
Abstract
Carbon monoxide (CO) is extremely poisonous to the Pt catalyst resulting in the available reaction surface reduction and fuel cell performance degradation. Therefore, the reform of natural gas or other hydrocarbons is avoided to be used as the CO is inevitable. CO-tolerant catalysts are required to overcome this bottleneck. Herein, MXene (Ti3C2Tx) and carbon nanotube (CNT) hybrid materials are used as the catalytic support to load Pt nanoparticles (NPs) to achieve the anode catalyst for proton-exchange-membrane fuel cell (PEMFC). The Pt/Ti3C2Tx-CNT catalyst performs higher hydrogen oxidation reaction (HOR) activities and stability compared with the Pt/C catalysts, and the peak power density of PEMFC based on the Pt/Ti3C2Tx-CNT catalyst achieves 0.86 W cm−2 with H2/air and 1.6 W cm−2 with H2/O2. Furthermore, the Pt/Ti3C2Tx-CNT catalyst exhibits superior electrocatalyst activity and remarkable CO-tolerant performance, which could be attributed to the metal-support interactions between Pt nanoparticles and Ti3C2Tx-CNT hybrid supports.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
PEMFC is attractive energy storage equipment for vehicles and other applications because of its high energy efficiency and being environmentally friendly [1, 2]. Hydrocarbons and other liquid fuels (for example, methanol) store a lot of energy [3, 4]. Nowadays, the majority of hydrogen production comes from the steam reforming of natural gas or other hydrocarbons, which makes hydrogen at a lower cost [5]. However, one problem associated with the reformer is the incomplete reaction resulting in inevitable amounts of carbon monoxide (CO) [6]. CO is extremely poisonous to the Pt catalyst because of easier absorption of CO to Pt surface rather than H2, which will result in a reduction of activity area and degradation of the fuel cell performance [7,8,9]. There are many significant efforts have been made to overcome the CO poisoning problem; one approach is Pt-based alloy to fabricate CO-tolerant bimetallic electrocatalysts (PtM, where M = Fe, Ni, Co, Mo, Ru, Sn), [10,11,12,13,14,15,16,17] that the non-noble metals improve the CO tolerance of bimetallic electrocatalysts through the electronic effect and bifunctional or oxyphilic effect (accelerating the removal of adsorbed CO by supplying species containing oxygen) [18,19,20]. On the other hand, the application of metal-support interactions represents a promising strategy to perfectly solve the problem of catalyst poisoning problem, which not only improves the catalyst activity and stability but also alters the electronic properties of metal and thus weakens the metal-CO interaction [21].
Metal oxides are the main support for metal-support interactions with metal NPs [22, 23]. However, the poor electronic conductivity of metal oxides restricts their application as catalyst support for PEMFC. MXene, a two-dimensional (2D) material, has attracted great attention as catalyst supports due to its kinetic-favorable layered nanostructure, high conductivity, and good surface reactivity [24,25,26,27,28,29]. Recently, Li et al. [30] reported a platinum nanoparticle catalyst supported on Nb2CTx MXene for water–gas shift reaction, which exhibited weaker CO adsorption than monometallic platinum due to the formation of a Pt–Nb surface alloy. Moreover, MXene as the support of metal catalyst (Pt, Pb, Co, Cu) can significantly improve CO oxidation reaction even at a lower temperature [31,32,33]. Unfortunately, two-dimensional MXene sheets demonstrated would suffer from aggregation and restacking due to van der Waals interactions and hydrogen bonds. However, the combination of MXene nanosheets and carbon nanomaterials to design MXene-carbon hybrid support is a considerable approach to preventing restacking of MXene nanosheets and improving electrochemical performance [34, 35]. Herein, Pt is loaded on Ti3C2Tx-CNT hybrid support by microwave reduction. This configuration enhanced the reaction activity and stability because of the synergistic effect between Pt and Ti3C2Tx-CNT. The Pt/Ti3C2Tx-CNT electrocatalysts demonstrate a much better CO tolerance than the Pt/C catalyst.
