
Abstract
Electrocatalytic CO2 reduction to fuels powered by renewable electricity is a potential clean strategy to replace fossil fuels and address climate change caused by increasing CO2 emissions. In this work, the electrocatalytic CO2 reduction process was briefly introduced; the latest literature on the electrocatalytic CO2 reduction to CO by cobalt phthalocyanine (CoPc) was summarized. The structure, catalytic activity, and improvement strategies of CoPc were analyzed in detail. Then, the possible mechanism for the electrocatalytic CO2 reduction to CO was elucidated. Finally, the challenges and possible solutions of cobalt-based catalysts in electrocatalytic CO2 reduction were discussed. We believe it will serve as an up-to-date reference for the design of more efficient and stable cobalt-based catalysts for CO2 reduction, providing new insights and perspectives for further development of excellent catalytic materials.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The burning of large amounts of fossil fuels has led to a dramatic increase in anthropogenic carbon dioxide emissions, and the resulting greenhouse effect has become a global concern [1,2,3]. Various environmental problems caused by global climate change have made human intervention in carbon emissions urgent [4, 5]. For several years, the geological storage [6, 7] and molecular transformation of CO2 [8,9,10,11] have been the main treatment technologies. However, geological storage may face leakage, resulting in uncertain long-term environmental impacts. Moreover, the high cost due to the difficulties inherent in the storage process hinders the application of storage technologies [12].
Conversely, conversion/utilization or recycling schemes may be a potential alternative from the point of view of energy demand and sustainable development. If the recycling technology is powered by inexhaustible solar energy, CO2 is used as a redox medium through which the diffused solar energy is stored in the form of chemical bonds by reduction, and the reduction products are oxidized to obtain electrical energy [13].
In general, CO2 can be converted into useful compounds, including urea [14], alcohols [15], carboxylic acids [16], lactones [17], heterocyclic compounds, and polymer materials [18,19,20] by a variety of processes, such as chemical [21, 22], photochemical [23,24,25,26], electrochemistry [27,28,29,30], biological [31,32,33], or inorganic transformation [34]. Among them, electrocatalytic CO2 reduction reaction (CO2RR) is one of the most promising conversion technologies since the required electricity can be derived from renewable energy sources [35]. Electrocatalytic reduction of CO2 also has many inherent advantages over other conversion processes, such as adjustable electrode potential, controllable selectivity, environmentally friendly catalytic process at ambient pressure and temperature, simple reaction unit, and high potential for industrial applications [35,36,37,38]. Electrolytic equipment for electrocatalytic CO2 reduction has four components, including an electrolyte with high electrical conductivity and rapid mass transfer of reactants to the product, a proton membrane to reduce oxidation of the liquid product, a cathode and an anode coated with a highly active and durable catalyst [35].
The key to CO2 conversion is the selection of a catalyst [39], which aims to decrease the energy barriers and accelerate the rate of the redox [40]. Many noble metals with high selectivity and catalytic activity, such as gold [41, 42], silver [43,44,45], platinum [46, 47], iridium [48, 49], and ruthenium [50, 51], have been used in the electrocatalytic reduction of CO2. However, the scarcity, high cost, and possible losses in the reduction process of precious metals limit their large-scale application. As an abundant transition metal on earth with the aforementioned catalytic properties, cobalt is recommended as one of the most promising ideal substitutes for noble metals. Cobalt is a group VIII B element, which is very readily available in its oxidized state, including Co0, CoI, CoII, CoIII, and CoIV[52]. CO2RR mediated by cobalt catalysts typically involve a CoII to CoI intermediate state transition [53]. A range of cobalt-based materials including atomic cobalt [54,55,56,57], metals [58,59,60], oxides [61,62,63,64,65], nitrogen-doped carbons [66,67,68,69], and molecular complexes [70,71,72,73,74,75,76,77] have been explored for CO2RR.
The major classes of molecular complexes explored for CO2 reduction to date include metal centers with macrocyclic ligands [78,79,80], bipyridine ligands [81,82,83], and phosphine ligands [84,85,86]. Among them, cobalt-containing macrocyclic ligands, such as cobalt phthalocyanine (CoPc) and cobalt tetraphenylporphyrin (CoTPP) constitute an attractive class of materials with distinct advantages in easy accessibility, chemical stability, and structural tunability at the molecular level [87,88,89,90]. Therefore, this work lays out the latest updated literature on the electrocatalytic CO2 reduction to CO by CoPc. The structure, catalytic activity, performance improvement, and catalytic mechanism of CoPc were analyzed in detail, so as to provide theoretical support for the preparation of efficient catalysts and CO2 emission control.
Electrocatalytic CO2 Reduction
CO2 is one of the carbon products produced by the combustion of organic matter. From the thermodynamic viewpoint, it is a gaseous substance that does not easily destroy its structure [91, 92]. Its conversion is kinetically challenging due to the high energy barrier during the reduction process [93], the high overpotential, and the low Faradaic efficiency (FE) [55]. FE is defined as the percentage of electrons in the target product, which represents the selectivity of the product to some extent [53].
Specifically, the thermodynamic potential for the single-electron reduction of CO2 to CO2•– is very high, reaching –1.90 V vs. standard hydrogen electrode (SHE) in neutral aqueous media [94, 95]. However, electrocatalytic CO2 reduction is a multiple proton-coupled electron transfer process that produces thermodynamically supported species that allow the reduction reaction to occur at lower potentials. It has been demonstrated that various C1 and C2 products (e.g., CO, HCOOH, CH3CH2OH, C2H4) can be produced by CO2 reduction with different catalysts and electrolyte media [96,97,98,99].
Figure 1 provides a model diagram of the equipment used for electrocatalytic CO2 reduction. It usually consists of four main components: an inorganic salt electrolyte with high electrical conductivity and allowing rapid mass transfer of reaction substrates and products, a proton membrane capable of reducing the oxidation of liquid products, a cathode, and an anode prepared from catalysts with high catalytic activity and durability and other materials[35, 100]. The CO2 RR occurs at the catalyst-electrolyte interface of the cathode when a specific voltage is applied to the working electrode [100]. Generally, the catalytic reduction process consists of four main steps. Firstly, CO2 is adsorbed onto the surface of the catalyst-coated anode by physicochemical interactions; secondly, CO2 is chemically activated to CO2•–; then, multiple electron/proton transfer processes occur to facilitate electrocatalytic CO2 reduction; finally, various products are generated from the catalyst surface.

A typical electrocatalytic CO2 reduction reaction cell (Reprinted from Ref. [100])
Table 1 lists the thermodynamic redox potentials of the reactions that reduce CO2 to various products [101]. These half-reaction potentials, which depend on the electrolyte medium, only illustrate the minimum thermodynamic potential that enables the reaction to proceed [102, 103]. However, reaction kinetics, including reaction activation energies, reaction rates, and reaction pathways cannot be derived simply from thermodynamic potentials [36].
Structure of Cobalt Phthalocyanine
Phthalocyanine compounds include three polymorphic forms, α-form, γ-form, and β-form, of which β-form is the most stable polymorph [104]. Since the development of metal phthalocyanines (MPcs) in 1907, they have been widely used in new molecular conductors, molecular magnets, molecular electronic components, electrochromic, photoelectric conversion, and liquid crystals functional materials because of their unique optical, electrical, thermal, and magnetic properties [105].
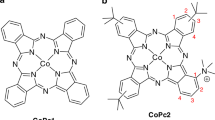
In fact, many common metals (Fe, Cu, Ni, Co, Zn) can be neutrally coordinated with Pc molecules to form thermodynamically and chemically stable complexes. The valence and spin states of the transition metal ions determine their electronic properties, especially magnetism. Therefore, the natures of these complexes change when the transition metal ions are substituted or doped into the system by electrons or holes [106]. Figure 2 shows a single CoPc molecule, the complex consisting of a phthalocyanine (Pc) ring with a Co metal ion in the center [107]. The CoPc has a D4h point-symmetric planar structure (Fig. 2b), with eight nitrogen atoms near the Co atom in the center of the molecule, surrounded by four pyrrole (N1) nitrogen atoms; the other four nitrogen atoms—bridging aza (N2). In addition, there are 32 carbon atoms derived from pyrrole (C1) and benzene (C2, C3, and C4), respectively [108].

The crystal structure of the β-Pc polymorph is classified as monoclinic, belonging to space group P21/a, with two centrosymmetric molecules present in each unit cell. Table 2 provides the unit cell dimensions of CoPc, NiPc, and ZnPc [104].
Electrocatalytic CO2 Reduction to CO
Homogeneous Electrocatalytic Activity of Cobalt Phthalocyanine
As early as 1965, the water-soluble Co(II)-terephthalocyanine was successfully synthesized [109], followed by the preparation of Co(II)-octacarboxyphthalocyanine and Co(II) tetracarboxyphthalocyanine [110]. Water-soluble CoPc was earlier used for the catalytic oxidation of thiols in petroleum fractions and for the removal of alkali sulfides from industrial wastewater. However, the dimer formed by CoPc in the aqueous phase is inactive leading to a significant decrease in its catalytic capacity [111]. Group substitution or the introduction of ligand additives in the solution can weaken the polymerization of phthalocyanines, including carboxylic acids, benzenesulfonyl, and amino groups [112,113,114]. Moreover, pristine phthalocyanines are insoluble in common organic solvents. However, additional solubility centers can be formed by group substitution at their peripheral and non-peripheral positions, thus increasing solubility [115].
