
Abstract
Anion exchange membrane fuel cell is a new chance to produce a functional and portable fuel cell; however, the studies are still at an early stage with few reports regarding the AEMFC. PtRh/C electrocatalysts with different Pt:Rh atomic ratios were prepared by an alcohol reduction process. X-ray diffraction patterns for all PtRh/C materials indicated no shift in Pt(fcc) peaks showing that Rh did not incorporated into Pt lattice; however, the analysis of lattice parameter showed that some Pt atoms are added to Rh(fcc) structure. The mean particle sizes were in the range of 4–5 nm. Electrochemical experiments showed that PtRh/C electrocatalyst with Pt:Rh atomic ratio of 70:30 had superior performance exhibiting a current density of 5.0 mA mgmetal −1. From in situ ATR-FTIR experiments, it was observed that PtRh/C electrocatalyst with Pt:Rh atomic ratio of 70:30 produced more acetate ions than other ones, while the material prepared with Pt:Rh atomic ratio of 50:50 was more selective to CO2 as observed in acid media.

Integrated acetaldehyde, carbonate, acetate, and CO2 band intensity as a function of the electrode potential for Pt/C, Rh/C, and PtRh/C electrocatalysts
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In theory, fuel cells show high efficiency to convert fuels into electricity when compared to conventional combustion or power generation [1]. Among the types of fuel cells, the direct liquid fuel cell (DLFC) is a technology that still is development to be applied as portable electronic devices and for stationary applications due to its high energy density, modest operating conditions, and safety [2, 3].
Among the liquid fuels, ethanol is an excellent candidate, because it is not toxic, it is produced by renewable sources (biomass), and it is characterized by high energy density (∼8.0 kWh/kg) [1–4]. In the last two decades, a large number of investigations have been focusing about ethanol oxidation, mainly in acid media, with the purpose of enhancing the electrocatalyst activities for applications in direct ethanol fuel cells (DEFC) [1, 5–11]. However, the complete and efficient oxidation of ethanol is a challenge yet [4, 12].
The products of ethanol oxidation reaction (EOR) obtained were mainly the acetaldehyde and acetic acid, and small amounts of CO2 [2, 12]. The ideal electrocatalyst for this reaction must promote the adsorption and dehydrogenation of ethanol, the cleavage of the C–C bond and the oxidation of the resulting CO and CHx intermediates to CO2 [12]. For this, a wide variety of materials are still studied [1, 5, 13–15].
PtSn electrocatalysts have higher activity than other Pt metals for ethanol oxidation in acid medium [12, 16]; however, this material does not facilitate the breaking of C–C bond as described for PtRh electrocatalyst despite its lower activity [4, 8]. In this case, Pt provides sites for ethanol dehydrogenative adsorption while Rh contributes to C–C bond breaking of these intermediates. Recently, Adzic [8, 17] reported that Rh sites are available to adsorb ethanol in alkaline medium which makes it interesting for fuel cell applications in this medium.
The development of solid anion exchange membranes (AEMs) [16, 18–22] as the use of Nafion/KOH [23, 24] has made the alkaline fuel cell more attractive because kinetics in alkaline medium are improved in comparison with acidic medium [16, 25]. On the other hand, the cleavage of C–C bond of ethanol molecule in alkaline medium is also a difficult task [26–28]. Recently, Fontes et al. [29] reported the synthesis of Pt/C, Rh/C, and PtRh/C electrocatalyst using borohydride reduction and tested for ethanol electro-oxidation in alkaline medium. The obtained PtRh/C electrocatalysts showed predominantly the presence of PtRh(fcc) alloy phases. The electrochemical and alkaline DEFC experiments showed that PtRh/C with Pt:Rh atomic ratio of 50:50 was the most active; however, the CO2 production only started from −0.35 V vs (Ag/AgCl 3 mol−1 KCl) that coincides with the decrease in the signal of acetate and carbonate ions. Despite of Rh/C electrocatalyst showing good activity in the first minutes of ethanol electro-oxidation, it deactivates more quickly than Pt/C and PtRh/C electrocatalysts.