Experimental Section
Synthesis of Ti3C2Tx MXene
The preparation of Ti3C2Tx nanosheets was carried out as follows [36]: 0.8 g LiF was injected into 10 ml 9 M HCl solution with stirring at 20 °C for 5 min, and then 0.5 g Ti3AlC2 was added into the solution with 24 h magnetically stirring at 35 °C. The precipitates were rinsed with deionized water several times until the pH was around 7. Finally, the collected supernatant was collected and vacuum dried.
Synthesis of Ti3C2Tx-CNT Hybrid Support
Ti3C2Tx-CNT hybrid support was prepared by a self-assembly process. A total of 100-ml CNT aqueous (1 mg ml−1) and 100-ml CTAB aqueous (2 mg ml−1) were ultrasonically mixed for 2 h in ambient conditions to fabricate CTAB-CNT, and then CTAB-CNT solution (1 mg ml−1) was added to Ti3C2Tx suspension (1 mg ml−1).
Synthesis of Pt/Ti3C2Tx-CNT Catalyst
Pt/Ti3C2Tx-CNT catalyst was prepared by a microwave-assisted polyol reduction method as shown in Scheme S1. Fifty milligrams of Ti3C2Tx-CNT hybrid support and 1.24 ml of H2PtCl6·6H2O aqueous (40 mg ml−1) were mixed with 50 ml of ethylene glycol solution in a custom-built flask with 2 h of magnetically stirring and adjusting the mixture pH to 10. Finally, the flask was placed in a microwave oven and heated to 160 °C at a power of 500 W for 5 min and filtering precipitates with enough deionized water. After being dried at 60 °C in a vacuum oven overnight, the Pt/Ti3C2Tx-CNT catalyst sample was collected.
Characterizations
X-ray diffraction (XRD) patterns were examined by X-ray diffraction (XRD, Philips X’Pert Pro diffractometer) with a CuKα X-ray source (λ = 1.5405 Å). Raman patterns were characterized using a HR Evolution Microscopic confocal laser Raman spectrometer. X-ray photoelectron spectroscopy (XPS) data were recorded using an ESCALAB250Xi model. The morphologies of the materials were obtained by JEM1400FLASH and high-resolution transmission electron microscopy (HRTEM). The Pt loading amount on the CNT modified MXene was measured by Agilent 5110 ICP-OES plasma spectrometer, and the Pt loading amount in the measured Pt/Ti3C2TX-CNT is 20%.
Electrochemical Measurement
Electrochemical test was under a three-electrode system (working electrode: a glassy carbon rotating disk electrode (area: 0.1963 cm2); reference electrode: Ag/AgCl electrode; counter electrode: graphite rod), and the electrochemical experiments were tested in 0.1 M HClO4 solution (the reversible hydrogen electrode potential is calculated according to E(RHE) = E(Ag/AgCl) +0.0591 * pH +0.197). The working electrode ink was prepared through dissolving 20% Pt/Ti3C2Tx-CNT and 20%Pt/C in solution (500 μL of deionized water: 450 μL of isopropanol: 50 μL of 5 wt% Nafion solution), and 18 μl catalyst ink was loaded onto the glassy carbon electrode (the Pt loading was 0.092 mg cm−2). The electrochemical surface areas (ECSA) were calculated by the following equation:
where \({\mathrm{S}}_{H}\) is the integrated area of hydrogen desorption peak or hydrogen adsorption peak of the CV, v is for the scanning speed of the CV, and \({M}_{\mathrm{pt}}\) represents the loading mass of Pt on the WE.
CV, LSV, and CO-stripping measurements were worked in 0.1 M HClO4 solution. CV was measured between 0 and 1.2 V (vs. RHE) at a scan rate of 50 mV s−1, and LSV was measured at the potential range of −0.1–0.6 V (vs. RHE) with 10 mV s−1 and 1600 rpm. The catalysts in the electrolyte solution were saturated with CO at 0.1 V (vs. RHE) for 30 min and purged with N2 to remove excess CO from the solution for CO stripping. Accelerated stability tests of the Pt/Ti3C2Tx-CNT catalysts and the commercial Pt/C catalysts were carried out by a consecutive sweep from 0 to 1.2 V at 50 mV s−1 for 1000 potential cycles in a 0.1 M HClO4 solution.