Soluble electrocatalysts act as electron transfer agents in homogeneous electrocatalytic CO2 reduction to improve the reduction reaction [116]. Usually, the difference between applied electrode potential and the product/substrate cannot be counteracted. Therefore, a significant overvoltage and an effective catalytic constant are required to improve the conversion efficiency in direct electroreduction of CO2 [117]. Wu et al. utilized dissolved CoPc for homogeneous electrocatalytic CO2 reduction in 0.1 M KHCO3 electrolyte at − 0.8 V vs. reversible hydrogen electrode (RHE). After 30 min of reaction, the average current density for the conversion of CO2 to CO was less than 1 mA·cm−2 and the Faradaic efficiency of CO (FECO) was only 50% [80]. In conclusion, the catalytic performance of water-soluble CoPc is unstable and low due to its polymerization properties. In addition, the separation of the catalyst from the product under homogeneous conditions is severely consistent.
Heterogeneous Electrocatalytic CO2 Reduction via Cobalt Phthalocyanine
Cobalt Phthalocyanine Anchored on Carbon-Based Material
The catalytic performance of CoPc molecules can be significantly enhanced by heterogeneity. A common strategy is anchoring on carbon-based carriers to stabilize the catalyst molecules by π-π stacking [118]. Generally, CoPc molecules need to be attached to a suitable carrier to form a hybrid electrode for CO2RR. The current carriers reported in the literature include activated carbon (AC), graphite, carbon, carbon black (CB), activated carbon fiber (ACF), carbon nanotube (CNT), carbon cloth (CC), reduced graphene oxide (rGO), and glassy carbon (GC). The CO selectivity, current density, and durability of the catalysts were greatly improved by homogeneous immobilization of CoPc molecules on carbon-based carriers.
As early as 1974, the electrocatalytic reduction of CO2 to CO was performed by CoPc immobilized on graphite in an aqueous medium with an overpotential of − 1.6 V vs. saturated calomel electrode (SCE), which is about 200 mV more positive than the proton reduction reaction under N2 [119]. Table 3 summarizes the performance of various CoPc for electrocatalytic CO2 reduction to CO as reported in the literature in recent years.
In the process of electrocatalytic CO2 reduction by CoPc, electrons must pass through the aggregate bulk with lower electrical conductivity to reach surface molecules, which may hinder the reduction of Co(II) to Co(I). Compared with the CoPc/CNT catalyst, the lower product selectivity of CoPc was attributed to fewer Co(I) sites on its surface and the slower cycling rate of Co(II) reduction or Co(I) oxidation [90]. The CoPc/CNT hybrid prepared by Zhang et al. was subjected to electrocatalytic experiments in 0.1 M KHCO3 electrolyte at pH 6.8. The FECO was achieved at 98% when the current density was 15.0 mA∙cm−2 and the turnover frequency (TOF) was 4.1 s−1 at an overpotential of 0.52 V [90].
The electrocatalytic CO2 reduction performance of CoPc immobilized by Vulcan XC-72 and CNT showed significantly higher catalytic activity compared to pure CoPc. The electrocatalytic stability of CoPc/XC-72 was 15 h with a turnover number (TON) of only 48,600, while that for CoPc/CNT was more than 45 h, with a TON of up to 180,000, and the FECO reached 88%. The excellent catalytic stability of the latter is attributed to the strong π-π interaction between CoPc and CNT [11]. Jiang et al. demonstrated that CoPc/CNT hybrids can achieve high FECO of over 90% at potentials below −0.60 V. However, the FECO of pure CoPc was higher than that of CoPc/CNT at low overpotentials. For example, the FECO of CoPc was about 85% at −0.45 V, while that for CoPc/CNT was 64%. This phenomenon may be because Co(II) was reduced to Co(I) under low overpotential, which was considered to be the active site for CO2 reduction [120].
Han et al. conducted CO2RR on CNT by template-directed polymerization of CoPc. They found that phthalocyanines in the form of polymers supported on conductive scaffolds exhibited a larger surface area for electrocatalytic activity and better physical and chemical stability than molecular phthalocyanines. The hybrid electrocatalyst showed a selective reduction of CO2 to CO at −0.61 V vs. RHE with FECO and TOF of 90% and 4900 h−1, respectively [121]. Wu et al. anchored CoPc on CNT as a catalyst for the electrocatalytic CO2 reduction to CO. Electrochemical tests showed that it has excellent catalytic performance, with TOF and FECO reaching 97% and 83.9 s−1, respectively, at a current density of −200 mA·cm−2 [122]. Furthermore, Li et al. found that the activity and stability of the catalysts could be enhanced by coordination engineering strategies for application in heterogeneous catalysis. They introduced amino, hydroxyl, and carboxyl groups into CNT for the immobilization of CoPc. Compared with the absence of the groups, the abovementioned groups significantly improved the TOF by changing the coordination environment of Co at the center of the molecule, with a TOF of 31.4 s−1 at − 0.6 V vs. RHE. The degree of enhancement of the groups was: CoPc/NH2-CNT > CoPc/OH-CNT > CoPc/COOH-CN > CoPc/CNT. Among them, the FECO of CoPc/NH2-CNT was up to 100% at a high current density of −225 mA·cm−2 [123].
In addition, conventional carbon materials can also significantly improve the catalytic capacity of CoPc. CO2 could be electrocatalytically reduced to CO via CoPc supported on nanoporous activated carbon fibers with FECO up to 70% [124]. The FECO of the electrocatalytic CO2 reduction to CO reached more than 95% through CoPc immobilized on carbon powder in a zero-gap membrane flow reactor with a current density of 150 mA⋅cm−2 [125].
CoPc was supported on oxygen-functionalized carbon paper by Zhu et al. as a catalyst for the electroreduction of CO2, and the TOF of CO increased about 3 orders of magnitude with the increase of the dispersion of CoPc on the support [107]. Magdesieva et al. found that the bulky molecules tetra-tert-butyl substituted phthalocyanine immobilized on the activated carbon had a higher efficiency for electrocatalytic CO2 reduction with a current efficiency of up to 85% for CO [126]. Ma et al. immobilized CoPc on acetylene black (CoPc/C) as a model catalyst for the CO2RR. After 10 h of electrochemical testing, the corresponding FECO of CoPc/C was consistently above 97% with a calculated TOF value of 14,040 h−1 at −0.7 V vs. RHE, indicating its good stability and selectivity for CO [127].
A modified graphite electrode coated with poly(4-vinylpyridine) (PVP) film containing CoPc was used for CO2RR by Abe et al. The catalyst film in the aqueous phase of 0.1 M NaH2PO4 (pH 4.4) was more selective for CO than the pure CoPc coating. The ratio of CO to H2 produced was about 6 at −1.20 V vs. Ag/AgCl [128]. Similarly, graphite electrodes loaded with cobalt octacyanophthalocyanine (CoPc(CN)8) were used for electrocatalytic CO2 reduction. The reduction of CO2 to CO at –1.20 V vs. Ag/AgCl yields a CO/H2 ratio of about 10 at an electrolyte pH of 9.3 [129].
Furthermore, the two-dimensional material graphene may also be an excellent carrier. Gu et al. used graphdiyne/graphene (GDY/G) heterostructures as two-dimensional conductive scaffolds to anchor monodispersed CoPc for selective electrocatalytic CO2 reduction. Electrocatalytic experiments in the H-cell demonstrated an FECO of 96% at a current density of 12 mA⋅cm−2, while that for liquid flow cell was 97% at 100 mA⋅cm−2. At −1.0 V vs. RHE, the TOF achieved 37 s−1, which exceeds most of the reported phthalocyanine-based catalysts [130]. Similarly, the introduction of intrinsic defects in graphene (DrGO) can accelerate the π-electron transfer between graphene and CoPc, and the greater exposure of electrocatalytically active Co sites leads to a positive shift of the Co2+/Co+ reduction potential, which enhances CO2 chemisorption. Therefore, compared with the defect-free counterpart rGO-CoPc, the maximum FECO of DrGO-CoPc for electrocatalytic CO2 reduction to CO reached 90.2% at a current density (JCO) of 73.9 mA cm−2 [131]. Tian et al. obtained a hybrid catalyst by immobilizing CoPc on reduced graphene oxide (rGO). The corresponding FECO, TOF, and overpotentials for the electrocatalytic CO2 reduction to CO in 0.75 M NaHCO3 electrolyte were 95.5%, 9.4 s−1, and −0.60 V vs. RHE, respectively [132].
Cobalt Phthalocyanine Anchored on Gas Diffusion Electrode
The catalytic activity of CoPc can be affected by various loaded electrodes. As early as 1987, Masheder et al. have demonstrated that cobalt (I) phthalocyanine was a reactive intermediate for the electrocatalytic CO2 reduction to CO at cobalt (II) phthalocyanine-impregnated electrodes [133]. Savinova et al. found that CoPc supported on a carbon gas diffusion electrode exhibited 100% selectivity for the electrocatalytic CO2 reduction to CO at current densities up to 80 mA⋅cm2 [134]. Furthermore, Wan et al. prepared a CoPc-based gas diffusion electrode (GDE) for the electrocatalytic CO2 reduction to CO. The current density and FECO were as high as 110 mA⋅cm−2 and 99%, respectively [135].
Group-Substituted Cobalt Phthalocyanine
At the molecular level, the catalytic activity of CoPc will be further improved by introducing substitution groups into the molecule [136]. The electrocatalytic CO2 reduction to CO by tetra-tert-butyl-substituted phthalocyanines loaded on activated carbon reached a production efficiency of 85% [124]. Perfluorinated cobalt phthalocyanine (CoFPc) was a new type of electrocatalyst, which was suitable for bipolar configuration with FECO up to 93%. The resistance to wide potential voltages from − 0.9 V to + 2.2 V vs. RHE during both reduction and oxidation catalysis makes CoFPc attractive for catalytic CO2 conversion [137]. De Riccardis et al. reported a cobalt-based phthalocyanine with pyridine moieties (CoPc-Py) supported on a carbon electrode, in which FECO (95%) was much higher than CoPc (86%) [138]. Zhu et al. immobilized CoPc on CNT containing pyridine functional groups as a catalyst (CoPc-py-CNT). The FECO, TOFCO of the electrocatalytic CO2 reduction to CO process were 98.7% and 34.5 s−1 at − 0.67 V vs. RHE, respectively. In addition, the reaction mechanism revealed that the axial coordination of the pyridine group with Co not only improved the dispersion of CoPc but also adjusted the electronic structure of Co sites and improved the TOF of the target product [118].
