
Abstract
This study offers a unique insight into the use of high surface area metallic tin as support material for platinum catalysts for fuel cell application. We have synthesized high surface area metallic β-tin nanostructures (TNSs) in aqueous solutions by novel one-pot process and used it as a platinum catalyst support in methanol electrooxidation reaction. Rigorous study of parameters controlling the size and shape of TNSs was performed, including selected surfactant molecules at various concentrations, tin salts, and the addition of sodium citrate. Rod-shaped particles with a 50-nm diameter and 500-nm length were obtained from solutions of selected surfactant in concentrations of 1–20 mM by sodium borohydride reduction. These particles had a β-Sn crystalline core with a main lattice plane of (101) and were covered by a 4-nm oxide shell. A maximal surface area of 170 m2 g−1 was measured from a sample prepared by using low concentration of sodium dodecyl sulfate (SDS) (1 mM). This sample is composed of nanorods and nano semi-spherical shape tin particles. Addition of sodium citrate, which acts as a Sn2+ ion ligand, yields longer rods. Electrochemical oxidation of methanol on platinum catalyst, supported on metallic Sn nanostructure, exhibits a high activity, which is comparable to commercial carbon-supported platinum catalysts. In situ surface-enhanced Raman (SER), emphasizing the role of surface oxides on the methanol oxidation activity, further studied methanol oxidation on Pt/TNS, Pt/C, and Pt-Sn alloy catalyst.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nano tin materials have a wide range of potential applications that can be envisioned in various scientific and technological areas, for example, electronic connections in microelectronics and catalysts for chemical sensors [1–3]. The use of tin particles has been demonstrated successfully in the detection of many gases, including nitric oxide [4, 5], ethanol [6, 7] acetone vapors [8], CO [5], H2 [7–9], and isobutene [10]. More recently, dye-sensitized solar cells (DSCs) were fabricated from mesoporous SnO2 electrodes that were prepared from nanosize SnO2 particles [11]. Tin and its oxides are also known to have a high charge capacity as negative electrode materials in Li-ion batteries [12–14].
Several synthesis techniques have been proposed for preparing Sn and SnO2 nanomaterials and, specifically, nanowires or nanorods, including solid-state methods [15] and a template-free vapor transport process [16]. The vapor solid method has been employed to fabricate single-crystalline Sn nanowires with diameters of 10–20 nm [16]. Structural characterization using high-resolution transmission electron microscopy showed that the nanowires and nanosquares grew in a preferential direction of 200. Hydrothermal [17] and non-hydrothermal [18] synthesis of SnO2 nanorods were reported at moderate temperatures (320 or 700 °C). Wang reported the formation of Sn/SnOx core-shell nanospheres (45 nm) via a modified polyol process for Li anode batteries [19]. Metallic tin nanorods were also synthesized by a sonochemical method employing a polyol process yielding rod-like particles with widely dispersed diameter sizes from 50 to 200 nm, a length of less than 3 μm, and a Brunauer-Emmett-Teller (BET) area of 53 m2 g−1 [20].
In the present work, we report the study of a facile synthesis strategy of single-crystalline metallic tin nanostructures (TNSs) for use as a platinum catalyst support in methanol electrooxidation [21–23]. The motivation is driven by several properties of tin, including a cooperative effect of Sn (in Pt-Sn alloy) and SnO2 support on lowering CO poisoning of platinum catalysts [24, 25]. CO oxidation is known as the rate-determining step (rds) of methanol electrooxidation reactions [26]. TNSs are also expected to provide a high specific area, good stability in acid solutions due to the presence of a stabilizing SnO2 passivation layer, and a low melting point (231 °C) which enables low temperature sintering of the nanostructures into an electroconducting Sn network [27].
It should be emphasized that Pt-Sn catalysts and Pt/SnO2 have been widely reported as fuel cell catalyst. However, to the best our knowledge, Pt directly supported on metallic TNSs has not been reported before. The issues associated with the durability and stability of Pt catalysts supported on carbon, caused by carbon degradation in acid solutions, especially at high anodic potentials, are well documented in the literature. In this context, Pt supported on metallic TNSs reported in this work could possibly serve as an alternative to Pt/C catalysts.
The aims of this work are to study the reaction conditions that influence the shape of Sn nanoparticles synthesized in an aqueous solution and to measure the methanol electrooxidation of a Pt catalyst supported on these materials.