Fuel Cell Performance Test
The catalyst ink was obtained by mixing commercial 20 wt% Pt/C or 20 wt% Pt/Ti3C2Tx-CNT catalysts for the anode and commercial 60 wt% Pt/C catalyst for the cathode, respectively. The homogeneous ink was sprayed to the surface of a 5 cm2 membrane by ultrasonic spraying. The Pt loadings of cathode and anode were 0.3 mg cm−2 and 0.1 mg cm−2, respectively. H2 or a mixture of H2 with CO and Air/O2 were fed into the cell at flow rates of 150 cm3 min−1 and 300 cm3 min−1 (air) or 150 cm3 min−1 (O2), respectively. For the CO-tolerance test, the current density of 0.8 A cm−2 was supplied constantly, and the drop in the voltage was recorded.
Results and Discussions
The XRD of Pt/Ti3C2Tx-CNT catalysts is exhibited in Fig. 1a. A characteristic peak of Ti3C2Tx left shift from 9.66 to 6.74° was observed, which indicates the increased interlayer space because of the successful etching and intercalation. The diffraction peaks of Pt at 39.96°, 46.37°, and 67.77° corresponded to (111), (200), and (220), respectively. These results demonstrated that Pt/Ti3C2Tx-CNT had been successfully synthesized. Meanwhile, the disappearance of peak (002) of Ti3C2Tx in Pt/Ti3C2Tx-CNT represents the incorporation of CNT, which led to an increased interplanar spacing of the MXene flakes when the hybrid support was achieved. All catalysts exhibit obvious peaks at 1360 cm−1 (D band) and 1580 cm−1 (G band) in Raman spectra of the Pt/Ti3C2Tx-CNT and Pt/C catalysts as shown in Fig. 1b. The Pt/Ti3C2Tx-CNT catalyst showed a higher ID/IG ratio, which indicates the Pt/Ti3C2Tx-CNT catalyst had a higher degree of graphite domain or defects, with a better load capacity of Ti3C2Tx-CNT hybrid support. The XPS spectra in Fig. 1c reveal the binding energy of C 1 s (284 eV) and Pt 4f (75 eV) in the Pt/C, and the XPS survey of the Pt/Ti3C2Tx-CNT catalyst presented the elements of Pt 4f, C 1 s, Ti 2p and O 1 s with binding energies of 74 eV, 284 eV, 459 eV, and 531 eV, respectively. The Pt 4f5/2 and Pt 4f7/2 binding energy of Pt/Ti3C2Tx-CNT in Fig. 1d were 71.93 eV and 74.93 eV, which 0.45 eV and 0.3 eV left shifted from Pt/C, respectively. These shifts indicated a stronger interaction between the Ti3C2Tx-CNT and Pt nanoparticles than that between carbon and Pt. The peaks of Pt 4f5/2 and Pt 4f7/2 corresponded to Pt0 and Pt2+ peaks, respectively (as shown in Fig. S2 and Table S1), which indicated the formation of the oxidized Pt species.

a XRD patterns of Pt/Ti3C2Tx-CNT, Ti3C2Tx, and Ti3AlC2; b Raman spectra of and Pt/C; c XPS spectra of Pt/Ti3C2Tx-CNT and Pt/C; and d Pt 4f regions of Pt/C and Pt 4f regions of Pt/C and Pt/Ti3C2Tx-CNT
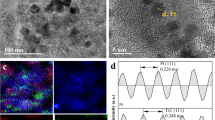
TEM and HRTEM images as shown in Fig. 2a, b indicated continuous substrates formed by lamellar Ti3C2Tx and tubular CNTs. Pt nanoparticles were uniformly distributed on the Ti3C2Tx-CNT hybrid support and the Pt particles with a diameter of around 3.36 nm. Figure S1 presents the TEM of layered Ti3C2Tx. Figure 2c exhibits that the fringe spacing was around 0.23 nm, which could be recognized as Pt (111) plane in face-centered cubic (fcc) structure. Moreover, the energy-dispersive system (EDS) mapping of the Pt/Ti3C2Tx-CNT catalyst in Fig. 2d further confirmed the uniform distribution of characteristic elements, Ti and O in Ti3C2Tx, and the C element from both Ti3C2Tx and CNT. These proved the good dispersibility of Pt nanoparticles on the ultrathin Ti3C2Tx-CNT hybrid support.