Luangchaiyaporn et al. obtained the catalyst 2,9,16,23-tetra(2,2′-bithiophen-5-yl) phthalocyaninato-cobalt(II) (CoPc-1) by substituting CoPc with 4-(2, 2′-bithiophen-5-yl) phthalonitrile, the product of heterogeneous electrocatalytic CO2 reduction in 0.5 M KHCO3 solution was mainly CO. At an overpotential of –0.54 V vs. RHE, the FECO, TONCO, and TOFCO of CO produced after 2 h of reaction were 94%, 2.10 × 103, and 0.29 s–1, respectively [139]. The hybrid electrode obtained by immobilizing cobalt(II) octaalkoxyphthalocyanine on graphene was used for the electrocatalytic CO2 reduction to CO with FECO exceeding 77% at −0.59 V vs. RHE [140]. Lu et al. found that the electrocatalytic performance and O2 tolerance could be further improved by introducing cyano and nitro substituents into the phthalocyanine ligands. A hybrid electrode (PIMCoPc/CNT) was formed by combining a thin layer of intrinsically microporous polymer (PIM) with CoPc molecules anchored on CNT. FECO was 75.9% at a battery voltage of 3.1 V and 5% O2 intake, and 96.9% without any O2 (jtotal = 29.3 mA⋅cm2) [141].
Wang et al. found that CoPc substituted with trimethylammonium group was able to efficiently electrocatalyze the reduction of CO2 to carbon monoxide in water at pH 4 to 14. FECO and maximum current density reached 95% and 165 mAcm−2 at −0.92 V vs. RHE, respectively [142]. Tian et al. prepared cobalt tetranitrophthalocyanine (CoTNPc), cobalt octacyanophthalocyanine (CoOCPc), and cobalt tetraaminophthalocyanine (CoTAPc) containing different functional groups to study the relationship between the electronic structure of the catalyst and CO2RR. They found that the modified phthalocyanine-based catalysts could significantly enhance the CO2 reduction with the order of activity CoTNPc > CoOCPc > CoPc > CoTAPc. The nitro-substituted cobalt phthalocyanine showed the best catalytic performance, the maximum FECO and current density were 94% and 12.6 mA⋅cm−2 at −0.877 V vs. RHE respectively [143].
In addition, another promising technique to improve the efficiency of CO2 capture and conversion is the coupling of metal–organic frameworks (MOFs) with CoPc. The current density and FECO of the hybrid CoTAPc-ZIF-90 catalyst prepared by decorating CoPc on the outer surface of ZIF-90 for electrocatalytic CO2 reduction to CO reached 13 mA⋅cm−2 and 90%, respectively. Moreover, CoTAPc-ZIF-90 exhibited significantly higher catalyst stability compared to free CoPc molecules [144]. Two-dimensional polyimide-linked phthalocyanine COFs (CoPc-PI-COF) were devised and prepared through solvothermal reaction of tetraanhydrides of octacarboxyphthalocyaninato cobalt(II) with 1,4phenylenediamine and 4,4′-biphenyldiamine. The electrocatalytic CO2 reduction to CO in 0.5 M KHCO3 solution resulted in FECO and current density up to 95% and 21.2 mA⋅cm−2 at −0.90 V vs. RHE, respectively [145].
Metal–Carbon-Nitrogen-Doped Cobalt Phthalocyanine
Metal-nitrogen active sites immobilized in porous carbon (Me-N–C) are considered a novel and efficient catalyst for CO2RR [146, 147]. Lin explored a CoPc and zinc-nitrogen-carbon (Zn-N–C) tandem catalyst for electrocatalytic CO2 reduction. This tandem catalyst has improved CH4/CO productivity by more than 100 times compared to using CoPc or Zn-N–C alone [148]. The electrocatalytic CO2RR performance was enhanced by cross-linking Fe–N sites on CoPc (CoPc©Fe–N-C). Compared with Fe-NC, the starting potential of CoPc©Fe-NC for RHE is as low as −0.13 V, while the potential window for CO Faraday efficiency of 93% is significantly widened from 0.18 V to 0.71 V, while the CO current density is increased by up to 10 times and the stability is significantly enhanced.
The electrocatalytic performance was enhanced by doping Fe–N sites on CoPc (CoPc©Fe–N-C). Compared with Fe-NC, the onset potential of CoPc©Fe-NC was as low as −0.13 V versus RHE, while the potential window of CO Faraday efficiency of 93% has been significantly broadened from 0.18 to 0.71 V, accompanied with a maximum tenfold increase in CO current density [149]. Ultrathin nitrogen-doped hollow carbon sphere with a high surface area of 653.3 m2⋅g−1 was used to anchor CoPc for electrocatalytic CO2 reduction to CO. A Faraday efficiency of 96% was achieved at a Co atom content of 0.49 wt.%, which was about 4 times that of unsupported CoPc (FECO = 24.54%); and a current density of 20.47 mA⋅cm−2 for CO at −0.82 V vs. RHE, which was 60 that of the unsupported CoPc (0.34 mA⋅cm−2) [150].
The N–C-CoPc catalyst prepared by Ma et al. exhibited excellent catalytic performance for the conversion of CO2 to CO. FECO achieved 90% at −0.58 to −0.78 V vs. RHE, as well as an excellent TOF of 3.89 s−1 and an ultra-low overpotential of 170 mV [151]. Similarly, a CoPc molecule-implanted graphitic carbon nitride nanosheets (CoPc/g-C3N4) electrocatalyst was used for electrocatalytic CO2 reduction. Electrochemical tests in natural seawater showed that an FECO of 89.5% could be achieved at a current density of 16.0 mA⋅cm−2, while the maximum FECO in simulated seawater was 98.1% after 25 h of reaction [152].
Zhu et al. proposed a nanoarray electrode formed by immobilizing CoPc in N-doped porous carbon nanorod (N–C-CoPc NR) by hydrothermal method. The prepared N–C-CoPc NR catalyst exhibited an FECO of 85.3% for the electrocatalytic CO2 reduction to CO in 0.1 M KHCO3 electrolyte [153]. Compared with pristine CoPc, non-peripheral octamethyl-substituted CoPc (N-CoMe2Pc) catalyst can activate the Co surface and significantly enhance CO2 adsorption at low overpotentials. Li et al. fabricated a nanocomposite catalyst (N-CoMe2Pc/NRGO) by immobilizing N-CoMe2Pc nanorods and nitrogen-doped reduced graphene oxide (NRGO) together by a non-covalent strategy. Electrocatalytic CO2 reduction experiments were carried out in a flow cell injected with neutral electrolyte. When the catalyst to NRGO ratio was 6:10, the current density, TOFCO and FECO were 56.4 mA⋅cm−2, 6.2 s−1, and 94.1% at –0.6 V vs. RHE, respectively [154].
Polymeric Cobalt Phthalocyanine
Moreover, the formation of polymers from CoPc monomers is another promising approach to alleviate catalyst aggregation and improve stability. For example, Li et al. immobilized polymerized CoPc on a three-dimensional (3D) g-C3N4 nanosheet-carbon nanotube scaffold to achieve electrocatalytic CO2 reduction to CO. The results showed that the electrocatalytic performance of the polymerized catalytic material maintained good stability over 24 h, with TOFCO and FECO reachED 95% and 4.9 s−1 at −0.8 V vs. RHE, respectively [155]. Chen et al. supported polymerized CoPc (CoPPc) on CNT and deposited 45 μg⋅cm−2 of CoPPc-CNT catalyst on carbon paper as a working electrode. Cyclic voltammetry (CV) measurements showed that the onset potential of CoPPc-CNT was −0.35 V vs. RHE. Compared with pure CNT, CoPPc-CNT significantly increased current density under negative potential, indicating that the catalytic process was mainly induced by CoPPc. FECO reached 96% at a wide range of potentials [156].
Mechanism of Electrocatalytic Reduction CO2 to CO
Zhang et al. systematically investigated the mechanism of electrocatalytic CO2 reduction by CoPc with a well-defined metal-N4 structure through DFT calculations. Theoretically, CoPc exhibits better CO formation activity due to the moderate binding energy of *CO at the Co site. The thermodynamic requirements of the original process are overcome by sacrificing the important reaction steps of *COOH formation and *CO desorption. [160]. Under laboratory conditions, efficient CO2 reduction catalysts can achieve high selectivity and low overpotential, but bottlenecks remain in commercial practice at high current densities.
Figure 3 shows the mechanism of electrocatalytic CO2 reduction to CO via CoPc[107]. Both the mechanism and the reaction rate-determining step (RDS) of the electrocatalytic CO2 reduction via CoPc are slightly different under high (> 0.3 M) and low (< 0.3 M) bicarbonate conditions. Since previous studies have confirmed that Co (I) is the stationary state of cobalt during the electroreduction of CO2, it is suggested that the first step involves the irreversible reduction of Co (II) to Co (I) [161, 162]. The RDS at low bicarbonate concentrations involves electron transfer, while that at high bicarbonate concentrations involves concerted proton-electron transfer. It is possible that the SO2 molecule are pre-bound to the active site before the rate-determining step involves electron transfer.

Proposed mechanism for electrocatalytic CO2 reduction to CO by CoPc (Reprinted from Ref. [107])
When the concentration of HCO3− is below 0.3 M, the possible reactions for the electrocatalytic CO2 reduction to CO are shown in Eqs. (1) to (5).