Experimental
TNS Synthesis
Sodium dodecyl sulfate (CH3(CH2)11OSO3Na, SDS, Acros 85 %), sodium stearate (C18H35NaO2, SS, Sigma >99 %), dodecyl amine (CH3(CH2)11NH2, DA, Acros 98 %), and hexadecyltrimethyl ammonium bromide (CH3(CH2)15N(Br)(CH3)3, HDTAB, Sigma >98 %) were dissolved in water (18 MΩ cm at 20 °C) and were used as templating surfactants. Stock solutions of 8 mg/mL tin chloride (SnCl2) and tin sulfate (SnSO4) from Sigma-Aldrich (ACS grade 99.9 %) were prepared. Five milliliters of the tin salt solution was added to 30 mL of the surfactant solution and stirred for few minutes followed by the addition of 5 mL (4.8 mg/mL) of a freshly prepared sodium borohydride solution (Strem Chemicals, 98 %) and allowed to react for 30 min. The suspended solid product was centrifuged and washed three times with water and then with ethanol and dried under vacuum for 2 h at room temperature. Sodium citrate (NaC6H7O7, Merck 99 %) was used as a cation-stabilizing agent in some of the syntheses [28].
Preparation of Pt-Supported TNS Catalysts and Pt-Sn
Platinum catalysts supported on TNSs (Pt/TNS) and on carbon black (Pt/C) were prepared by two methods. The first was impregnation of the substrate with commercial Pt black (BASF HP) or with Pt particles prepared by polyol-assisted microwave method [29–31]. The second method applied direct chemical reduction of H2PtCl6 (Strem chemicals 99.9 %, 38–40 % Pt) on TNSs or on XC72 carbon black (Cabot) by a sodium borohydride solution to produce 50 % (wt) loading of Pt.
Pt-Sn alloy catalyst was prepared by polyol-assisted microwave (MW) method for comparative purposes [32]. MW synthesis system was carried out using an adapted domestic MW (1000 W) equipped with a reflux condenser. The precursors, i.e., H2PtCl6 and SnCl2, in the proper stoichiometries are first dissolved in ethylene glycol. The mixture is then purged with pure Ar gas and irradiated by MW for few minutes under Ar atmosphere. After cooling, the remaining powder is washed few times with pure ethanol, separated by centrifuge, and mildly heat dried.
TNS Characterization
Scanning electron microscopy (SEM) measurements were performed using a JEOL GSM-6510LV system. The images were collected using secondary electron imaging (SEI), back scattering electron imaging (BEI), and back electron composition (BEC) detectors. SEM samples were prepared by spray coating from an ethanol suspension onto a conductive silicon wafer. High-resolution transmission electron microscopy (HRTEM) measurements were carried out on a Tecnai F20 Field Emission Gun TEM. The structure of the TNS samples was characterized by X-ray diffraction (XRD) using a PANalytical X-ray diffractometer (X’Pert PRO) with Cu Kα radiation (λ = 0.154 nm) and phase analysis identification at 40 kV and 40 mA. Full pattern identification was carried out with the X’Pert High Score Plus software package, version 2.2e (2.2.5), by PANalytical. In addition, BET measurements were performed using a ChemBET pulsar from Quantachrome. Inductively coupled plasma (ICP) measurements were performed using a Varian EIS 710 system.
Electrochemical Measurements
Electrodes were prepared from slurries of suspended particles that were applied to the electrode surface. Each suspension contained 5 mg of prepared supported catalyst, 19.6 μL of Nafion® suspension (ion power) in a mixture of 30 % isopropanol (Frutarom Ltd., 99.5 %), and 70 % water (v/v). A 5 μL aliquot of this suspension was pipetted onto a 5-mm diameter glassy carbon disk electrode (Pine Instruments) that was used as a working electrode. After drying in air for 20 min, the Pt loading on the disk electrode was 10 μg cm−2. A three-electrode standard glass cell equipped with the working electrode, a Pt wire counter electrode, and an Ag/AgCl reference electrode (Metrohm) was used for the electrochemical measurements. A fresh electrolyte solution of 0.5 M H2SO4 and 1 M methanol (Merck 99.9 %) in water was prepared before each experiment. The electrolyte was purged with N2 (99.999 %) for 20 min prior to each run and continued to flow above the solution during the measurement, in order to avoid oxygen penetration into the cell.