Silva et al. [30] prepared PtSn/C electrocatalysts by polymeric precursor method (PPM), where predominantly PtSn(fcc) alloy was formed, while that PtSn/C electrocatalysts prepared by a sol gel method (SGM) was observed a non-alloyed with phases of Pt(fcc) and SnO2 coexisting separately. Chronoamperometric experiments in acid medium showed that higher ethanol electro-oxidation activity was obtained on the PPM material than on the SGM material; however, FTIR experiments indicate that C–C bond breaking on ethanol molecule occurred preferentially on the material prepared by SGM.
In this study, Pt/C, Rh/C, and PtRh/C electrocatalysts were prepared by an alcohol-reduction process [31–33] and, in this case, predominantly segregated phases of Pt(fcc) and Rh(fcc) were formed for these materials. These materials were tested for ethanol electro-oxidation in alkaline medium, while the products formed were determined by in situ FTIR spectroscopy.
Experimental
PtRh/C electrocatalysts (20 wt% of metals loading) with Pt:Rh atomic ratios of 100:0, 90:10, 70:30, 50:50, and 0:100) were prepared using H2PtCl6 · 6H2O (chloroplatinic acid—Aldrich), RhCl3 · xH2O as metal sources, ethylene glycol as a solvent and reducing agent, and Carbon Vulcan XC72 as the support [31–33]. The metal sources were dissolved in ethylene glycol/water (75/25, v/v), and Carbon Vulcan XC72 support was added. The resulting mixture was stirred in an ultrasonic bath for 5 min and then refluxed for 3 h. After, the mixture was filtered, and the obtained electrocatalysts were washed with water and dried at 70 °C for 2 h.
Powder X-ray diffraction (XRD) patterns of the catalysts were recorded on a Rigaku diffractometer model Miniflex II using a Cu Kα source (λ = 1.54056 Å) at 2θ = 20° to 90° with a step size of 0.05° and a scan time of 2 s per step. A JEOL JEM-2100 electron microscope operated at 200 kV was used to determine the morphology, distribution, and the size of the nanoparticles.
All electrochemical measurements were performed on a Microquimica potentiostat using standard three-electrode cells where the counter electrode was a Pt and the reference electrode was an Ag|AgCl (in 3 mol L-1 KCl). The working electrode (geometric area of 0.3 cm2 with a depth of 0.3 mm) was prepared using the thin porous coating technique [34]. The ink was prepared with 10 mg of catalyst and 100 μL of PTFE solution in 50 ml of water, and then dispersed in an ultrasonic bath for 5 min. After that, the ink was filtered, and the entire solid was deposited on the working electrode.
The spectroelectrochemical ATR-FTIR in situ measurements were performed on a Nicolet 6700 FT-IR spectrometer equipped with an MCT detector cooled with liquid N2, ATR accessory (MIRacle with a Diamond/ZnSe Crystal Plate Pike®), and an electrochemical cell better explained in the literature [30, 35, 36]. The working electrodes were the same as the electrochemical experiments, in the presence of 1.0 mol L−1 ethanol + 1 mol L−1 KOH. The absorbance spectra were collected as the ratio R:R 0, where R represents a spectrum at a given potential and R 0 is the spectrum collected at −0.85 V. Positive and negative directional bands represent gain and loss of species at the sampling potential, respectively. The spectra were computed from 128 interferograms averaged from 3000 to 850 cm−1 with the spectral resolution set to 8 cm−1, and the sample spectra were collected after applying potential successive steps from 0.1 from −0.75 to −0.05 V.