When the concentration of HCO3− is higher than 0.3 M, the possible reactions for the electrocatalytic CO2 reduction to CO are shown in Eqs. (6) to (9).
The exact mechanism of the electrocatalytic CO2 reduction to CO via CoPc remains discrepant (Fig. 4). Figure 4 a shows the mechanism of electrocatalytic CO2 reduction by CoPc in a competitive HER pathway. Firstly, CoPc is reduced to (CoPc)− during the reaction process, and then (CoPcH) is formed by complex protonation on the Pc ring, followed by a second reduction to produce (CoPcH)−. A branching mechanism is that (CoPcH)− can react with H+ to release H2 and regenerate the CoPc starting material in step (iv), or (CoPcH)− react with CO2 to form CO2 adduct in step (i), the subsequent protonation step (iii) produces CO. This indicates that the catalytic activity is mainly generated in the second reduction process. However, electrocatalytic CO2 reduction in DMSO solution via CoPc also requires a third reduction event at low H+ activity conditions. This suggests that the (CoPc-CO) adduct requires further reduction to release CO and re-enter the catalytic cycle at (CoPc)− (Fig. 4b). In addition, the coordination of CO2 may occur at the 1 e−-reduced species confirmed by the reduction of CO2 by adsorbed CoPc in bicarbonate solution; this pathway also supported by Tafel analysis and DFT studies (Fig. 4c) [163].

Proposed CO2 reduction mechanisms of CoPc in the literature. a A proposed mechanism for CO2 reduction by CoPc showing pathway for competitive H2 generation. b Other proposed CO2 reduction by CoPc in organic solutions, c low concentration bicarbonate buffer in aqueous solution (Reprinted from Ref. [163])
Cooperative proton-electron transfer (CPET) is the theoretical basis of the CHE methodology. However, some researchers believe that the reduction of CO2 on Co porphyrin molecules is carried out by sequential proton-electron transfer (SPET) under high pH conditions [164,165,166]. Han et al. studied the SPET step of electrocatalytic CO2 reduction by polymeric cobalt phthalocyanine (CoPPc) through density functional theory to reveal the reaction mechanism [121]. The Co in the center of CoPPc is reduced from Co(II) to Co(I) during the first electron transfer process, and the injected electrons are mainly located in the dz2 orbital and partially in the C-pz orbital (Fig. 5B). According to the Bader charge population analysis, the charge of Co (I) (+ 0.86 |e|) on CoPPc− is much lower than that of Co(II) (+ 1.15|e|). The formation of the anionic adduct of CO2 when CO2 is added to Co(I) is evidenced by the apparent charge transfer (0.54|e|) from CoPPc− to CO2 (Fig. 5C). Then, the intermediate becomes COOH* via proton transfer (Fig. 5D). Finally, COOH* is transformed into CO* by a CPET step (Fig. 5E). Figure 4F summarizes the abovementioned whole process of electrocatalytic CO2 reduction to CO by CoPPc.

The optimized geometric structure of various states (CoPPc, CoPPc−, CO2−*, COOH*, and CO*) along the reaction path of CO2RR on a 2D CoPPc monolayer. Co, N, O, C, and H atoms are presented by orange, blue, red, gray, and green spheres, respectively. For CoPPc− and CO2.−*, the electron density differences caused by electron injection and CO2 adsorption are also plotted. Cyan and purple correspond to electron accumulation and depletion regions, respectively. Also shown are Co–N bond lengths (A, B) and Co–C bond lengths (C–E) in Ångstroms. F Proposed mechanistic scheme for the CO2RR on CoPPc (Reprinted from Ref. [121])
Conclusion and Perspectives
Based on cost-effective and earth-abundant catalysts, CO2 reduction can decrease its atmospheric concentration and produce carbonaceous value-added fuel molecules and chemical feedstocks. From the perspective of long-term economic and environmental benefits, catalysts prepared from non-precious transition metals such as cobalt are more potential for electrocatalytic CO2 reduction than noble metals such as gold, silver, platinum, and rhodium. The elemental cobalt has the characteristics of moderate CO2 adsorption strength, polyvalency, high coordination, unsaturated electronic d-orbital. Furthermore, CoPc exhibits size-dependent dissociative adsorption of CO2 molecules on CO and O ligands, which more actively promotes subsequent CO2 activation. The reaction process overpotential, i.e., the energy barrier for electrocatalytic CO2 reduction can be significantly reduced through stabilizing the -*COOH and *OCHO intermediates structural modifications, ligand modifications, hydrogen-bonding modifications, and coordinating anions. Ultimately, the selectivity of CO is increased accordingly.
The metal center affects the polymerization kinetics and polymer morphology. On the other hand, the chemical nature of the catalyst metal center is the most important factor affecting the electrocatalytic CO2 reduction and the associated products. Since electrocatalytic CO2 reduction is a multiple electron transfer process leading to multiple reduction pathways and various product production, this limits the selectivity and catalytic efficiency of the target product. The conversion of CO2 to CO can be understood as a double reduction process with coordinated proton-assisted electron transfer, rather than a single-electron process. Therefore, avoiding the formation of intermediates with high energy barriers is the key to enhance the catalytic rate of CoPc.
Direct loading of metal phthalocyanine molecules on the electrodes results in their catalytic performance being blocked by molecular aggregates. In contrast to similar hybrid electrocatalysts, the aggregated form of molecular catalysts immobilized on carbonaceous materials has an interconnected network that expands the electrocatalytic active surface area and improves structural and operational stability [155]. Therefore, the catalytic efficiency parameters including current density, stability, and selectivity of the hybrid catalysts can be significantly improved by hybridization with CNT, CB, AC, ACF, rGO, and other materials compared to their molecular counterparts. In addition, strategies such as anchoring on gas diffusion electrodes, group substitution, metal–carbon-nitrogen doping, and monomer polymerization can further improve the stability and CO selectivity of CoPc to varying degrees.
Although some advancements have been made in electrocatalytic CO2 reduction to CO, further improvements still need to be investigated as follows:
In the future, strategies to control and reduce CO2 emissions should focus on the development of efficient, durable, stable, and low-cost catalysts. In situ manipulations and measurements need to be explored to provide more specific and precise details of electrocatalytic CO2 reduction reactions, including reaction sites, types of intermediates, reaction pathways, etc. In addition, theoretical research should further elucidate the reduction reaction process to provide more valuable experimental directions. Based on the full elucidation of the CO2 reduction mechanism, the catalytic performance of the catalyst can be improved.
Then, DFT studies showed that pyridine moiety-induced enhancement of CO2 adsorption energy and the greater electron affinity of the Co center brought about a reduction in overpotential. The electron affinity has an antagonistic effect. On the one hand, the presence of electron-withdrawing groups can effectively adjust the electron affinity of the catalyst and improve the catalytic activity of CoPc derivatives for CO2. On the other hand, a sharp increase of electron affinity also inhibits the binding of CO2 and hinders the CO2RR. Therefore, finding the optimal balance between these two effects is the key to developing novel and efficient catalysts for CO2RR [138].
Furthermore, as one of the main components of air, CO2 is often mixed with other gases. Currently, experimental data on catalytic CO2 reduction from mixed gases are lacking. Therefore, the realization of highly selective adsorption and catalytic CO2 conversion from atmospheric is of great significance for future practical applications.
Finally, in order to improve the selectivity of target products, catalysts with appropriate compositions and structures as well as appropriate reaction schemes must be designed and developed. In addition, stabilization of carbonaceous intermediates by rare earth metal-doped catalyst materials may effectively limit the competing HER. Several effective means, including the formation of metal alloys, dopants, defects, and mixed oxide supports are all good options for improving CO selectivity.