In Situ Surface-Enhanced Raman Spectroscopic Measurements of Methanol Oxidation
In situ surface-enhanced Raman (SER) spectroscopic measurements were performed in a home-built single-compartment three-electrode plastic cell covered by an optically flat glass disc window (Fig. 1). Gold-coated quartz crystal supplied by CH instruments was used as working electrode. A Pt wire and Ag wire (potential of Ag wire was calibrated using a standard reference electrode prior to in situ measurements) were used as counter and reference electrodes, respectively. An XploRA Raman confocal microscope (HORIBA) was applied, utilizing laser excitation of 548 nm and ×50 microscope objective lens. The laser power was set to 20 mW, and the spectrum acquisition time was 60 s. Collected spectra were subjected to a multi-point baseline correction using LabRAM program.

Electrochemical cell used for in situ Raman measurements
Results and Discussion
The Surfactant Effect
Surfactants are widely used as templating and capping agents that determine the shape and diameter of produced particles [33–37]. In this part, surfactants with selected cationic and anionic head groups and chain lengths, at selected concentrations, were employed in the synthesis process. Figure 2a–c shows SEM images of solid tin particles produced by sodium borohydride reduction of 5-mM SnCl2 solutions. The application of 1 mM SDS (Fig. 2a) in this synthesis yields mostly rod-shaped particles with a diameter of 50 nm and length of about 300 nm. Inevitable residues of the surfactant are also observed as gray shapeless matter between the particles and were confirmed by FTIR spectra of these samples (not shown). At a higher concentration of 5 mM SDS, smaller-size particles with higher abundance of round shapes are seen (Fig. 2b). These round particles grow up to 100 nm in diameter at a SDS concentration of 10 mM (Fig. 2c).

SEM images of TNSs produced from SnCl2 and SDS: a 5 mM SnCl2 and 1 mM SDS (inset picture scale bar is 0.5 μm), b 5 mM SnCl2 and 5 mM SDS, and c 5 mM SnCl2 and 10 mM SDS and from d 5 mM SnSO4 and 1 mM SDS, e 5 mM SnSO4 and 5 mM SDS, and f 5 mM SnSO4 and 10 mM SDS
An opposite behavior is seen upon replacing SnCl2 by SnSO4. The effect of the SDS concentration on Sn particle growth (Fig. 2d–f) shows no elongated TNSs when using 1 mM SDS, but rather small spherical particles with a diameter of about 100 nm (Fig. 2d). When the SDS concentration is increased to 5 mM, small nanorods of 30-nm diameter and 150-nm length start to appear (Fig. 2e). However, 10 mM SDS was needed to attain TNSs of similar dimensions to those seen in a sample prepared in 1 mM SDS in a SnCl2 solution (Fig. 2a).
These opposite trends seen in Fig. 2a–c using SnCl2 versus Fig. 2d–f with SnSO4 are attributed to different particle growth mechanisms which are induced by the salt anion. The chloride anions allow the particle to grow by a direct growth mechanism, utilizing the surfactant as a capping agent [38]. After initial formation of Sn as round-shaped seeds, due to disoriented nucleation, further growth of the particles proceeds in a unidirectional process which leads to the formation of nanorods. This process is controlled by the SDS concentration as well as by the salt anion charge and concentration [36]. Particles prepared in the presence of a Cl− have a smaller tendency to interfere with the surface coverage of SDS, which facilitates the elongation process. Conversely, the SO4 2− anion has stronger adsorption to the fresh Sn surface than Cl−, thereby resulting in lower coverage of the capping agent’s dodecyl sulfate anion. Thus, these particles grow equally in all directions and result in round particles. When the SDS concentration is higher (>5 mM), the equilibrium adsorption of the dodecyl sulfate shifts to higher coverage, which direct the particle growth. The surfactant tends to adsorb on planes with higher surface energy [39]. In the case of TiO2 nanorod arrays (TNRs), nanorod growth along the (101) plane (Fig. 5b) is induced by the relatively low surface energy of the (111) plane on which the surfactant is poorly adsorbed.
The Effect of SC
Sodium citrate (SC) is used as a chelating agent to stabilize Sn2+ ions in aqueous solutions [28]. Mildly acidic Sn(II) produces predominantly SnL2− and SnHL− (L = tetravalent citrate ligands) complexes [40] which are known to have high association constants that prevent the formation of Sn(OH)2 [41].
