinflammatory reaction has to our knowledge not been systematically investigated until now, but it is plausible that peripheral immune alterations might also affect central neuroinflammation. Indeed, a previous study performing a face-to-face comparison of the extent of cerebral leukocyte invasion, microglial activation, and cytokine secretion in three common models of brain ischemia of differing lesion size detected crucial differences among models with a manifold stronger inflammatory reaction in small permanent ischemia models than in extensive hemispheric lesions using TMVO models [18]. This discrepancy in the extent of the neuroinflammatory response between stroke models has also been confirmed in a recent multicenter experimental stroke trial [19]. The impact of brain-invading leukocytes on the local neuroinflammatory milieu became particularly evident in a previous study where inhibition of cerebral leukocyte invasion by antibodies against the alpha4-integrin (Anti-CD49d) resulted also in reduced microglial cell activation [64]. Therefore, it is plausible that Treg has an inferior role in stroke models, with only minor bystander inflammation. This comparably low neuroinflammatory response might be due to the secondary immunosuppression after extensive brain lesions or due to currently unknown factors (such as differences in skull trepanation, surgical wound, pain, and food intake) also in other stroke models, which have not been systematically characterized in respect to their inflammatory component. Moreover, the extent of secondary neuroinflammation and contribution to outcome has until now not been characterized in human stroke subtypes. These studies are urgently needed to get a better understanding of the specific inflammatory milieu and its role in stroke outcome depending on lesion size and location.
Secondary Microthrombosis in TMVO Stroke Models
The most commonly used acute brain ischemia models in experimental stroke research are transient mechanical vascular occlusion (TMVO) models with potential endothelial damage and induction of secondary microthrombosis [27, 65, 66]. Notably, such an experimental stroke model was used in both studies detecting a deleterious role of Treg in stroke [26, 44]. A decisive feature of this model is the occurrence of delayed neuronal damage due to secondary microthrombosis [65]. In contrast, secondary microthrombosis was not observed in animal models with gradual reperfusion [66]. Furthermore, the rate of secondary microthrombosis after recanalization in stroke patients is currently still unclear; however, microthrombosis might substantially contribute to pathophysiology particularly after mechanical recanalization of proximal arteries. Several potential mechanisms leading to microthrombosis and thromboinflammation have been suggested: (1) the extent of endothelial activation induced by the prompt reperfusion, (2) endothelial damage and thereby activation of the contact-dependent coagulation system by the intravascular manipulation, and (3) the occlusion time itself and thereby potential endothelial ischemic injury [27, 28, 67]. These methodological differences might have a major impact on the presence of thromboinflammation and secondary microthrombosis. Considering that microthrombosis is at least partially an inflammation-independent mechanism, the occurrence or absence of such a confounder to a cerebrovascular disease model has justifiably a critical impact on outcome. Moreover, the occurrence of secondary microthrombosis will shift the importance of local tissue-specific pathophysiological cascades toward the role of intravascular events, hence underestimating the contribution of inflammation in tissue homeostasis in cases where no microthrombosis/thromboinflammation occurs. The cascade of endothelial activation, thromboinflammation, and microvascular dysfunction in stroke models and potentially also in patients with pronounced endothelial damage not only leads to secondary ischemic infarct progression but will also alter the neuroinflammatory response to brain ischemia as such.
Milieu-Dependent Treg Function
Moreover, the above-stated differences between different experimental models as well as potentially between clinical stroke subtypes—including the occurrence of secondary microthrombosis, induction of peripheral immune alterations, and the extent of local neuroinflammation—might have a direct impact on Treg function. It is known from other disease paradigms that Treg function in vivo might depend on the specific immunologic milieu in order to preserve tissue homeostasis. In cancer immunology, this phenomenon has been termed the “Janus-faced function of Treg” [68, 69]. The concept that Treg adapt their suppressive function to a particular inflammatory milieu is based on functional studies of transcription factors, prominently the members of interferon regulatory factor (IRF) among others. It has been demonstrated that several transcription factors such as IRF, BLIMP-1, and GATA-3 are associated with Foxp3 regulation and contribute in a tissue-specific manner to Treg activation and function [70]. Therefore, the same genetic program in Treg might on one side contribute to tissue homeostasis (e.g., preventing an inflammatory collateral damage in the brain parenchyma after stroke) and at the same time has a detrimental function at a different site associated with the same disease (e.g., promoting thromboinflammation within the cerebral microvasculature). This hypothesis is well in line with the recent finding that Treg effector function is tightly controlled and differentially regulated by metabolic cues such as extracellular ATP and hypoxia [71], which evidently differs between post-stroke vasculature and the brain parenchyma. Therefore, it is conceivable that differences in oxygen partial pressure, glucose supply, and consequently ATP concentrations will differentially affect Treg function within the different compartments in the post-ischemic brain.