a TEM image of Pt/Ti3C2Tx-CNT, b, c HRTEM image of Pt/Ti3C2Tx-CNT, and d EDS mapping of Pt/Ti3C2Tx-CNT
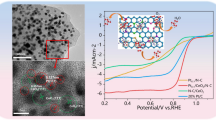
As shown in Fig. 3a, b, the solid line indicated the CV and LSV curves for pure H2 circumstance, and the ECSAs of Pt/Ti3C2Tx-CNT and Pt/C catalysts were calculated as 68.65 m2 g−1 and 49.19 m2 g−1, respectively. The Pt/Ti3C2Tx-CNT catalysts exhibited a slightly larger ECSA than the Pt/C catalyst. Figure 3b shows all of the catalysts exhibited the overpotential for H2 oxidation obviously in pure H2; the Pt/Ti3C2Tx-CNT catalyst provided a higher current density, indicating a higher HOR catalytic activity. A total of 1000 ppm CO with an H2 mixture was applied to examine the CO tolerance performance of the catalyst; the ECSAs of Pt/Ti3C2Tx-CNT and Pt/C catalysts were 65.5 m2 g−1 and 41.86 m2 g−1, respectively. The performance decrease of Pt/Ti3C2Tx-CNT catalyst (4.6% ECSA lost) is negligible compared to Pt/C (14.9% ECSA lost). Moreover, the limit current of Pt/Ti3C2Tx-CNT in Fig. 3b decreased much less than that of Pt/C. As shown in Fig. 3d, the onset potential and the maximum peak for CO oxidation acquired from the Pt/C catalyst are at 0.53 V and 0.85 V, while the onset potential and the maximum peak for CO oxidation are 0.41 V and 0.73 V for Pt/Ti3C2Tx-CNT catalyst, respectively. These lower values indicate that the Pt/Ti3C2Tx-CNT catalyst has superior CO oxidation activity than Pt/C because MXene could promote the oxidation of adsorbed CO at lower potentials [31,32,33]. As shown in Fig. S3, it could be seen that an obvious ECSA drop (about 35.6%) of Pt/C catalyst after 1000 cycles of potential sweeping according to the dissolution and sintering of Pt particles and/or the corrosion of the carbon support. However, the Pt/Ti3C2Tx-CNT catalyst exhibited a smaller ECSA drop; this superior stability may be attributed to the strong interaction between the Pt nanoparticles and the Ti3C2Tx-CNT hybrid supports. Moreover, the CNT could also prevent the agglomeration of MXene and reduce the deformation of Pt nanoparticles [34, 35].

a CV curves for Pt/Ti3C2Tx-CNT and Pt/C, b LSV curves for Pt/Ti3C2Tx-CNT and Pt/C, c The ECSA of Pt/Ti3C2Tx-CNT and Pt/C, and d CO stripping voltammograms of Pt/Ti3C2Tx-CNT and Pt/C
As shown in Fig. 4a, the peak power densities of fuel cells based on Pt/Ti3C2Tx-CNT were 0.86 W with H2/Air cm−2 and 1.6 W cm−2 with H2/O2, respectively. The fuel cell based on Pt/Ti3C2Tx-CNT and commercial Pt/C catalysts both exhibit similar open-circuit voltages (OCVs) with H2/air in Fig. 4b. Moreover, it can be seen that the current density based on Pt/C anode decreased 29.4% at 0.65 V when CO mixture gas was filled in, while only an 11.1% reduction at 0.65 V based on Pt/Ti3C2Tx-CNT anode. Pt/C anode also exhibits larger decaying at 0.5 V and 0.8 V than Pt/Ti3C2Tx-CNT anode in Fig. 4c. These results identified that the Pt/Ti3C2Tx-CNT catalyst could provide better performance than the commercial Pt/C catalyst both in the pure H2 and CO/H2 mixture gas. CO-tolerance durability evaluation of the Pt/Ti3C2Tx-CNT catalysts is demonstrated in Fig. 4d. A total of 5 ppm CO/H2 results in a fast decrease of the relative voltage. The durability was enhanced to over 80 min with the Pt/Ti3C2Tx-CNT catalyst. These results clearly demonstrate that the Pt/Ti3C2Tx-CNT catalyst has a much better CO tolerance than that of commercial Pt/C.