The effect of the addition of SC on the morphology of synthesized Sn particles was therefore investigated. Standard solutions containing 5 mM SnCl2 and 1 mM SDS were used with SC concentrations of 1, 10, and 100 mM to prepare TNSs (Fig. 3). At a SC concentration of 1 mM, only small-size nanorods and shapeless large Sn particles of more than 2 μm in diameter are formed (Fig. 3a). Using 100 mM, SC results in large oval-shaped particles with an average length of 800 nm and a width of about 250–350 nm as the main product (Fig. 3c). Ten millimolars of SC yields well-defined rectangular-shaped elongated 500–700-nm- long and about 70-nm-wide Sn particles, together with only few spherical particles (Fig. 3b). The high SC concentration (10 mM) facilitates the formation of a thinner surfactant layer on the facets of the particles, thereby providing a shorter path for mass transport of Sn2+ through this layer. In addition, the SC coordinated Sn2+ ion can promote transport of ions through the SDS bilayer, resulting in larger particle dimensions as seen in Fig. 3c [28]. However, in the presence of a very high concentration of SC (100 mM), SDS has minor influence on the particle’s dimensions, which results in significant growth in all directions and especially in their width (Fig. 3c).

SEM images of TNSs produced from 5 mM SnCl2 and 1 mM SDS in a 1, b 10, and c 100 mM sodium citrate; d 10 mM sodium citrate added to 1 mM SDS and 5 mM SnCl2 solution and e 1 mM SDS added to 5 mM SnCl2 and 10 mM sodium citrate solution
The role of SC was further studied by examining its influence on the morphology of the formed TNSs when added at different stages of the synthesis. In the first experiment, a SC solution was added to a solution of SnCl2 and SDS to give final concentrations of 10, 5, and 1 mM, respectively. In a second experiment, SnCl2 was first equilibrated with SC followed by adding SDS to yield solutions with the same final concentrations. An identical reduction step by NaBH4 was applied in both experiments. SEM images of TNSs formed in each synthesis are presented in Fig. 3d–e. Well-resolved tetragonal-shaped TNSs are seen in Fig. 3d. These particles grew by the method described in the first experiment. The typical length of the TNSs is 700 nm with a diameter of 70–100 nm. However, some particles display a width of more than 170 nm. Smaller TNSs grew in the second experiment, with a length of 500–700 nm and a diameter of 50–70 nm (Fig. 3e). These results signify the importance of SC as a chelating agent in influencing the growth process and the final structure.
The crystalline structures of the synthesized TNSs were characterized by XRD and HRTEM measurements. Figure 4 shows a typical XRD pattern of TNSs synthesized from SnCl2 and 1 mM SDS. An almost identical diffraction pattern was measured from TNSs synthesized with 10 mM SC added to the SDS and SnCl2 solution. These well-defined diffraction peaks are attributed to single-phase β-Sn TNRs and nanoparticles [42]. Nanorods and spherical particles have various crystalline orientations but demonstrate strongest peaks in hkl directions of (101), (200), and (211). It is important to note that no traces of crystalline forms of SnO or SnO2 can be identified in any of the diffraction patterns.

XRD pattern of TNSs produced from 5 mM SnCl2 and 1 mM SDS and reference pattern as per [42]
HRTEM of the same TNR sample is shown in Fig. 5. The TNSs visible in TEM images is representative of tetragonal-shaped particles and rods. These two structures have a diameter of 80 and 40 nm, respectively. Small semi-spherical particles of size 15 ± 2 nm can also be observed in the vicinity of the TNRs (Fig. 5a). Zooming on the horizontal particle reveals an extremely well-resolved image composed of a core covered by a 4-nm shell. The HRTEM and FFT diffraction images (Fig. 5b) show a well-organized atomic structure of the core with (101) lattice plane of a tetragonal β-Sn structure, with d-spacing of 0.27 nm that is compatible with the XRD results. The outer shell layer of the TNR is also seen in the image. It is a much less-ordered material with lower electron reflectivity assumed to be tin oxides (SnOx).