Results and Discussion
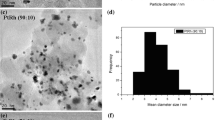
Figure 1 showed the TEM micrographs and histograms of Pt/C, Rh/C, and PtRh/C electrocatalysts determined by counting 100 nanoparticles in randomly selected regions, and all the sizes were measured according to similar materials in the literature [37–39]. For all materials, a good dispersion of the nanoparticles on the carbon support was observed, even though some small particles agglomeration can be observed, particularly when the amounts of Rh were increased. The average diameter of the nanoparticles determined were 4.8 +/−1.4 nm, 4.1 +/−0.9 nm, 3.7 +/−0.8 nm, 3.6 +/−1.1 nm, and 4.74 +/−1.4 nm for Pt/C, Pt90Rh10/C, Pt70Rh30/C, Pt50Rh50/C, Rh/C, respectively, and a narrow particle size distribution was observed for all cases.

TEM image and particles size distribuition for Pt/C, Pt-Rh/C and Rh/C materials
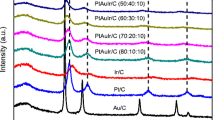
Powder XRD patterns of Pt/C, Rh/C, and PtRh/C electrocatalyts are shown in Fig. 2. For all materials, it was observed peaks at approximately 2θ ≈ 40°, 47°, 68°, 82°, and 87° are associated with the (111), (200), (220), and (311) planes, characteristic of the face-centered cubic (fcc) structure of Pt and Rh crystals.

a X-ray diffractograms of the Pt/C, Rh/C, and PtRh/C electrocatalysts. b Pawley refinement of peak (2 2 0) in all diffractograms with dots for experimental measure, traced modeled, black for individual contribution, and gray for residual line
For PtRh/C binary electrocatalysts it was observed a broadening of the fcc diffraction peaks that could be attributed to the product of the convolution of two PseudoVoigt line forms related to Pt(fcc) and RhPt(fcc) phases present in these materials. These patterns analysis were made by the Pawley method with the Fityk 0.98 software [40, 41] and are show in Fig 2b.
It is possible to note the changes in the asymmetric peak shape as a result of the contribution of Pt(fcc) lattice parameters (a = 0.392 nm (JCPDS # 04 802)). In the case of Rh(fcc) is not observed change the lattice parameters (a = 0.381 nm (JCPDS # 88 2334)); however, for PtRh was observed some phases with lattice parameters shifted near to 0.381, this behavior could be explained considering that Pt atoms are added to Rh crystalline structure.
Figure 3 shows the cyclic voltammetry (CV) in alkaline media for Pt/C, Rh/C, and PtRh/C electrocatalysts in the presence of 1 mol L−1 KOH. PtRh/C and Rh/C electrocatalysts do not have a well-defined hydrogen adsorption-desorption region (−0.85 to −0.45 V) as observed for Pt/C electrocatalyst. The poor definition of this region is associated with the presence of Rh that activates water molecules leading to the formation of layers of oxides and hydroxides [42]. PtRh/C and Rh/C electrocatalysts also showed a redox pair in the current values of −0.3 V/−05 V at −0.3/−0.7 V, following the increase of Rh loading. This effect may be attributed to the increase of the rhodium oxides species [42].

Cyclic voltammograms of Pt/C, Rh/C, and PtRh/C electrocatalysts in 1 mol L−1 KOH solution with a scan rate of 10 mV s−1 at 25 °C
Figure 4a shows the CV of Pt/C, Rh/C, and PtRh/C electrocatalysts in the presence of 1.0 mol L−1 of ethanol and 1 mol L−1 of KOH. For Pt70Rh30/C and Pt50Rh50/C electrocatalysts, it was possible to observe that the onset potential for ethanol electro-oxidation started at −0.65 V. The current values are greater than the ones observed for Pt90Rh10C, Pt/C, and Rh/C, where the electro-oxidation was started only at −0.55 V. Rh/C electrocatalyst was active for ethanol electro-oxidation in alkaline medium confirming the results of Adzic’s group [8, 17] and Fontes et al. [29].