References
R.J. Andres, T.A. Boden, F.M. Bréon, P. Ciais, S. Davis, D. Erickson, J.S. Gregg, A. Jacobson, G. Marland, J. Miller, T. Oda, J.G.J. Olivier, M.R. Raupach, P. Rayner, K. Treanton, A synthesis of carbon dioxide emissions from fossil-fuel combustion. Biogeosciences 9, 1845–1871 (2012)
D.C. Frank, J. Esper, C.C. Raible, U. Buntgen, V. Trouet, B. Stocker, F. Joos, Ensemble reconstruction constraints on the global carbon cycle sensitivity to climate. Nature 463, 527–530 (2010)
S.E. Schwartz, Uncertainty in climate sensitivity: Causes, consequences, challenges. Energy Environ. Sci. 1, 430–453 (2008)
W. da Silva Freitas, A. D’Epifanio, B. Mecheri, Electrocatalytic CO2 reduction on nanostructured metal-based materials: Challenges and constraints for a sustainable pathway to decarbonization, Journal of CO2 Utilization 50, 101579 (2021)
S. Piao, P. Ciais, Y. Huang, Z. Shen, S. Peng, J. Li, L. Zhou, H. Liu, Y. Ma, Y. Ding, P. Friedlingstein, C. Liu, K. Tan, Y. Yu, T. Zhang, J. Fang, The impacts of climate change on water resources and agriculture in China. Nature 467, 43–51 (2010)
A. Sanna, M. Uibu, G. Caramanna, R. Kuusik, M.M. Maroto-Valer, A review of mineral carbonation technologies to sequester CO2. Chem. Soc. Rev. 43, 8049–8080 (2014)
J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han, C. Yang, Enzymatic conversion of carbon dioxide. Chem. Soc. Rev. 44, 5981–6000 (2015)
D.T. Whipple, P.J.A. Kenis, Prospects of CO2 utilization via direct heterogeneous electrochemical reduction. The Journal of Physical Chemistry Letters 1, 3451–3458 (2010)
A. Goeppert, M. Czaun, J.P. Jones, G.K. Surya Prakash, G.A. Olah, Recycling of carbon dioxide to methanol and derived products - Closing the loop, Chemical Society Reviews 43, 7995–8048 (2014)
D.R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P.R. Ohodnicki, C. Zeng, R. Jin, Efficient electrochemical CO2 conversion powered by renewable energy. ACS Appl. Mater. Interfaces. 7, 15626–15632 (2015)
M. Huai, Z. Yin, F. Wei, G. Wang, L. Xiao, J. Lu, L. Zhuang, Electrochemical CO2 reduction on heterogeneous cobalt phthalocyanine catalysts with different carbon supports. Chem. Phys. Lett. 754, 137655 (2020)
W.J.J. Huijgen, R.N.J. Comans, G.-J. Witkamp, Cost evaluation of CO2 sequestration by aqueous mineral carbonation. Energy Convers. Manage. 48, 1923–1935 (2007)
G.F. Manbeck, E. Fujita, A review of iron and cobalt porphyrins, phthalocyanines and related complexes for electrochemical and photochemical reduction of carbon dioxide. J. Porphyrins Phthalocyanines 19, 45–64 (2015)
C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan, G. Yu, Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nature Sustainability 4, 868–876 (2021)
L. Ji, L. Chang, Y. Zhang, S. Mou, T. Wang, Y. Luo, Z. Wang, X. Sun, Electrocatalytic CO2 reduction to alcohols with high selectivity over a two-dimensional Fe2P2S6 nanosheet. ACS Catal. 9, 9721–9725 (2019)
J. Annie Modestra, B. Navaneeth, S. Venkata Mohan, Bio-electrocatalytic reduction of CO2: Enrichment of homoacetogens and pH optimization towards enhancement of carboxylic acids biosynthesis. J. CO2 Utiliz. 10, 78–87 (2015)
L. Ponsard, E. Nicolas, N.H. Tran, S. Lamaison, D. Wakerley, T. Cantat, M. Fontecave, Coupling electrocatalytic CO2 reduction with thermocatalysis enables the formation of a lactone monomer. Chemsuschem 14, 2198–2204 (2021)
E.E. Benson, C.P. Kubiak, A.J. Sathrum, J.M. Smieja, Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 38, 89–99 (2009)
Y. Qu, X. Duan, Progress, challenge and perspective of heterogeneous photocatalysts. Chem. Soc. Rev. 42, 2568–2580 (2013)
J. Qiao, Y. Liu, F. Hong, J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 43, 631–675 (2014)
J. Tang, W. Sun, H. Tang, M. Radosz, Y. Shen, Enhanced CO2 absorption of poly(ionic liquid)s. Macromolecules 38, 2037–2039 (2005)
B.E. Gurkan, d.l.J.C. Fuente, E.M. Mindrup, L.E. Ficke, B.F. Goodrich, E.A. Price, W.F. Schneider, J.F. Brennecke, Equimolar CO2 absorption by anion-functionalized ionic liquids. J. Am. Chem. Soc. 132, 2116–2117 (2010)
Y. Oh, X. Hu, Organic molecules as mediators and catalysts for photocatalytic and electrocatalytic CO2 reduction. Chem. Soc. Rev. 42, 2253–2261 (2013)
A. Razzaq, A. Sinhamahapatra, T.-H. Kang, C.A. Grimes, J.-S. Yu, S.-I. In, Efficient solar light photoreduction of CO2 to hydrocarbon fuels via magnesiothermally reduced TiO2 photocatalyst. Appl. Catal. B 215, 28–35 (2017)
I.I. Alkhatib, C. Garlisi, M. Pagliaro, K. Al-Ali, G. Palmisano, Metal-organic frameworks for photocatalytic CO2 reduction under visible radiation: A review of strategies and applications. Catal. Today 340, 209–224 (2020)
V. Butera, H. Detz, Photochemical CO2 conversion on pristine and Mg-doped gallium nitride (GaN): A comprehensive DFT study based on a cluster model approach. Mater. Chem. Front. (2021)
Y.Y. Birdja, E. Pérez-Gallent, M.C. Figueiredo, A.J. Göttle, F. Calle-Vallejo, M.T.M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nature. Energy 4, 732–745 (2019)
K. Torbensen, B. Boudy, D. Joulié, N. von Wolff, M. Robert, Emergence of CO2 electrolyzers including supported molecular catalysts. Curr. Opin. Electrochem. 24, 49–55 (2020)
P. Gotico, W. Leibl, Z. Halime, A. Aukauloo, Shaping the electrocatalytic performance of metal complexes for CO2 reduction. ChemElectroChem 8, 3472–3481 (2021)
W. Ma, X. He, W. Wang, S. Xie, Q. Zhang, Y. Wang, Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts. Chem. Soc. Rev. (2021)
S. Bajracharya, S. Srikanth, G. Mohanakrishna, R. Zacharia, D.P. Strik, D. Pant, Biotransformation of carbon dioxide in bioelectrochemical systems: State of the art and future prospects. J. Power Sources 356, 256–273 (2017)
J. Zabranska, D. Pokorna, Bioconversion of carbon dioxide to methane using hydrogen and hydrogenotrophic methanogens. Biotechnol. Adv. 36, 707–720 (2018)
K.K. Sahoo, G. Goswami, D. Das, Biotransformation of methane and carbon dioxide into high-value products by methanotrophs: Current state of art and future prospects. Front. Microbiol. 12, 636486 (2021)
A.A. Olajire, A review of mineral carbonation technology in sequestration of CO2. J. Petrol. Sci. Eng. 109, 364–392 (2013)
J. Hao, W. Shi, Transition metal (Mo, Fe Co, and Ni)-based catalysts for electrochemical CO2 reduction. Chin. J. Catal. 39, 1157–1166 (2018)
B. Kumar, J.P. Brian, V. Atla, S. Kumari, K.A. Bertram, R.T. White, J.M. Spurgeon, New trends in the development of heterogeneous catalysts for electrochemical CO2 reduction. Catal. Today 270, 19–30 (2016)
F.P. Garcia de Arquer, O.S. Bushuyev, P. De Luna, C.T. Dinh, A. Seifitokaldani, M.I. Saidaminov, C.S. Tan, L.N. Quan, A. Proppe, M.G. Kibria, S.O. Kelley, D. Sinton, E.H. Sargent, 2D metal oxyhalide-derived catalysts for efficient CO2 electroreduction. Adv. Mater. 30, e1802858 (2018)
R. He, A. Zhang, Y. Ding, T. Kong, Q. Xiao, H. Li, Y. Liu, J. Zeng, Achieving the widest range of syngas proportions at high current density over cadmium sulfoselenide nanorods in CO2 electroreduction. Adv. Mater. 30, 1705872 (2018)
A. Chapovetsky, M. Welborn, J.M. Luna, R. Haiges, T.F. Miller 3rd., S.C. Marinescu, Pendant hydrogen-bond donors in cobalt catalysts independently enhance CO2 reduction. ACS Cent. Sci. 4, 397–404 (2018)
X. Yang, Hydrogenation of carbon dioxide catalyzed by pnp pincer iridium, iron, and cobalt complexes: A computational design of base metal catalysts. ACS Catal. 1, 849–854 (2011)
Y. Hori, K. Kikuchi, S. Suzuki, Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 14, 1695–1698 (1985)
M. Dunwell, Q. Lu, J.M. Heyes, J. Rosen, J.G. Chen, Y. Yan, F. Jiao, B. Xu, The central role of bicarbonate in the electrochemical reduction of carbon dioxide on gold. J. Am. Chem. Soc. 139, 3774–3783 (2017)
T. Hatsukade, K.P. Kuhl, E.R. Cave, D.N. Abram, T.F. Jaramillo, Insights into the electrocatalytic reduction of CO2 on metallic silver surfaces. Phys. Chem. Chem. Phys. 16, 13814–13819 (2014)
K.G. Schmitt, A.A. Gewirth, In situ surface-enhanced raman spectroscopy of the electrochemical reduction of carbon dioxide on silver with 3,5-diamino-1,2,4-triazole. J. Phys. Chem. C 118, 17567–17576 (2014)
L. Zhang, Z. Wang, N. Mehio, X. Jin, S. Dai, Thickness- and particle-size-dependent electrochemical reduction of carbon dioxide on thin-layer porous silver electrodes. Chemsuschem 9, 428–432 (2016)