HRTEM and FFT images of TNSs prepared from SnCl2/SDS with addition of 10 mM sodium citrate. Two magnifications: a scale bar = 50 nm and (b) scale bar = 2 nm. An FFT electron diffraction image is shown as an insert
Characterization of Sn powder surface area by BET measurements was performed for selected synthesized materials of interest. A summary of these results and their corresponding particle size and morphology based on SEM images is presented in Table 1. The highest BET surface area of 170 m2 g−1 was measured from samples without SC (Table 1), composed of TNSs and nanoparticles with a size of 15 ± 2 nm (Fig. 5a). The high surface area may have resulted from the combination of considerable number of smaller semi-spherical particles along with relatively larger nanorods. A slightly lower surface area (152 m2 g−1) was measured in samples prepared by a similar method but in SnSO4 solution. Samples with TNSs and some smaller spherical particles clearly have a higher surface area. However, smaller-size TNSs prepared without SC (synthesis N1, N4) have about twice the surface area of particles prepared with SC (synthesis N2, N3). This result is explained by the relatively larger diameter of the TNSs attained in the presence of SC and fewer very small spherical particles observed in the SEM images of these materials (Fig. 3).
Methanol Oxidation on Pt-Supported TNSs
TNSs were deposited on a glassy carbon disk electrode and were used as a support material for Pt catalysts. Due to a relatively low standard reduction potential of Sn2+/Sn redox (Eo = −0.14 V) [43], it was important to compare the oxidation current of the bare support with the normal electrochemical behavior of a Pt coat on the TNS. Cyclic voltammetry of a bare TNS support (sample N2) and platinum on the TNS is measured in sulfuric acid solution under an oxygen-free environment (Fig. 6). The bare TNS presents capacitive currents in the range of a few microamperes per square centimeter with no significant redox, while the Pt on the TNS has a classical hydrogen adsorption/desorption peak shape of polycrystalline Pt in the potential range of −0.2–0.1 V (vs. Ag/AgCl) and clear surface oxide formation reduction waves above 0.5 V [44]. The low measured currents of the bare TNS support are in line with the calculated current density, which yields currents of 3.8 μA cm−2 using Eq. (1):
where S BET is the measured BET surface area of TNS in square centimeter per gram, m is the mass loading of TNS on the electrode in grams, ν is the voltage scan rate (volts s−1), and F is the nominal typical capacitance, 20 μF cm−2, in aqueous solutions [45].

Cyclic voltammetry (20 mV/s) of a TNS (N2 synthesis) and b 50 % (wt) Pt on TNS in 0.5-M H2SO4 solution under N2 atmosphere
These comparable calculated and measured currents suggest that the passive oxide layer formed on Sn and seen in Fig. 6 makes no contribution to the electrode’s interfacial resistance.
Platinum catalysts supported on various TNS materials, specified in Table 1, were prepared using chemical reduction by sodium borohydride or polyol-assisted microwave methods. Samples N1–N3 in Table 1 were impregnated by Pt black particles synthesized by the microwave synthetic method and by reducing dissolved H2PtCl6. The precious metal loading of 50 % weight percent on these selected TNSs or XC72 carbon support was determined by ICP measurements.
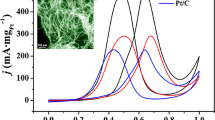
Figure 7 plots the voltammogram of selected supported catalysts, and Table 2 summarizes the values of the onset potential (E i ), potential at peak current (E p), and peak current (I p) of methanol oxidation extracted from the experimental graphs of all prepared electrodes and materials. Anodic peaks that appear at 0.7 V vs. Ag/AgCl in Fig. 7 correspond to methanol oxidation on Pt, followed by oxidation of COad to CO2 as the rate reaction determining step [46, 47].

Methanol oxidation on (a) Pt reduced on N1 NS, (b) Pt impregnated to N1 NS, (c) Pt reduced by sodium borohydride on carbon, (d) Pt reduced by sodium borohydride impregnated to carbon, and (e) Pt impregnated to carbon, measured by CV (20 mV/s)
All measured materials have a comparable voltammogram shape, E i values around 0.3–0.4 V, and an oxidation peak potential E p around 0.65 V, which suggest that an identical reaction mechanism occurs at somewhat different rates, as indicated by the I p values. Nevertheless, Pt catalysts produced by direct NaBH4 reduction on high surface area substrates exhibit a higher I p compared to catalysts prepared by impregnation of Pt catalysts into carbon or a TNS support.