a Linear sweep voltammograms with a scan rate of 10 mV s−1. b Current-time curves at −0.3 V for Pt/C, Rh/C and PtRh/C electrocatalysts in 1 mol L−1 ethanol solution in 1 mol L−1 KOH at 25 °C
Figure 4b shows the current time curves obtained by chronoamperometry with the potential set at −0.35 V for 30 min. It observed for all electrocatalysts a fast current decay in the first 3 min and a slow decay for PtRh/C and Pt/C after this period. The final current values observed for all electrocatalysts were 5 mA mgPt −1 for Pt70Rh30/C > 3.6 mA mgPt −1 for Pt50Rh50/C > ≈3.3 mA mgPt −1 for Rh/C, and Pt90Rh10/C > 3.0 mA mgPt −1 for Pt/C. Rh/C electrocatalyst is apparently more active than Pt/C taking into account these first 30 min; however, the decrease of activity of Rh/C is greater than the Pt/C electrocatalyst indicating that for great reaction times, it could become less active than Pt/C. To better understand the reasons for the different activities, in situ ATR-FTIR experiments were performed.
Figure 5 displays in situ ATR-FTIR spectra in the range of 3000 cm−1 at 850 cm−1 of ethanol electro-oxidation on Pt/C, Rh/C, and PtRh electrocatalysts at potentials range from −0.75 to −0.05 V with an interval of 0.1 V. It is possible to observe the appearance of two intense bands at approximately 1553 and 1410 cm-1, corresponding asymmetric and symmetric vibrations of C–O bond related to acetate ions [2, 43]. The carbonate band (1370 cm−1) [43] was not clearly visible in the spectrum probably due to the overlapping with the acetate band 1410 cm−1. Bands related to ethanol being consumed appeared at 1080, 1036, and 874 cm−1 [2]. In these spectra, bands also appeared at 926 cm−1 associated with the C–C–O stretch of acetaldehyde [25] and bands at 2343 cm−1 related to CO2 [2]. These species would not normally be expected to be found in alkaline media due to the instability of aldehydes in this medium [44], and CO2 under alkaline conditions is precipitated as carbonate [16]. Nevertheless, these species can be observed due to the variation of interfacial pH which occurred during ethanol electro-oxidation as recently reported by Figueiredo et al. [45].

In situ ATR-FTIR spectra taken at several potentials (indicated) in 1 mol L−1 KOH + 1.0 mol L−1 ethanol for Pt/C, Rh/C, and Pt-Rh/C electrocatalysts. The backgrounds were collected at −0.85 V (Ag/AgCl scale)
Figure 6 shows the integrated bands corresponding to acetaldehyde, carbonate ions, acetate, and CO2 deconvoluted by Lorentzian line forms [46]. Although unstable in alkaline medium, acetaldehyde is produced by all electrocatalysts at low over potentials and is important observe that only for the materials Pt50Rh50/C and Rh/C it is possible to observe a decrease in the intensity of this band to less negative potentials, since for Pt50Rh50/C, this band disappears at −0.05 V.

Integrated acetaldehyde, carbonate, acetate, and CO2 band intensity as a function of the electrode potential for Pt/C, Rh/C, and Pt-Rh/C electrocatalysts
The Pt70Rh30/C material produces small relative amounts of carbonates compared to the others, mainly at −0.35 V, potential that chronoamperometric experiments were conducted, which is one clue to explain the high activity of this material, while others accumulate carbonate that can create a physical barrier between the catalytic sites thus reducing the ability to get more alcohol to oxidize.