N. Hoshi, Y. Hori, Electrochemical reduction of carbon dioxide at a series of platinum single crystal electrodes. Electrochim. Acta 45, 4263–4270 (2000)
Y. Tomita, S. Teruya, O. Koga, Y. Hori, Electrochemical reduction of carbon dioxide at a platinum electrode in acetonitrile-water mixtures. J. Electrochem. Soc. 147, 4164–4167 (2000)
P. Kang, T.J. Meyer, M. Brookhart, Selective electrocatalytic reduction of carbon dioxide to formate by a water-soluble iridium pincer catalyst. Chem. Sci. 4 (2013)
P. Kang, S. Zhang, T.J. Meyer, M. Brookhart, Rapid selective electrocatalytic reduction of carbon dioxide to formate by an iridium pincer catalyst immobilized on carbon nanotube electrodes. Angew Chem Int Ed Engl 53, 8709–8713 (2014)
H. Nagao, T. Mizukawa, K. Tanaka, Carbon-carbon bond formation in the electrochemical reduction of carbon dioxide catalyzed by a ruthenium complex. Inorg. Chem. 33, 3415–3420 (1994)
S. Ramakrishnan, C.E.D. Chidsey, Initiation of the electrochemical reduction of CO2 by a singly reduced ruthenium(ii) bipyridine complex. Inorg. Chem. 56, 8326–8333 (2017)
A.K. Singh, R. Mukherjee, Cobalt(II) and cobalt(III) complexes of thioether-containing hexadentate pyrazine amide ligands: C-S bond cleavage and cyclometallation reaction. Dalton Transac. 260–270 (2008)
C. Li, X. Tong, P. Yu, W. Du, J. Wu, H. Rao, Z.M. Wang, Carbon dioxide photo/electroreduction with cobalt. Journal of Materials Chemistry A 7, 16622–16642 (2019)
S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang, Y. Xie, Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 529, 68–71 (2016)
P. Su, K. Iwase, T. Harada, K. Kamiya, S. Nakanishi, Covalent triazine framework modified with coordinatively-unsaturated Co or Ni atoms for CO2 electrochemical reduction. Chem. Sci. 9, 3941–3947 (2018)
J.-H. Liu, L.-M. Yang, E. Ganz, Electrochemical reduction of CO2 by single atom catalyst TM–TCNQ monolayers. Journal of Materials Chemistry A 7, 3805–3814 (2019)
H. Yang, Q. Lin, Y. Wu, G. Li, Q. Hu, X. Chai, X. Ren, Q. Zhang, J. Liu, C. He, Highly efficient utilization of single atoms via constructing 3D and free-standing electrodes for CO2 reduction with ultrahigh current density. Nano Energ. 70, 104454, (2020)
J.-P. Grote, A.R. Zeradjanin, S. Cherevko, A. Savan, B. Breitbach, A. Ludwig, K.J.J. Mayrhofer, Screening of material libraries for electrochemical CO2 reduction catalysts – Improving selectivity of Cu by mixing with Co. J. Catal. 343, 248–256 (2016)
Y. Takatsuji, I. Nakata, M. Morimoto, T. Sakakura, R. Yamasaki, T. Haruyama, Highly selective methane production through electrochemical CO2 reduction by electrolytically plated Cu-Co Electrode. Electrocatalysis 10, 29–34 (2018)
C. Dai, L. Sun, J. Song, H. Liao, A.C. Fisher, Z.J. Xu, Selective electroreduction of carbon dioxide to formic acid on cobalt-decorated copper thin films. Small Methods 3, 1900362 (2019)
R. Parajuli, J.B. Gerken, K. Keyshar, I. Sullivan, N. Sivasankar, K. Teamey, S.S. Stahl, E.B. Cole, Integration of anodic and cathodic catalysts of earth-abundant materials for efficient, scalable CO2 reduction. Top. Catal. 58, 57–66 (2014)
V.S.K. Yadav, M.K. Purkait, Electrochemical studies for CO2 reduction using synthesized Co3O4 (anode) and Cu2O (cathode) as electrocatalysts. Energy Fuels 29, 6670–6677 (2015)
V.S.K. Yadav, M.K. Purkait, Electrochemical reduction of CO2 to HCOOH using zinc and cobalt oxide as electrocatalysts. New J. Chem. 39, 7348–7354 (2015)
S. Gao, X. Jiao, Z. Sun, W. Zhang, Y. Sun, C. Wang, Q. Hu, X. Zu, F. Yang, S. Yang, L. Liang, J. Wu, Y. Xie, Ultrathin Co3O4 layers realizing optimized CO2 electroreduction to formate. Angew. Chem. Int. Ed. 55, 698–702 (2016)
D. Pan, X. Ye, Y. Cao, S. Zhu, X. Chen, M. Chen, D. Zhang, G. Li, Photoanode driven photoelectrocatalytic system for CO2 reduction to formic acid by using CoOx cathode. Appl. Surf. Sci. 511, 145497 (2020)
H. Ogihara, T. Maezuru, Y. Ogishima, I. Yamanaka, Electrochemical reduction of CO2 to CO by a Co-N-C electrocatalyst and PEM reactor at ambient conditions. ChemistrySelect 1, 5533–5537 (2016)
J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu, G. Wang, Nitrogen-doped porous carbon supported nonprecious metal single-atom electrocatalysts: From synthesis to application. Small Methods 3, 1900159 (2019)
J. Li, P. Pršlja, T. Shinagawa, A.J. Martín Fernández, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou, R. García-Muelas, N. López, J. Pérez-Ramírez, F. Jaouen, Volcano trend in electrocatalytic CO2 reduction activity over atomically dispersed metal sites on nitrogen-doped carbon. ACS Catalysis 9, 10426–10439 (2019)
W. Liu, Z. Miao, Z. Li, X. Wu, P. Zhou, J. Zhao, H. Zhao, W. Si, J. Zhou, S. Zhuo, Electroreduction of CO2 catalyzed by Co@N-C materials. J. CO2 Utiliz. 32, 241–250 (2019)
T. Atoguchi, A. Aramata, A. Kazusaka, M. Enyo, Cobalt(II)–tetraphenylporphyrin–pyridine complex fixed on a glassy carbon electrode and its prominent catalytic activity for reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 156–157 (1991)
G. Ramírez, G. Ferraudi, Y.Y. Chen, E. Trollund, D. Villagra, Enhanced photoelectrochemical catalysis of CO2 reduction mediated by a supramolecular electrode of packed CoII(tetrabenzoporphyrin). Inorg. Chim. Acta 362, 5–10 (2009)
D. Quezada, J. Honores, M. García, F. Armijo, M. Isaacs, Electrocatalytic reduction of carbon dioxide on a cobalt tetrakis(4-aminophenyl)porphyrin modified electrode in BMImBF4. New J. Chem. 38, 3606–3612 (2014)
K. Alenezi, Electrocatalytic study of carbon dioxide reduction by Co(TPP)Cl complex. J. Chem. 2016, 1–7 (2016)
C.L. Yao, J.C. Li, W. Gao, Q. Jiang, Cobalt-porphine catalyzed CO2 electro-reduction: A novel protonation mechanism. Phys Chem Chem Phys 19, 15067–15072 (2017)
Y. Bochlin, E. Korin, A. Bettelheim, Different pathways for CO2 electrocatalytic reduction by confined CoTMPyP in electrodeposited reduced graphene oxide. ACS Applied Energy Materials 2, 8434–8440 (2019)
B. Hu, W. Xie, R. Li, Z. Pan, S. Song, Y. Wang, How does the ligands structure surrounding metal-N4 of Co-based macrocyclic compounds affect electrochemical reduction of CO2 performance? Electrochim. Acta 331, 135283 (2020)
S. Fernández, S. Cañellas, F. Franco, J.M. Luis, M.A. Pericas, J. Lloret‐Fillol, The dual effect of coordinating –NH groups and light in the electrochemical CO2 reduction with pyridylamino Co complexes Chem. Electro. Chem. 8, 4456–4465 (2021)
J. Shen, R. Kortlever, R. Kas, Y.Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K.J. Schouten, G. Mul, M.T. Koper, Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 6, 1–8 (2015)
M. Abdinejad, A. Seifitokaldani, C. Dao, E.H. Sargent, X.-A. Zhang, H.B. Kraatz, enhanced electrochemical reduction of CO2 catalyzed by cobalt and iron amino porphyrin complexes. ACS Applied Energy Materials 2, 1330–1335 (2019)
Y. Wu, G. Hu, C.L. Rooney, G.W. Brudvig, H. Wang, Heterogeneous nature of electrocatalytic CO/CO2 reduction by cobalt phthalocyanines. Chemsuschem 13, 6296–6299 (2020)
J. Hawecker, J.-M. Lehn, R. Ziessel, Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine), J. Chem. Soc., Chem. Commun. 984, 328–330 (1984)
C.M. Bolinger, N. Story, B.P. Sullivan, T.J. Meyer, electrocatalytic reduction of carbon dioxide by 2,2’-bipyridine complexes of rhodium and iridium. Inorg. Chem. 27, 4582–4587 (1988)
H. Ishida, K. Tanaka, T. Tanaka, Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the Formation of CO and HCOO, Organometallics 6, 181–186 (1987)
D.L. DuBois, A. Miedaner, R.C. Haltiwanger, Electrochemical reduction of CO2 catalyzed by [Pd(triphosphine)(solvent)](BF4)2 complexes: Synthetic and mechanistic studies. J. Am. Chem. Soc. 113, 8753–8764 (1991)
S. Slater, J.H. Wagenknecht, Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J. Am. Chem. Soc. 106, 5367–5368 (1984)
J.W. Raebiger, J.W. Turner, B.C. Noll, C.J. Curtis, A. Miedaner, B. Cox, D.L. DuBois, electrochemical reduction of CO2 to CO catalyzed by a bimetallic palladium complex. Organometallics 25, 3345–3351 (2006)
K. Takahashi, K. Hiratsuka, H. Sasaki, S. Toshima, Electrocatalytic behavior of metal porphyrins in the reduction of carbon dioxide. Chem. Lett. 8, 305–308 (1979)
N. Kornienko, Y. Zhao, C.S. Kley, C. Zhu, D. Kim, S. Lin, P. Yang, Metal–organic frameworks for electrocatalytic reduction of carbon dioxide. J. Am. Chem. Soc. 137, 14129–14135 (2015)