The electrochemical surface area (ECSA) of the Pt catalysts was calculated from CV measurements in 0.5-M H2SO4 solutions in the potential range of −0.2–0.15 V vs. Ag/AgCl (e.g., Fig. 5) and is listed in Table 2. These calculations were basically performed by integration of the hydrogen oxidation charge of Pt divided by the theoretical value of the electrical charge of monolayer adsorbed hydrogen atoms on Pt, 210 μCcm−2 [48].
It is generally expected that a higher surface area support should provide better catalyst dispersion and higher specific current. Despite the lower specific surface area of the N1 TNS compared to carbon XC72R black (170 vs. 250 m2 g−1, respectively), the electrochemical catalytic surface area of Pt reduced directly on the TNS material has the same ECSA of 25 m2 g−1 as in commercial loaded Pt on XC72R. In line with this result, the methanol oxidation peak current on various Pt-supported catalysts listed in Table 2 is plotted against the ECSA measured in the CV (Fig. 8). A very good agreement was found between the hydrogen active surface area of Pt catalysts detrained by ECSA and the activity of methanol oxidation. Moreover, the synthesis method adopted for preparing Pt catalyst on XC72R carbon black has a lower ECSA (0.77 m2/g) as compared to commercial Pt catalysts on XC72R carbon black (24.9 m2/g). This is possibly due to the large size and/or aggregation of Pt particles produced as a result of direct NaBH4 reduction of Pt precursor solution onto XC72R carbon black. From this figure and from Table 2, it can be concluded that high reactivity should be attributed only to the Pt surface and not to the formation of a Pt-Sn intermetallic interface or alloy. Formation of Pt-Sn is expected to enhance the catalytic methanol oxidation current and reduce the overpotential which is frequently attributed to a bifunctional mechanism of CO oxidation to CO2 on Pt-supported or alloyed materials. However, this behavior does not occur on any of the supported catalysts in this study. Pt/TNS has the same catalytic behavior as commercial Pt supported on high surface area carbon, which is controlled primarily by the activity of Pt alone.

Methanol oxidation specific peak (I p) and electrochemical surface area (ECSA) of Pt catalysts supported on synthesized TNS and activated carbon (XC72R)
Figure 9 shows in situ SER spectra of three representative samples, Pt black (Fig. 9a), Pt-TNS (Fig. 8b), and microwave-synthesized Pt-Sn alloy (Fig. 9c), in 1 M methanol/0.5 M H2SO4. Electrochemical oxidation of methanol leading to the formation of CO on Pt black, Pt-TNS, and Pt-Sn alloy occurs at −0.1 V as evident by the appearance of bands corresponding to the stretching of metal-CO bond at 351, 372, and 344 cm−1 and CO stretching at 2062, 2076, and 2082 cm−1, respectively [49]. At 0.5 V, the relatively less-intense CO stretching band found in Pt black and Pt-TNS indicates the oxidation of most of the adsorbed CO. However, it is important to note that this occurred at a lower overpotential of 0.3 V in the case of Pt-Sn alloy. This may be attributed to the presence of Sn bonded to Pt which facilitates the oxidation of CO adsorbed on the Pt-Sn alloy surface, either by electronic effect [50] or by bifunctional mechanism [51]. This observation reinforces our supposition that Pt-TNS behave similar to Pt/C and not Pt-Sn alloy; namely, there is no cooperative effect between the Pt particle and the supporting Sn. At higher applied potential (0.6 V and above), the appearance of strong band at 2452 cm−1 in Pt black, at 2484 cm−1 (at all applied potential) in Pt-TNS, and at 2602 cm−1 (at all applied potential) in Pt-Sn alloy is attributed to the formation of corresponding metal oxides. It is also important to mention that the band at 2452 cm−1 assigned to metal oxide stretching vibration appears at higher potential only in Pt black, and further, the same is not visible at potential ≤0.5 V. This may be due to the relatively faster formation of CO causing the poisoning of Pt black upon electrochemical oxidation of methanol [49].