Particularly, the currently prevalent perception of post-stroke neuroinflammation as too much of a bad thing should be revisited. Several previous reports in primary autoimmune diseases as well as acute brain injury models have established the concept of “protective autoimmunity” (see Schwartz and Raposo [72] for a review). This concept ascribes secondary inflammation as a generally physiological and protective mechanism in which excessive immune activation as well as immunosuppression might be deleterious. Hence, in certain situations of post-stroke neuroinflammation—determined by the complex interplay of model-dependent features, kinetics, and the tissue-specific milieu—both Treg depletion as well as expansion might have a negative effect on stroke outcome [73–75]. Therefore, the immunological properties of the used stroke model, the targeted mechanisms of Treg function and potentially differing tissue-specific Treg functions need to be carefully considered before ascribing Treg in a potentially oversimplified view as good or bad immune cells after stroke.
References
Sakaguchi S. Regulatory T, cells: key controllers of immunologic self-tolerance. Cell. 2000;101(5):455–8.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Ochs HD, Ziegler SF, Torgerson TR. FOXP3 acts as a rheostat of the immune response. Immunol Rev. 2005;203:156–64.
Baecher-Allan C, Anderson DE. Regulatory cells and human cancer. Semin Cancer Biol. 2006;16(2):98–105.
Chatila TA. Role of regulatory T cells in human diseases. J Allergy Clin Immunol. 2005;116(5):949–59.
Battaglia M, Roncarolo MG. Induction of transplantation tolerance via regulatory T cells. Inflamm Allergy Drug Targets. 2006;5(3):157–65.
Jiang H, Chess L. Regulation of immune responses by T cells. N Engl J Med. 2006;354(11):1166–76.
Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol. 2013;14(10):1007–13.
Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–95.
Ding Y, Xu J, Bromberg JS. Regulatory T cell migration during an immune response. Trends Immunol. 2012;33(4):174–80.
Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73.
Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–65.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4(+)CD25(+) regulatory T cells. Nat Immunol. 2003;4(4):330–6.
Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445(7129):766–70.
Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–57.
Llovera G, Roth S, Plesnila N, Veltkamp R, Liesz A. Modeling stroke in mice: permanent coagulation of the distal middle cerebral artery. J Vis Exp. 2014;89:e51729.
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15(2):192–9.
Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, et al. Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol. 2013;23(1):34–44.
Llovera G, Hofmann K, Roth S, Salas-Perdomo A, Ferrer-Ferrer M, Perego C, et al. Results of a preclinical randomized controlled multicenter trial (pRCT): anti-CD49d treatment for acute brain ischemia. Sci Transl Med. 2015;7(299):299ra121.
Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2013;33(1):37–47.
Liesz A, Zhou W, Na SY, Hammerling GJ, Garbi N, Karcher S, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013;33(44):17350–62.
Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176(11):6523–31.
Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009;40(8):2849–58.
Li P, Mao L, Zhou G, Leak RK, Sun BL, Chen J, et al. Adoptive regulatory T-cell therapy preserves systemic immune homeostasis after cerebral ischemia. Stroke. 2013;44(12):3509–15.
Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP Signaling is a Key Pathway Inducing Immune Modulation after Brain Injury. J Neurosci. 2015;35(2):583–98.
Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Gobel K, Schuhmann MK, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121(4):679–91.
Bai J, Lyden PD. Revisiting cerebral postischemic reperfusion injury: new insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke. 2015;10(2):143–52.
Nieswandt B, Kleinschnitz C, Stoll G. Ischaemic stroke: a thrombo-inflammatory disease? J Physiol. 2011;589(Pt 17):4115–23.
Chen CJ, Ding D, Starke RM, Mehndiratta P, Crowley RW, Liu KC, et al. Endovascular vs medical management of acute ischemic stroke. Neurology. 2015;85(22):1980–90.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490–500.
Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett. 1998;251(3):189–92.
Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, et al. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci. 2000;12(7):2265–72.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis. 2013;28(3):375–86.
Liesz A, Bauer A, Hoheisel JD, Veltkamp R. Intracerebral interleukin-10 injection modulates post-ischemic neuroinflammation: an experimental microarray study. Neurosci Lett. 2014;579:18–23.
Na SY, Mracsko E, Liesz A, Hunig T, Veltkamp R. Amplification of Regulatory T Cells Using a CD28 Superagonist Reduces Brain Damage After Ischemic Stroke in Mice. Stroke. 2015;46(1):212–20.
Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci. 2005;233(1-2):125–32.
Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, et al. Interleukin-10 in the brain. Crit Rev Immunol. 2001;21(5):427–49.
Xie L, Sun F, Wang J, Mao X, Xie L, Yang SH, et al. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol. 2014;192(12):6009–19.
Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, et al. Essential role of program death 1-ligand 1 in regulatory T-cell-afforded protection against blood-brain barrier damage after stroke. Stroke. 2014;45(3):857–64.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. PD-L1 enhances CNS inflammation and infarct volume following experimental stroke in mice in opposition to PD-1. J Neuroinflammation. 2013;10:111.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74(3):458–71.
Ishibashi S, Maric D, Mou Y, Ohtani R, Ruetzler C, Hallenbeck JM. Mucosal tolerance to E-selectin promotes the survival of newly generated neuroblasts via regulatory T-cell induction after stroke in spontaneously hypertensive rats. J Cereb Blood Flow Metab. 2009;29(3):606–20.