Another clue that may help explain the increase of activity of Pt70Rh30/C is the fact that at a potential of −0.35 V, it presents greater intensity of acetate than other materials, indicating that activity can be linked to kinetic production of this species non-completely oxidized [47]. Considering the CO2 production, PtRh/C materials present lower onset potential to produce this species compared to Pt/C or Rh/C. This could suggest that Pt phase adsorbs ethanol molecule and Rh phases promotes the cleavage of the C–C bond, as it was described for an acid medium. Nevertheless, the increase in CO2 production is not associated with an increase in the current values, as described for acidic medium too [4, 8, 17, 48].
From the results shown in Fig. 6, it is noteworthy that Pt50Rh50/C carbonate bands decay in intensity at −0.35 V, which is the same potential that the acetaldehyde band began to decrease in intensity and the acetate production is attenuated, while the CO2 production increase significantly. As described by other authors [8, 17, 48], probably this electrocatalyst could promote an acidification near the surface of the electrode that facilitates the C–C bond breaking of ethanol molecule.
Xu et al. [49] concluded that the enhanced electrocatalytic activity of the Pt2Rh/C catalyst for ethanol oxidation in alkaline medium could be ascribed to the improvement of the C–C bond cleavage in the presence of Rh. These authors also observed that Rh improved oxidation kinetics of COads to CO2 in an alkaline medium.
Bhattacharya et al. [50] also concluded that the C–C bond of ethanol was broken to a greater extent with the increasing of Rh in Ni-supported Pt-Rh nanocomposite. The complete oxidation of ethanol occurs through some chemical reactions between electrochemically generated M-OH (M = Pt, Rh) and ethanol oxidation followed by acetaldehyde and acetate leading to the final product carbonate. These authors showed that eletrocatalyst containing 35 atom% of Rh in Pt-Rh co-deposit was the best in providing more current, stability, and poison-resisting capability among the different mutual binary prepared for ethanol oxidation in alkaline medium.
Conclusions
PtRh/C electrocatalysts prepared by an alcohol reduction process showed the presence of Pt(fcc) phase and Rh rich RhPt(fcc) alloy. Pt/C, Rh/C, and PtRh/C electrocatalysts were active for ethanol electro-oxidation, being the material prepared with Pt:Rh atomic ratio of 70:30 the most active. Probably, this could be explained by the smaller quantities of carbonate ions produced when compared to the other electrocatalyts. Carbonate ions could create a physical barrier between the catalytic sites thus reducing the ability to get more alcohol to be oxidized. Its superior activity is due the production of acetate ions. Interestingly, the material prepared with Pt:Rh atomic ratio of 50:50 was more selective to CO2 production, probably due the interfacial shift of pH which enables the selectivity of Rh for breaking the C-C bond, despite its inferior performance. Nevertheless, further work is now necessary to investigate the surface of these materials as the studies in direct ethanol alkaline fuel cell.
References
S.P.S. Badwal, S. Giddey, A. Kulkarni, J. Goel, S. Basu, Applied Energy 145, 80–103 (2015)
Z.-Y. Zhou, Q. Wang, J.-L. Lin, N. Tian, S.-G. Sun, Electrochim. Acta 55, 7995–7999 (2010)