Selected SER spectra on Pt black (a), Pt-TNS (b), and Pt-Sn alloy (c) in 1 M methanol/0.5-M H2SO4 solution under N2 atmosphere simultaneously measured with the application of constant potential
Summary and Conclusions
This study reports various aspects of the synthesis of metallic tin structures (TNSs). A newly developed synthesis method yields β-Sn nanorods in a one-pot procedure using a surfactant as the templating agent and sodium citrate (SC) as a Sn2+ stabilizer. The effects of the surfactant, tin salt, and SC were investigated. Morphological inspection of each parameter shows a strong effect of the salt anion and surfactant concentration on the shape, length, and diameter of the produced Sn particles. Some products show TNSs with a length of 500–700 nm and a diameter of about 50–70 nm. Nanosize sphere-shaped Sn particles, formed in some of the TNS syntheses, provide the highest BET area. XRD and HRTEM/FFT measurements clearly show that nanorods and Sn particles produced by these methods have a pure crystalline β-Sn and form a core-shell structure with a SnOx shell of 4 nm. Electrochemical oxidation of methanol on Pt-supported TNSs was measured compared to commercial Pt or Pt reduced on carbon black (XC72). The results of these experiments clearly present high specific oxidation activity of Pt/TNS which was similar to the commercial product on carbon. This is albeit the lower BET area of the TNS compared to that of carbon. It was demonstrated that improving the Pt deposition method leads to significant enhancement of the methanol oxidation. It is believed that the content of the Pt catalyst could be further reduced with the use of other deposition methods on the TNSs (e.g., temperature programed reduction).
References
P.G. Harrison, M.J. Willett, Nature 332, 337 (1988)
D.-S. Lee, C.-H. Shim, J.-W. Lim, J.-S. Huh, D.-D. Lee, Y.-T. Kim, Sens. Actuators B 83, 250 (2002)
Z. Zhang, R. Zou, G. Song, L. Yu, Z. Chen, J. Hu, J. Mater. Chem. 21, 17360 (2011)
N.S. Ramgir, I.S. Mulla, K.P. Vijayamohanan, Sens. Actuators B 107, 708 (2005)
P. Candeloro, A. Carpentiero, S. Cabrini, E. Di Fabrizio, E. Comini, C. Baratto, G. Faglia, G. Sberveglieri, A. Gerardino, Microelectron. Eng. 78–79, 178 (2005)
Y.-D. Wang, C.-L. Ma, X.-H. Wu, X.-D. Sun, H.-D. Li, Talanta 57, 875 (2002)
Y. Wang, X. Wu, Y. Li, Z. Zhou, Solid-State Electron. 48, 627 (2004)
S.B. Patil, P.P. Patil, M.A. More, Sens. Actuators B 125, 126 (2007)
Y.-D. Wang, C.-L. Ma, X.-H. Wu, X.-D. Sun, H.-D. Li, Sens. Actuators B 85, 270 (2002)
C.-H. Shim, D.-S. Lee, S.-I. Hwang, M.-B. Lee, J.-S. Huh, D.-D. Lee, Sens. Actuators B 81, 176 (2002)
Y. Fukai, Y. Kondo, S. Mori, E. Suzuki, Electrochem. Commun. 9, 1439 (2007)
J.M. Tarascon, M. Armand, Nature 414, 359 (2001)
J.L. Tirado, Mater. Sci. Eng. R 40, 103 (2003)
W.W. Lee, J.-M. Lee, J. Mater. Chem. A 2, 1589 (2014)
J.Q. Sun, J.S. Wang, X.C. Wu, G.S. Zhang, J.Y. Wei, S.Q. Zhang, H. Li, D.R. Chen, Cryst. Growth Des. 6, 1584 (2006)
Y.-J. Hsu, S.-Y. Lu, J. Phys. Chem. B 109, 4398 (2005)
K. Quang Trung, V. Xuan Hien, D. Duc Vuong, N. Duc Chien, Adv. Nat. Sci.: Nanosci. Nanotechnol. 1, 025010 (2010)
Y. Wang, J.Y. Lee, J. Phys. Chem. B 108, 17832 (2004)
X.-L. Wang, M. Feygenson, M.C. Aronson, W.-Q. Han, J. Phys. Chem. C 114, 14697 (2010)