C. Costentin, S. Drouet, M. Robert, J.-M. Save´ant, A local proton source enhances CO2 electroreduction to CO by a molecular Fe catalyst, Sci. 338, 90–94 (2012)
X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang, H. Wang, Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 8, 14675 (2017)
J. Schneider, H. Jia, J.T. Muckerman, E. Fujita, Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2 reduction catalysts. Chem. Soc. Rev. 41, 2036–2051 (2012)
D.A. Tryk, T. Yamamoto, M. Kokubun, K. Hirota, K. Hashimoto, M. Okawa, A. Fujishima, Recent developments in electrochemical and photoelectrochemical CO2 reduction: involvement of the (CO2)2− dimer radical anion. Appl. Organomet. Chem. 15, 113–120 (2001)
R. B. A., S.-K. A., T.M. R., Z. W., W.D. T., K.P. J., M.R. I., Ionic liquid-mediated selective conversion of CO2 to CO at low overpotentials, Science 334, 643–644 (2011)
K. Chandrasekaran, L.M. Bockris, In-situ spectroscopic investigation of adsorbed intermediate radicals in electrochemical reactions: CO2− on platinum. Surf. Sci. 185, 495–514 (1987)
Z. Chen, J.J. Concepcion, M.K. Brennaman, P. Kang, M.R. Norris, P.G. Hoertz, T.J. Meyer, Splitting CO2 into CO and O2 by a single catalyst. Proc. Natl. Acad. Sci. 109, 15606–15611 (2012)
J.E. Pander, A. Fogg, A.B. Bocarsly, Utilization of electropolymerized films of cobalt porphyrin for the reduction of carbon dioxide in aqueous media. ChemCatChem 8, 3536–3545 (2016)
V.S.K. Yadav, M.K. Purkait, Effect of copper oxide electrocatalyst on CO2 reduction using Co3O4 as anode. Journal of Science: Advanced Materials and Devices 1, 330–336 (2016)
Y.Y. Birdja, J. Shen, M.T.M. Koper, Influence of the metal center of metalloprotoporphyrins on the electrocatalytic CO2 reduction to formic acid. Catal. Today 288, 37–47 (2017)
S. Abner, A. Chen, Design and mechanistic study of advanced cobalt-based nanostructured catalysts for electrochemical carbon dioxide reduction. Appl. Catal. B 301, 120761 (2022)
M. Abdinejad, M.N. Hossain, H.-B. Kraatz, Homogeneous and heterogeneous molecular catalysts for electrochemical reduction of carbon dioxide. RSC Adv. 10, 38013–38023 (2020)
W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang, Z. Jin, Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Advanced Science 5, 1700275 (2018)
M. Todoroki, K. Hara, A. Kudo, T. Sakata, Electrochemical reduction of high pressure CO2 at Pb, Hg and In electrodes in an aqueous KHCO3 solution. J. Electroanal. Chem. 394, 199–203 (1995)
L. Sun, G.K. Ramesha, P.V. Kamat, J.F. Brennecke, Switching the reaction course of electrochemical CO2 reduction with ionic liquids. Langmuir 30, 6302–6308 (2014)
J.M. Assour, W.K. Kahn, Electron spin resonance of α-and β-cobalt phthalocyanine. J. Am. Chem. Soc. 87, 207–212 (1965)
M. Wojdyła, M. Rębarz, W. Bała, B. Derkowska, Z. łukasiak, Optical properties of vacuum sublimed cobalt phthalocyanine (CoPc) thin layers. Molecul. Cryst. Liquid Cryst. 485, 974–989 (2008)
T. Kroll, V.Y. Aristov, O.V. Molodtsova, Y.A. Ossipyan, D.V. Vyalikh, B. Buchner, M. Knupfer, Spin and orbital ground state of Co in cobalt phthalocyanine. J. Phys. Chem. A 113, 8917–8922 (2009)
M. Zhu, R. Ye, K. Jin, N. Lazouski, K. Manthiram, Elucidating the reactivity and mechanism of CO2 electroreduction at highly dispersed cobalt phthalocyanine. ACS Energy Lett. 3, 1381–1386 (2018)
V.V. Maslyuk, V.Y. Aristov, O.V. Molodtsova, D.V. Vyalikh, V.M. Zhilin, Y.A. Ossipyan, T. Bredow, I. Mertig, M. Knupfer, The electronic structure of cobalt phthalocyanine. Appl. Phys. A 94, 485–489 (2008)
J.H. Weber, D.H. Busch, Complexes derived from strong field ligands. XIX. Magnetic properties of transition metal derivatives of 4, 4′, 4″, 4‴-tetrasulfophthalocyanine, Inorganic Chem. 4, 469–471 (1965)
K. Sakamoto, E. Ohno, Synthesis of cobalt phthalocyanine derivatives and their cyclic voltammograms. Dyes Pigm. 35, 375–386 (1997)
Y. Pan, W. Chen, S. Lu, Y. Zhang, Novel aqueous soluble cobalt phthalocyanine: Synthesis and catalytic activity on oxidation of 2-mercaptoethanol. Dyes Pigm. 66, 115–121 (2005)
V. Iliev, V. Alexiev, L. Bilyarska, Effect of metal phthalocyanine complex aggregation on the catalytic and photocatalytic oxidation of sulfur containing compounds. J. Mol. Catal. A: Chem. 137, 15–22 (1999)
M. Hassanein, M. Abdo, S. Gerges, S. El-Khalafy, Study of the oxidation of 2-aminophenol by molecular oxygen catalyzed by cobalt(II) phthalocyaninetetrasodiumsulfonate in water. J. Mol. Catal. A: Chem. 287, 53–56 (2008)
A.B. Sorokin, Phthalocyanine metal complexes in catalysis. Chem. Rev. 113, 8152–8191 (2013)
Ü. Demirbaş, C. Göl, B. Barut, R. Bayrak, M. Durmuş, H. Kantekin, İ Değirmencioğlu, Peripherally and non-peripherally tetra-benzothiazole substituted metal-free zinc (II) and lead (II) phthalocyanines: Synthesis, characterization, and investigation of photophysical and photochemical properties. J. Mol. Struct. 1130, 677–687 (2017)
K.A. Grice, Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 336, 78–95 (2017)
D.-M. Feng, Y.-P. Zhu, P. Chen, T.-Y. Ma, Recent advances in transition-metal-mediated electrocatalytic CO2 reduction: From homogeneous to heterogeneous systems. Catalysts 7, 373 (2017)
M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang, Y.-F. Han, Cobalt phthalocyanine coordinated to pyridine-functionalized carbon nanotubes with enhanced CO2 electroreduction. Appl. Catal. B 251, 112–118 (2019)
S. Meshitsuka, M. Ichikawa, K. Tamaru, Electrocatalysis by metal phthalocyanines in the reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 158–159 (1974)
Z. Jiang, Y. Wang, X. Zhang, H. Zheng, X. Wang, Y. Liang, Revealing the hidden performance of metal phthalocyanines for CO2 reduction electrocatalysis by hybridization with carbon nanotubes. Nano Res. 12, 2330–2334 (2019)
N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D.J. Miller, J. Lu, S.-T. Lee, Y. Li, Supported cobalt polyphthalocyanine for high-performance electrocatalytic CO2 reduction. Chem. 3, 652–664 (2017)
X. Wu, J.W. Sun, P.F. Liu, J.Y. Zhao, Y. Liu, L. Guo, S. Dai, H.G. Yang, H. Zhao, Molecularly dispersed cobalt phthalocyanine mediates selective and durable CO2 reduction in a membrane flow cell. Adv. Func. Mater. 32, 2107301 (2022)
H. Li, Y. Pan, Z. Wang, Y. Yu, J. Xiong, H. Du, J. Lai, L. Wang, S. Feng, Coordination engineering of cobalt phthalocyanine by functionalized carbon nanotube for efficient and highly stable carbon dioxide reduction at high current density. Nano Res. 15, 3056–3064 (2022)
T.V. Magdesieva, T. Yamamoto, D.A. Tryk, A. Fujishima, Electrochemical reduction of CO2 with transition metal phthalocyanine and porphyrin complexes supported on activated carbon fibers. J. Electrochem. Soc. 149, D89 (2002)
S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert, C.P. Berlinguette, Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 365, 367–369 (2019)
T.V. Magdesieva, T. Yamamoto, D.A. Tryk, A. Fujishima, Electrochemical reduction of CO2 with transition metal phthalocyanine and porphyrin complexes supported on activated carbon fibers. J. Electrochem. Soc. 149, (2002)
J.-J. Ma, H.-L. Zhu, Y.-Q. Zheng, M. Shui, An insight into anchoring of cobalt phthalocyanines onto carbon: Efficiency of the CO2 reduction reaction. ACS Applied Energy Materials 4, 1442–1448 (2021)
T. Abe, T. Yoshida, S. Tokita, F. Taguchi, H. Imaya, M. Kaneko, Factors affecting selective electrocatalytic CO2 reduction with cobalt phthalocyanine incorporated in a polyvinylpyridine membrane coated on a graphite electrode. J. Electroanal. Chem. 412, 125–132 (1996)
T. Abe, H. Imaya, T. Yoshida, S. Tokita, D. Schlettwein, D. Wöhrle, M. Kaneko, Electrochemical CO2 reduction catalysed by cobalt octacyanophthalocyanine and its mechanism. J. Porphyrins Phthalocyanines 1, 315–321 (1997)
H. Gu, L. Zhong, G. Shi, J. Li, K. Yu, J. Li, S. Zhang, C. Zhu, S. Chen, C. Yang, Y. Kong, C. Chen, S. Li, J. Zhang, L. Zhang, Graphdiyne/graphene heterostructure: A universal 2D scaffold anchoring monodispersed transition-metal phthalocyanines for selective and durable CO2 electroreduction. J. Am. Chem. Soc. 143, 8679–8688 (2021)
F. Liang, J. Zhang, Z. Hu, C. Ma, W. Ni, Y. Zhang, S. Zhang, Intrinsic defect-rich graphene coupled cobalt phthalocyanine for robust electrochemical reduction of carbon dioxide. ACS Appl. Mater Interfaces. 13, 25523–25532 (2021)