a Polarization curves for MEA (anode: Pt/Ti3C2Tx-CNT, cathode: Pt/C), b polarization curves for Pt/C and Pt/Ti3C2Tx-CNT as anode catalysts in the presence of 100 ppm CO under H2/O2 conditions, c current density for Pt/C and Pt/Ti3C2Tx-CNT as anode catalysts in different voltages in the presence of 100 ppm CO under H2/O2 conditions, and d relative voltage vs. time curves of Pt/C and Pt/Ti3C2Tx-CNT as anode catalysts in the presence of 5 ppm CO at a current density of 0.8A cm−2
Conclusion
In summary, the 2D porous Ti3C2Tx-CNT hybrid supports were synthesized by self-assembly of oppositely charged CNT and Ti3C2Tx in an aqueous solution, and the Pt/Ti3C2Tx-CNT catalyst was obtained by integration of Pt metal and Ti3C2Tx-CNT composite supports through a microwave-assisted polyol reduction method. The Pt/Ti3C2Tx-CNT catalyst exhibited higher HOR activities and stability than that of the commercial Pt/C catalyst. The peak power density of Pt/Ti3C2Tx-CNT catalyst achieves 0.86 W cm−2 with H2/Air and 1.6 W cm−2 with H2/O2, respectively. Moreover, the interactions between Pt nanoparticles with the Ti3C2Tx-CNT hybrid support could reduce the adsorption of CO to Pt. Pt/Ti3C2Tx-CNT catalyst exhibits much less reduction of ECSA and more mitigating fuel cell performance degradation during CO presence.
References
Y.Y. Cong, B.L. Yi, Y.J. Song, Hydrogen oxidation reaction in alkaline media: From mechanism to recent electrocatalysts. Nano Energy 44, 288–303 (2018)
V. Mehta, J.S. Cooper, Review and analysis of PEM fuel cell design and manufacturing. J. Power Sources 114, 32–53 (2003)
J. Bae, S. Lee, S. Kim, J. Oh, S. Choi, M. Bae, I. Kang, S.P. Katikaneni, Liquid fuel processing for hydrogen production: a review. Int. J. Hydrogen Energy 41, 19990–20022 (2016)
A.N. Gennadiev, Y.I. Pikovskii, A.S. Tsibart, M.A. Smirnova, Hydrocarbons in soils: Origin, composition, and behavior (Review). Eurasian Soil Sci. 48, 1076–1089 (2015)
A. Basile, F. Dalena, J. Tong, T.N. Veziroğlu, Hydrogen production, separation and purification for energy, vol. 89, (IET, 2017)
G. Voitic, S. Nestl, K. Malli, J. Wagner, B. Bitschnau, F.-A. Mautner, V. Hacker, High purity pressurised hydrogen production from syngas by the steam-iron process. Rsc Adv. 6, 53533–53541 (2016)
A.A. Franco, Polymer electrolyte fuel cells: Science, applications, and challenges (CRC Press, 2013)
H. Igarashi, T. Fujino, M. Watanabe, Hydrogen electro-oxidation on platinum catalysts in the presence of trace carbon monoxide. J. Electroanal Chem. 391, 119–123 (1995)
N. Zamel, X. Li, Effect of contaminants on polymer electrolyte membrane fuel cells. Prog. Energy Combust Sci. 37, 292–329 (2011)
M. Ahmadi, F. Behafarid, C. Cui, P. Strasser, B. Cuenya, Long-range segregation phenomena in shape-selected bimetallic nanoparticles: chemical state effects Acs. NANO 7, 9195–9204 (2013)
J. Li, Z. Luo, F. He, Y. Zuo, C. Zhang, J. Liu, X. Yu, R. Du, T. Zhang, M.F. Infante-Carrió, Colloidal Ni–Co–Sn nanoparticles as efficient electrocatalysts for the methanol oxidation reaction. J. Mater. Chem. A 6, 22915–22924 (2018)