H. Takahashi, M. Sagihara, M. Taguchi, Int. J. Hydrogen Energy 39, 18424–18432 (2014)
B. Liu, Z.W. Chia, C.H. Cheng, J.Y. Lee, Energy & Fuels 25, 3135–3141 (2011)
Y. Zhou, Q.J. Xu, in: Advanced Materials Research, 797-800 (2014).
S. Beyhan, J.M. Léger, F. Kadirgan, Appl. Catal. B 130–131, 305–313 (2013)
S. García-Rodríguez, S. Rojas, M.A. Peña, J.L.G. Fierro, S. Baranton, J.M. Léger, Appl. Catal. B 106, 520–528 (2011)
M. Li, W.P. Zhou, N.S. Marinkovic, K. Sasaki, R.R. Adzic, Electrochim. Acta 104, 454–461 (2013)
C. Xu, P.K. Shen, J. Power Sources 142, 27–29 (2005)
J. Ribeiro, D.M. dos Anjos, K.B. Kokoh, C. Coutanceau, J.M. Léger, P. Olivi, A.R. de Andrade, G. Tremiliosi-Filho, Electrochim. Acta 52, 6997–7006 (2007)
F. Colmati, E. Antolini, E.R. Gonzalez, Appl. Catal. B 73, 106–115 (2007)
S.C. Zignani, V. Baglio, J.J. Linares, G. Monforte, E.R. Gonzalez, A.S. Aricò, Electrochim. Acta 70, 255–265 (2012)
S. Beyhan, J.-M. Léger, F. Kadırgan, Appl. Catal. B 144, 66–74 (2012)
J. Lobato, P. Cañizares, D. Ubeda, F.J. Pinar, M.A. Rodrigo, Appl. Catal. B 106, 174–180 (2011)
E. Antolini, J. Power Sources 170, 1–12 (2007)
Z. Zhang, L. Xin, K. Sun, W. Li, Int. J. Hydrogen Energy 36, 12686–12697 (2011)
A. Kowal, M. Li, M. Shao, K. Sasaki, M.B. Vukmirovic, J. Zhang, N.S. Marinkovic, P. Liu, A.I. Frenkel, R.R. Adzic, Nat. Mater. 8, 325–330 (2009)
G. Wang, Y. Weng, D. Chu, D. Xie, R. Chen, J. Membr. Sci 326(2009), 4–8 (2009)
G. Wang, Y. Weng, D. Chu, R. Chen, D. Xie, J. Membr. Sci. 332(2009), 63–68 (2009)
H. Herman, R.C.T. Slade, J.R. Varcoe, J. Membr, Sci. 218, 147–163 (2003)
T.N. Danks, R.C.T. Slade, J.R. Varcoe, J. Mater, Chem. 13, 712–721 (2003)
J.R. Varcoe, Phys Chem Chem Phys 9(2007), 1479–1486 (2007)
S.G. da Silva, M.H.M.T. Assumpcao, R.F.B. de Souza, G.S. Buzzo, E.V. Spinace, A.O. Neto, J.C.M. Silva, Electrocatal. 5, 438–444 (2014)
H. Hou, S. Wang, W. Jin, Q. Jiang, L. Sun, L. Jiang, G. Sun, Int. J. Hydrogen Energy 36, 5104–5109 (2011)
A.N. Geraldes, D.F. Da Silva, E.S. Pino, J.C.M. Da Silva, R.F.B. De Souza, P. Hammer, E.V. Spinacé, A.O. Neto, M. Linardi, M.C. Dos Santos, Electrochim. Acta 111, 455–465 (2013)
S.Q. Song, W.J. Zhou, Z.H. Zhou, L.H. Jiang, G.Q. Sun, Q. Xin, V. Leontidis, S. Kontou, P. Tsiakaras, Int. J. Hydrogen Energy 30, 995–1001 (2005)
C. Coutanceau, S. Brimaud, C. Lamy, J.M. Léger, L. Dubau, S. Rousseau, F. Vigier, Electrochim. Acta 53, 6865–6880 (2008)
A.O. Neto, L.A. Farias, R.R. Dias, M. Brandalise, M. Linardi, E.V. Spinacé, Electrochem. Comm. 10, 1315–1317 (2008)
E.H. Fontes, R.M. Piasentin, J.M.S. Ayoub, J.C.M. da Silva, M.H.M.T. Assumpção, E.V. Spinacé, A.O. Neto, R.F.B. de Souza, Mater. Renew. Sustain. Energy. 4, 1–10 (2015)