P. Tian, B. Zhang, J. Chen, J. Zhang, L. Huang, R. Ye, B. Bao, M. Zhu, Curvature-induced electronic tuning of molecular catalysts for CO2 reduction. Cat. Sci. Technol. 11, 2491–2496 (2021)
D. Masheder, K.P.J. Williams, Raman spectro‐electrochemistry. II. In situ Raman spectroscopic studies of the electrochemical reduction of CO2 at cobalt (II) phthalocyanine‐impregnated PTFE‐bonded carbon gas diffusion electrodes. J Raman Spectros. 18, 391–398 (1987)
E.R. Savinova, S.A. Yashnik, E.N. Savinov, V.N. Parmon, Gas-phase electrocatalytic reduction of CO2 to CO on carbon gas-diffusion electrode promoted by cobalt phthalocyanine. React. Kinet. Catal. Lett. 46, 249–254 (1992)
Q. Wan, Q. He, Y. Zhang, L. Zhang, J. Li, J. Hou, X. Zhuang, C. Ke, J. Zhang, Boosting the faradaic efficiency for carbon dioxide to monoxide on a phthalocyanine cobalt based gas diffusion electrode to higher than 99% via microstructure regulation of catalyst layer. Electrochim. Acta 392, 139023 (2021)
S. Aoi, K. Mase, K. Ohkubo, S. Fukuzumi, Selective electrochemical reduction of CO2 to CO with a cobalt chlorin complex adsorbed on multi-walled carbon nanotubes in water. Chem. Commun. 51, 10226–10228 (2015)
N. Morlanés, K. Takanabe, V. Rodionov, Simultaneous reduction of CO2 and splitting of H2O by a single immobilized cobalt phthalocyanine electrocatalyst. ACS Catal. 6, 3092–3095 (2016)
A. De Riccardis, M. Lee, R.V. Kazantsev, A.J. Garza, G. Zeng, D.M. Larson, E.L. Clark, P. Lobaccaro, P.W.W. Burroughs, E. Bloise, J.W. Ager, A.T. Bell, M. Head-Gordon, G. Mele, F.M. Toma, Heterogenized pyridine-substituted cobalt(II) phthalocyanine yields reduction of CO2 by tuning the electron affinity of the Co center. ACS Appl. Mater. Interfaces. 12, 5251–5258 (2020)
J. Luangchaiyaporn, D. Wielend, D. Solonenko, H. Seelajaroen, J. Gasiorowski, M. Monecke, G. Salvan, D.R.T. Zahn, N.S. Sariciftci, P. Thamyongkit, High-performance CoII-phthalocyanine-based polymer for practical heterogeneous electrochemical reduction of carbon dioxide. Electrochim. Acta. 367, (2021)
J. Choi, P. Wagner, S. Gambhir, R. Jalili, D.R. MacFarlane, G.G. Wallace, D.L. Officer, Steric modification of a cobalt phthalocyanine/graphene catalyst to give enhanced and stable electrochemical CO2 reduction to CO. ACS Energy Lett. 4, 666–672 (2019)
X. Lu, Z. Jiang, X. Yuan, Y. Wu, R. Malpass-Evans, Y. Zhong, Y. Liang, N.B. McKeown, H. Wang, A bio-inspired O2-tolerant catalytic CO2 reduction electrode. Science Bulletin 64, 1890–1895 (2019)
M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulie, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Isci, C.P. Berlinguette, M. Robert, CO2 electrochemical catalytic reduction with a highly active cobalt phthalocyanine. Nat. Commun. 10, 3602 (2019)
H. Tian, K. Wang, Z. Shui, M. Ali Raza, H. Xiao, M. Que, L. Zhu, X. Chen, Enhanced CO2 electroreduction on Co active site of cobalt phthalocyanine by electronic effect. Mater. Letters 310, 131482 (2022)
Z. Yang, X. Zhang, C. Long, S. Yan, Y. Shi, J. Han, J. Zhang, P. An, L. Chang, Z. Tang, Covalently anchoring cobalt phthalocyanine on zeolitic imidazolate frameworks for efficient carbon dioxide electroreduction. CrystEngComm 22, 1619–1624 (2020)
B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen, J. Jiang, Two-dimensional covalent organic frameworks with cobalt(ii)-phthalocyanine sites for efficient electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 143, 7104–7113 (2021)
A.S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H.S. Oh, P. Strasser, Metal-doped nitrogenated carbon as an efficient catalyst for direct CO2 electroreduction to CO and hydrocarbons, Angew. Chem. Int. Ed. Engl. 54, 10758–10762 (2015)
W. Ju, A. Bagger, G.P. Hao, A.S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl, P. Strasser, Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2. Nat. Commun. 8, 1–9 (2017)
L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang, X. Bao, Enhancing CO2 electroreduction to methane with a cobalt phthalocyanine and zinc-nitrogen-carbon tandem catalyst. Angew. Chem. Int. Ed. Engl. 59, 22408–22413 (2020)
L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang, X. Bao, Synergistic catalysis over iron-nitrogen sites anchored with cobalt phthalocyanine for efficient CO2 electroreduction. Adv. Mater. 31, 1903470 (2019)
S. Gong, W. Wang, X. Xiao, J. Liu, C. Wu, X. Lv, Elucidating influence of the existence formation of anchored cobalt phthalocyanine on electrocatalytic CO2-to-CO conversion. Nano Energy 84, 105904 (2021)
J. Ma, H. Zhu, Y. Zheng, Highly dispersed cobalt phthalocyanine on nitrogen-doped carbon towards electrocatalytic reduction of CO2 to CO. Ionics 27, 2583–2590 (2021)
X. Tan, C. Yu, X. Song, C. Zhao, S. Cui, H. Xu, J. Chang, W. Guo, Z. Wang, Y. Xie, J. Qiu, Toward an understanding of the enhanced CO2 electroreduction in NaCl electrolyte over copc molecule-implanted graphitic carbon nitride catalyst. Adv. Energy Mater. 11, 2100075 (2021)
H.-L. Zhu, Y.-Q. Zheng, M. Shui, Synergistic interaction of nitrogen-doped carbon nanorod array anchored with cobalt phthalocyanine for electrochemical reduction of CO2. ACS Applied Energy Materials 3, 3893–3901 (2020)
M. Li, C. Yan, R. Ramachandran, Y. Lan, H. Dai, H. Shan, X. Meng, D. Cui, F. Wang, Z.-X. Xu, Non-peripheral octamethyl-substituted cobalt phthalocyanine nanorods supported on N-doped reduced graphene oxide achieve efficient electrocatalytic CO2 reduction to CO. Chem. Eng. J. 430, 133050 (2022)
T.T. Li, Y. Mei, H. Li, J. Qian, M. Wu, Y.Q. Zheng, Highly selective and active electrochemical reduction of CO2 to CO on a polymeric Co(II) phthalocyanine@graphitic carbon nitride nanosheet-carbon nanotube composite. Inorg. Chem. 59, 14184–14192 (2020)
J. Chen, J. Li, W. Liu, X. Ma, J. Xu, M. Zhu, Y.-F. Han, Facile synthesis of polymerized cobalt phthalocyanines for highly efficient CO2 reduction. Green Chem. 21, 6056–6061 (2019)
C.M. Lieber, N.S. Lewis, Catalytic reduction of carbon dioxide at carbon electrodes modified with cobalt phthalocyanine. J. Am. Chem. Soc. 106, 5033–5034 (1984)
J. Chen, M. Zhu, J. Li, J. Xu, Y.-F. Han, Structure–activity relationship of the polymerized cobalt phthalocyanines for electrocatalytic carbon dioxide reduction. The J. Phys. Chem. C 124, 16501–16507 (2020)
H. Wu, M. Zeng, X. Zhu, C. Tian, B. Mei, Y. Song, X.-L. Du, Z. Jiang, L. He, C. Xia, S. Dai, Defect engineering in polymeric cobalt phthalocyanine networks for enhanced electrochemical CO2 reduction. ChemElectroChem 5, 2717–2721 (2018)
Z. Zhang, J. Xiao, X.J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng, Y. Wang, Z.Q. Tian, D. Deng, Reaction mechanisms of well-defined metal-N4 sites in electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. Engl. 57, 16339–16342 (2018)
S. Lin, C.S. Diercks, Y.B. Zhang, N. Kornienko, E.M. Nichols, Y. Zhao, C.J. Chang, Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208–1213 (2015)
N. Kornienko, Y. Zhao, C.S. Kley, C. Zhu, D. Kim, S. Lin, C.J. Chang, O.M. Yaghi, P. Yang, Metal-organic frameworks for electrocatalytic reduction of carbon dioxide. J. Am. Chem. Soc. 137, 14129–14135 (2015)
Y. Liu, C.C. McCrory, Modulating the mechanism of electrocatalytic CO2 reduction by cobalt phthalocyanine through polymer coordination and encapsulation. Nat. Commun. 10, 1–10 (2019)
I.M. Nielsen, K. Leung, Cobalt−porphyrin catalyzed electrochemical reduction of carbon dioxide in water. 1. A density functional study of intermediates. J. Phys. Chem. A 114, 10166–10173 (2010)
J. Shen, M.J. Kolb, A.J. Gottle, M.T. Koper, DFT study on the mechanism of the electrochemical reduction of CO2 catalyzed by cobalt porphyrins. J. Phys. Chem. C 120, 15714–15721 (2016)
A.J. Göttle, M.T. Koper, Proton-coupled electron transfer in the electrocatalysis of CO2 reduction: prediction of sequential vs. concerted pathways using DFT. Chem. Sci. 81, 458–465 (2017)
Acknowledgements
This work was supported by the Shandong Provincial Rizhao Eco-environment Monitoring Center, Hunan University of Technology, and Rizhao Ecological Environmental Protection Service Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Feng, Q., Sun, Y., Gu, X. et al. Advances of Cobalt Phthalocyanine in Electrocatalytic CO2 Reduction to CO: a Mini Review. Electrocatalysis 13, 675–690 (2022). https://doi.org/10.1007/s12678-022-00766-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-022-00766-y