Z. Liu, J.E. Hu, Q. Wang, K. Gaskell, A.I. Frenkel, G.S. Jackson, B. Eichhorn, PtMo alloy and MoO x@ Pt Core− shell nanoparticles as highly CO-tolerant electrocatalysts. J. Am. Chem. Soc. 131, 6924–6925 (2009)
Z. Liu, G.S. Jackson, B. Eichhorn, PtSn intermetallic, core–shell, and alloy nanoparticles as CO-tolerant electrocatalysts for H2 oxidation. Angew Chem. Int. Ed. 122, 3241–3244 (2010)
G. Shi, H. Yano, D.A. Tryk, A. Iiyama, H. Uchida, Highly active, CO-tolerant, and robust hydrogen anode catalysts: Pt–M (M= Fe, Co, Ni) alloys with stabilized Pt-skin layers. ACS Catal. 7, 267–274 (2017)
V.R. Stamenkovic, B.S. Mun, K.J. Mayrhofer, P.N. Ross, N. Markovic, Effect of surface composition on electronic structure, stability, and electrocatalytic properties of Pt-transition metal alloys: Pt-skin versus Pt-skeleton surfaces. J. Am. Chem. Soc. 128, 8813–8819 (2006)
J. Svendby, F. Seland, G. Singh, J.L.G. de la Fuente, S. Sunde, The potential of zero total charge and electrocatalytic properties of Ru@ Pt core-shell nanoparticles. J. Electroanal Chem. 833, 189–197 (2019)
B.W. Zhang, Z.C. Zhang, H.G. Liao, Y. Gong, L. Gu, X.M. Qu, L.X. You, S. Liu, L. Huang, X.C. Tian, R. Huang, F.C. Zhu, T. Liu, Y.X. Jiang, Z.Y. Zhou, S.G. Sun, Tuning Pt-skin to Ni-rich surface of Pt3Ni catalysts supported on porous carbon for enhanced oxygen reduction reaction and formic electro-oxidation. Nano Energy 19, 198–209 (2016)
K.-W. Park, J.-H. Choi, B.-K. Kwon, S.-A. Lee, Y.-E. Sung, H.-Y. Ha, S.-A. Hong, H. Kim, A. Wieckowski, Chemical and electronic effects of Ni in Pt/Ni and Pt/Ru/Ni alloy nanoparticles in methanol electrooxidation. J. Phys. Chem. B 106, 1869–1877 (2002)
K. Singh, E.B. Tetteh, H.-Y. Lee, T.-H. Kang, J.-S. Yu, Tailor-made Pt catalysts with improved oxygen reduction reaction stability/durability. ACS Catal. 9, 8622–8645 (2019)
Y. Wang, G. Wang, G. Li, B. Huang, J. Pan, Q. Liu, J. Han, L. Xiao, J. Lu, L. Zhuang, Pt–Ru catalyzed hydrogen oxidation in alkaline media: Oxophilic effect or electronic effect? Energy Environ. 8, 177–181 (2015)
L. Lin, S. Yao, R. Gao, X. Liang, Q. Yu, Y. Deng, J. Liu, M. Peng, Z. Jiang, S. Li, A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 14, 354–361 (2019)
R.M. Antoniassi, J. Quiroz, E. Barbosa, L.S. Parreira, R.A. Isidoro, E. Spinace, J. Silva, P.H. Camargo, Improving the electrocatalytic activities and CO tolerance of Pt NPs by incorporating TiO2 nanocubes onto carbon supports. ChemCatChem 13, 1931–1939 (2021)
Y. Wang, D. Widmann, M. Heenemann, T. Diemant, J. Biskupek, R. Schloegl, The role of electronic metal-support interactions and its temperature dependence: CO adsorption and CO oxidation on Au/TiO2 catalysts in the presence of TiO2 bulk defects. J. Catal. 354, 46–60 (2017)
W. Bao, L. Liu, C. Wang, S. Choi, D. Wang, G. Wang, Solar cells: Facile synthesis of crumpled nitrogen-doped MXene nanosheets as a new sulfur host for lithium–sulfur batteries. Adv. Energy Mater. 8, 1870060 (2018)