L. Qiu, V.G. Pol, J. Calderon-Moreno, A. Gedanken, Ultrason. Sonochem. 12, 243 (2005)
T.W. Chen, X.N. Yu, Z.H. Wang, J.L. Zhang, Adv. Mater. Res. 830, 426 (2014)
N.S. Veizaga, V.I. Rodriguez, T.A. Rocha, M. Bruno, O.A. Scelza, S.R. De Miguel, E.R. Gonzalez, J. Electrochem. Soc. 162, F243 (2015)
L. Zheng, L. Xiong, Q. Liu, K. Han, W. Liu, Y. Li, K. Tao, L. Niu, S. Yang, J. Xia, Electrochim. Acta 56, 9860 (2011)
W.-Z. Hung, W.-H. Chung, D.-S. Tsai, D.P. Wilkinson, Y.-S. Huang, Electrochim. Acta 55, 2116 (2010)
H. Zhang, C. Hu, X. He, L. Hong, G. Du, Y. Zhang, J. Power Sources 196, 4499 (2011)
A.O. Neto, R.R. Dias, M.M. Tusi, M. Linardi, E.V. Spinacé, J. Power Sources 166, 87 (2007)
J.C.M. Silva, R.F.B. De Souza, L.S. Parreira, E.T. Neto, M.L. Calegaro, M.C. Santos, Appl. Catal. B Environ 99, 265 (2010)
I. Hanzu, T. Djenizian, G.F. Ortiz, P. Knauth, J. Phys. Chem. C 113, 20568 (2009)
R. Harpeness, Z. Peng, X. Liu, V.G. Pol, Y. Koltypin, A. Gedanken, J. Colloid Interface Sci. 287, 678 (2005)
R. Harpeness, A. Gedanken, A.M. Weiss, M.A. Slifkin, J. Mater. Chem. 13, 2603 (2003)
C.R. Patra, S. Patra, A. Gabashvili, Y. Mastai, Y. Koltypin, A. Gedanken, V. Palchik, M.A. Slifkin, J. Nanosci. Nanotechnol. 6, 845 (2006)
H. Teller, H. Kornwietz, A. Schechter, Meet. Abstr. MA2012-01, 268 (2012)
C.J. Murphy, T.K. Sau, A.M. Gole, C.J. Orendorff, J. Gao, L. Gou, S.E. Hunyadi, T. Li, J. Phys. Chem. B 109, 13857 (2005)
N.R. Jana, L. Gearheart, C.J. Murphy, Adv. Mater. 13, 1389 (2001)
O. Krichevski, Y. Levi-Kalisman, D. Szwarcman, Y. Lereah, G. Markovich, J. Colloid Interface Sci. 314, 304 (2007)
J. Xiao, L. Qi, Nanoscale 3, 1383 (2011)
N. Du, H. Zhang, B. Chen, X. Ma and D. Yang, Chem. Commun., 3028 (2008)
J. Gao, C.M. Bender, C.J. Murphy, Langmuir 19, 9065 (2003)
G. Xi, J. Ye, Inorg. Chem. 49, 2302 (2010)
A. Survila, Z. Mockus, S. Kanapeckaitė, Electrochim. Acta 46, 571 (2000)
C. Han, Q. Liu, D.G. Ivey, Electrochim. Acta 53, 8332 (2008)
V.T. Deshpande, D.B. Sidreshmukh, Acta Crystallogr. 14, 335 (1961)
R.C. Weast, Handbook of Chemistry and Physics (CRC Press, Cleveland, 1976)
A.V. Tripković, R.R. Adžić, J. Electroanal. Chem. Interfacial Electrochem. 205, 335 (1986)
E. Gileadi, Physical electrochemistry (WILEY-VCH Verlag GmbH & Co, Weinheim, 2011)
T. Iwasita, Electrochim. Acta 47, 3663 (2002)
T. Frelink, W. Visscher, J.A.R. van Veen, Surf. Sci. 335, 353 (1995)
S.-A. Sheppard, S.A. Campbell, J.R. Smith, G.W. Lloyd, F.C. Walsh, T.R. Ralph, Analyst 123, 1923 (1998)
H. Yang, Y. Yang, S. Zou, J. Phys. Chem. C 111, 19058 (2007)
M. Arenz, V. Stamenkovic, B.B. Blizanac, K.J. Mayrhofer, N.M. Markovic, P.N. Ross, J. Catal. 232, 402 (2005)
E.M. Crabb, R. Marshall, D. Thompsett, J. Electrochem. Soc. 147, 4440 (2000)
Acknowledgments
The authors thank “The Israeli Ministry of National Infrastructures, Energy and Water” for the kind support of this work under project 211-11-005/2011-7-7.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Krichevski, O., Teller, H., Subramanian, P. et al. The Synthesis of Metallic β-Sn Nanostructures for Use as a Novel Pt Catalyst Support and Evaluation of Their Activity Toward Methanol Electrooxidation. Electrocatalysis 6, 554–562 (2015). https://doi.org/10.1007/s12678-015-0273-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-015-0273-y