Schuhmann MK, Kraft P, Stoll G, Lorenz K, Meuth SG, Wiendl H, et al. CD28 superagonist-mediated boost of regulatory T cells increases thrombo-inflammation and ischemic neurodegeneration during the acute phase of experimental stroke. J Cereb Blood Flow Metab. 2015;35(1):6–10.
Hug A, Liesz A, Muerle B, Zhou W, Ehrenheim J, Lorenz A, et al. Reduced Efficacy of Circulating Costimulatory Cells After Focal Cerebral Ischemia. Stroke. 2011;42(12):3580–6.
Yan J, Read SJ, Henderson RD, Hull R, O’Sullivan JD, McCombe PA, et al. Frequency and function of regulatory T cells after ischaemic stroke in humans. J Neuroimmunol. 2012;243(1-2):89–94.
Li Q, Wang Y, Yu F, Wang YM, Zhang C, Hu C, et al. Peripheral Th17/Treg imbalance in patients with atherosclerotic cerebral infarction. Int J Clin Exp Pathol. 2013;6(6):1015–27.
Ren X, Akiyoshi K, Vandenbark AA, Hurn PD, Offner H. CD4 + FoxP3+ regulatory T-cells in cerebral ischemic stroke. Metab Brain Dis. 2011;26(1):87–90.
Kleinschnitz C, Wiendl H. Con: regulatory T cells are protective in ischemic stroke. Stroke. 2013;44(8):e87–8.
Hu X, Li P, Chen J. Pro: regulatory T cells are protective in ischemic stroke. Stroke. 2013;44(8):e85–6.
Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009;158(3):1174–83.
Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191–7.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23.
Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140(6):845–58.
Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. 2008;39(5):1575–82.
Brea D, Agulla J, Rodriguez-Yanez M, Barral D, Ramos-Cabrer P, Campos F, et al. Regulatory T cells modulate inflammation and reduce infarct volume in experimental brain ischaemia. J Cell Mol Med. 2014;18(8):1571–9.
Chen Y, Ruetzler C, Pandipati S, Spatz M, McCarron RM, Becker K, et al. Mucosal tolerance to E-selectin provides cell-mediated protection against ischemic brain injury. Proc Natl Acad Sci U S A. 2003;100(25):15107–12.
Wang J, Xie L, Yang C, Ren C, Zhou K, Wang B, et al. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front Cell Neurosci. 2015;9:361.
Kleikers PW, Hooijmans C, Gob E, Langhauser F, Rewell SS, Radermacher K, et al. A combined pre-clinical meta-analysis and randomized confirmatory trial approach to improve data validity for therapeutic target validation. Sci Rep. 2015;5:13428.
Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6(10):775–86.
Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. 2012;8(7):401–10.
Liesz A, Ruger H, Purrucker J, Zorn M, Dalpke A, Mohlenbruch M, et al. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS One. 2013;8(9):e74839.
Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke. 2009;40(10):3226–32.
Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134(Pt 3):704–20.
Hossmann KA. The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J Cereb Blood Flow Metab. 2012;32(7):1310–6.
Gauberti M, Martinez de Lizarrondo S, Orset C, Vivien D. Lack of secondary microthrombosis after thrombin-induced stroke in mice and non-human primates. J Thromb Haemost. 2014;12(3):409–14.
Gob E, Reymann S, Langhauser F, Schuhmann MK, Kraft P, Thielmann I, et al. Blocking of plasma kallikrein ameliorates stroke by reducing thromboinflammation. Ann Neurol. 2015;77(5):784–803.
Levings MK, Allan S, d’Hennezel E, Piccirillo CA. Functional dynamics of naturally occurring regulatory T cells in health and autoimmunity. Adv Immunol. 2006;92:119–55.
Danese S, Rutella S. The Janus face of CD4 + CD25+ regulatory T cells in cancer and autoimmunity. Curr Med Chem. 2007;14(6):649–66.
Rudra D, de Roos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012;13(10):1010–9.
Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. 2015;21(6):638–46.
Schwartz M, Raposo C. Protective autoimmunity: a unifying model for the immune network involved in CNS repair. Neuroscientist. 2014;20(4):343–58.
Walsh JT, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J. Regulatory T cells in central nervous system injury: a double-edged sword. J Immunol. 2014;193(10):5013–22.
Kipnis J, Schwartz M. Controlled autoimmunity in CNS maintenance and repair: naturally occurring CD4 + CD25+ regulatory T-Cells at the crossroads of health and disease. Neuromolecular Med. 2005;7(3):197–206.
Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4 + CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A. 2002;99(24):15620–5.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This work was supported the Excellence Cluster for Systems Neurology (SyNergy) and by grants from the German Research Foundation (DFG), project LI2534/1-1 to A.L. and SFB 688, project A13 and KL2323/6-1 to C.K.
Conflict of Interest
Both authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Liesz, A., Kleinschnitz, C. Regulatory T Cells in Post-stroke Immune Homeostasis. Transl. Stroke Res. 7, 313–321 (2016). https://doi.org/10.1007/s12975-016-0465-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-016-0465-7