C. Chen, X. Xie, B. Anasori, A. Sarycheva, T. Makaryan, M. Zhao, P. Urbankowski, L. Miao, J. Jiang, Y. Gogotsi, MoS2-on-MXene heterostructures as highly reversible anode materials for lithium-ion batteries. Angew Chem. Int. Ed. 57, 1846–1850 (2018)
M.R. Lukatskaya, O. Mashtalir, C.E. Ren, Y. Dall’Agnese, P. Rozier, P.L. Taberna, M. Naguib, P. Simon, M.W. Barsoum, Y. Gogotsi, Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science 341, 1502–1505 (2013)
F. Shahzad, M. Alhabeb, C.B. Hatter, B. Anasori, S.M. Hong, C.M. Koo, Y. Gogotsi, Electromagnetic interference shielding with 2D transition metal carbides (MXenes). Science 353, 1137–1140 (2016)
S.H. Talib, S. Baskaran, X. Yu, Q. Yu, B. Bashir, S. Muhammad, S. Hussain, X. Chen, J. Li, Non-noble metal single-atom catalyst of Co 1/MXene (Mo 2 CS 2) for CO oxidation. Sci. China Mater. 64, 651–663 (2021)
X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo, A. Yamada, Pseudocapacitance of MXene nanosheets for high-power sodium-ion hybrid capacitors. Nat. Commun. 6, 1–6 (2015)
Z. Li, Y.R. Cui, Z.W. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E.Z. Shi, J.T. Miller, F.H. Ribeiro, Y. Wu, Reactive metal-support interactions at moderate temperature in two-dimensional niobium-carbide-supported platinum catalysts. Nat. Cata. 1, 349–355 (2018)
C. Cheng, X. Zhang, M. Wang, S. Wang, Z. Yang, Single Pd atomic catalyst on Mo 2 CO 2 monolayer (MXene): Unusual activity for CO oxidation by trimolecular Eley-Rideal mechanism. Phys. Chem. Chem. Phys. 20, 3504–3513 (2018)
C. Cheng, X. Zhang, Z. Yang, K. Hermansson, Identification of high-performance single-atom Mxenes catalysts for low-temperature CO oxidation. Adv. Theory Simul. 2, 1900006 (2019)
C. Cheng, X. Zhang, Z. Yang, Z. Zhou, Cu3-cluster-doped monolayer Mo2CO2 (MXene) as an electron reservoir for catalyzing a CO oxidation reaction. ACS Appl. Mater. Interfaces 10, 32903–32912 (2018)
X. Xie, M.-Q. Zhao, B. Anasori, K. Maleski, C.E. Ren, J. Li, B.W. Byles, E. Pomerantseva, G. Wang, Y. Gogotsi, Porous heterostructured MXene/carbon nanotube composite paper with high volumetric capacity for sodium-based energy storage devices. Nano Energy 26, 513–523 (2016)
C.X. Xu, C.C. Fan, X.L. Zhang, H.T. Chen, X.T. Liu, Z.M. Fu, R.R. Wang, T. Hong, J.G. Cheng, MXene (Ti3C2Tx) and carbon nanotube hybrid-supported platinum catalysts for the high-performance oxygen reduction reaction in PEMFC. ACS Appl. Mater. Interfaces 12, 19539–19546 (2020)
M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin, Y. Gogotsi, Guidelines for synthesis and processing of two-dimensional titanium carbide (Ti3C2T x MXene). Science 29, 7633–7644 (2017)
Funding
We thank the financial support from the National Key Research and Development Program of China under Grant No. 2021YFE0191700. We also thank the fund support by Hefei Municipal Natural Science Foundation (2022042).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chang, Z., Ren, W., Wang, Y. et al. A Highly CO-Tolerant Anode Pt/Ti3C2Tx-CNT Hybrid Catalysts for PEMFC. Electrocatalysis 14, 1–8 (2023). https://doi.org/10.1007/s12678-022-00768-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-022-00768-w