J.C.M. Silva, L.S. Parreira, R.F.B. De Souza, M.L. Calegaro, E.V. Spinacé, A.O. Neto, M.C. Santos, Appl. Catal. B 110(2011), 141–147 (2011)
V.A. Ribeiro, O.V. Correa, A.O. Neto, M. Linardi, E.V. Spinacé, Appl. Catal. A 372, 162–166 (2010)
E.G. Franco, A.O. Neto, M. Linardi, J Braz Chem Soc 13, 516–521 (2002)
A.O. Neto, A.Y. Watanabe, M. Brandalise, M.M. Tusi, R.M. de S. Rodrigues, M. Linardi, E.V. Spinacé, C.A.L.G.O. Forbicini, J. Alloys. Compd. 476, 288–291 (2009)
A.O. Neto, M. Brandalise, R.R. Dias, J.M.S. Ayoub, A.C. Silva, J.C. Penteado, M. Linardi, E.V. Spinacé, Int. J. Hydrogen Energy 35, 9177–9181 (2010)
R.F.B. De Souza, J.C.M. Silva, F.C. Simoes, M.L. Calegaro, A.O. Neto, M.C. Santos, Int. J. Electrochem. Sci. 7, 5356–5366 (2012)
R.S. Henrique, R.F.B. De Souza, J.C.M. Silva, J.M.S. Ayoub, R.M. Piasentin, M. Linardi, E.V. Spinacé, M.C. Santos, A.O. Neto, Int. J. Electrochem. Sci. 7, 2036–2046 (2012)
S. Sen Gupta, J. Datta, J. Electroanal, Chem. 594, 65–72 (2006)
L.C. Silva-Junior, G. Maia, R.R. Passos, E.A. de Souza, G.A. Camara, M.J. Giz, Electrochim. Acta 112, 612–619 (2013)
H.J. Kim, S.M. Choi, S.H. Nam, M.H. Seo, W.B. Kim, Appl. Catal. A 352, 145–151 (2009)
G.S. Pawley, J Appl Crystallogr 14, 357–361 (1981)
M. Wojdyr, J Appl Crystallogr 43, 1126–1128 (2010)
Y. Zhang, X.P. Gao, M.J. Weaver, J. Phys. Chem 97, 8656–8663 (1993)
X. Fang, L. Wang, P.K. Shen, G. Cui, C. Bianchini, J. Power Sources 195, 1375–1378 (2010)
Y. Kwon, S.C.S. Lai, P. Rodriguez, M.T.M. Koper, J. Am, Chem. Soc. 133, 6914–6917 (2011)
M.C. Figueiredo, R.M. Arán-Ais, V. Climent, T. Kallio, J.M. Feliu, Chem. Electro. Chem. 2, 1254–1258 (2015)
A.O. Neto, J. Nandenha, M.H.M.T. Assumpção, M. Linardi, E.V. Spinacé, R.F.B. de Souza, Int. J. Hydrogen Energy 38, 10585–10591 (2013)
B. Pierozynski, Int. J. Electrochem. Sci. 7, 6406–6416 (2012)
J.P.I. de Souza, S.L. Queiroz, K. Bergamaski, E.R. Gonzalez, F.C. Nart, J. Phys, Chem. B 106, 9825–9830 (2002)
S.Y. Shen, T.S. Zhao, J.B. Xu, Int J Hydrogen Energy 35, 12911–12917 (2010)
P. Mukherjee, P.S. Roy, S.K. Bhattacharya, Int J Hydrogen Energy 40, 13357–13367 (2015)
Acknowledgements
The authors thank the Laboratório de Microscopia do Centro de Ciências e Tecnologia de Materiais (CCTM) by TEM measurements and CAPES, CNPq, and FAPESP (2014/09087-4, 2014/50279-4) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fontes, E.H., da Silva, S.G., Spinace´, E.V. et al. In Situ ATR-FTIR Studies of Ethanol Electro-oxidation in Alkaline Medium on PtRh/C Electrocatalyst Prepared by an Alcohol Reduction Process. Electrocatalysis 7, 297–304 (2016). https://doi.org/10.1007/s12678-016-0308-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-016-0308-z