
Abstract
The aim of the present study is to investigate the effect of the hydrolysis process on the properties of nanocrystalline cellulose (NCC) isolated from different precursors and the subsequent use of the extracted NCC for the reduction of graphene oxide (GO). The raw materials (almond and peanut shells) chosen for the isolation of cellulose were selected on the basis of their abundance and their poorly investigation in the production of NCC. Microcrystalline cellulose (MCC) was firstly extracted by alkali and bleaching treatments, then hydrolyzed under different processes to produce NCC polymorphs with structure I (NCC-I) and NCC structure II (NCC-II). The Fourier transform infrared spectroscopy, the X-ray diffraction (XRD) and the 13C NMR studies of the alkali and bleached products confirmed the formation of cellulose type I with high purity and good crystallinity, while scanning electron microscopy (SEM) showed micrometric fibers with lengths reaching 80 µm. Sulfuric acid treatment of these microfibers results in NCC type I or II, depending on the hydrolysis process. SEM of the NCC samples exhibited nanorods with diameter and aspect ratio in the range of 20–40 and 20–25 nm, respectively. Thermogravimetric analysis (TGA) of the MCC and NCC products indicated stable materials with a degradation temperature reaching 240 and 200 °C for MCC and NCC, respectively. The other part of our work concerns the use of the obtained cellulose nanocrystals (type II) for the preparation of reduced graphene oxide composite (NCC/RGO), to demonstrate the reducing properties of the isolated NCCII.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose is one of the most abundant natural polymers. This material continues to attract increasing attention among researchers due to its interesting properties (renewability, biocompatibility, good biodegradability, high Young modulus, low cost, etc.) and its potential applications in a variety of domains (food industry, paper production, biomedical, pharmaceutics, etc.) [1,2,3,4,5]. Therefore, till now great efforts have been devoted to its extraction from a variety of natural sources using different biological, chemical, physical and mechanical methods [6, 7]. Recently, it has been realized that nanocrystalline cellulose, which can be obtained from cellulose fibers by acid hydrolysis or by mechanical or physical treatments, presents superior and even new properties compared to the microcrystalline one [8]. These properties depend on several parameters related especially to the synthesis conditions (hydrolysis process, acid concentration, hydrolysis time, hydrolysis temperature, sonication time, ultrasonic irradiation intensity, etc.) and eventually to the source of cellulose fibers [9].
In another side, graphene oxide (GO) has recently captured a great interest among physicists, chemists, and materials scientists and has brought significant progress to applied electrochemical fields [10,11,12,13,14]. The large delocalized π–electron system and high theoretical specific surface area of graphene make the material suitable for adsorption of heavy metals, organic dyes, and pesticide residues. In fact, graphene displays outstanding properties such as high surface area, good electrical conductivity, mechanical exibility, and chemical stability. Furthermore, chemically derived graphene and its derivatives exhibit a high density of edge–plane like defect sites, which might provide many active sites for electron transfer to chemical and biological species [15]. A plenty of electronic devices, adsorbents and sensors based on these materials have been constructed especially for the removal and detection of environmental pollutants [11, 16,17,18].
Despite the distinctive advantages of the wet chemical method when used in preparing GO and reduced graphene oxide (RGO), the most widely used reducing agents, such as hydrazine, dimethylhydrazine, and NaBH4, are highly toxic; trace amount of the poisonous agents could have detrimental effects, particularly for biological related applications. Moreover, handling the hazardous waste generated by the reduction reaction may significantly increase the cost on industrial scale [19, 20]. Another obstacle in practical applications of chemical RGO is the poor process ability, because rGO tends to form irreversible aggregation, which results from strong van der Waals attractive forces between the graphene planes, unless a modification step (covalent or noncovalent) is included in the process. Nevertheless, the surface functionalization process usually suffers from limited scalability, time-consuming and environmental problems. Very recently, solvothermal reduction in some high boiling point solvents has emerged as alternative for one-pot [21,22,23] reduction and functionalization of GO [19, 24,25,26]. However, refluxing at such high temperature could lead to the decomposition of the solvent molecules and release poisonous gases and wastes [24, 25]. Recently, efforts have been made toward addressing the above-mentioned problems by using natural products instead of toxic reducing agents (see Table S1 in supplementary information). For example, vitamin C [27,28,29,30,31] performs well in reduction of GO; however, in most cases, the product exhibited a highly agglomerated morphology in absence of external stabilizer [27,28,29]. Reducing sugar [32], protein bovine serum albumin [33], green tea [34] have also been employed in reducing GO, although the resultant rGO formed stable aqueous solution, an alkali is needed as a co-reductant because of their weak reducing capability. Therefore, an effective, low-cost, and green reducing agent for chemical synthesis of soluble graphene in bulk quantity is still highly desirable [20].
As a part of the goal of developing the chemistry of nanocellulose, this paper reports a protocol of isolation of microcrystalline cellulose (MCC) from almond and peanut shells (PS) and their sulfuric acid hydrolysis leading to cellulose II. Cotton and Alfa fibers were also used to prepare nanocrystalline cellulose (NCC) type II to further confirm the effect of the hydrolysis process on the structure and microstructure of the NCC. The as-extracted NCC was then tested as a green reducing and stabilizing matrix of GO to prepare NCC/reduced graphene oxide (NCC/RGO) composite. The synthesized materials have been characterized using various techniques such as X-ray diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, UV–Vis spectrophotometry and scanning electron microscopy (SEM).
Materials and Methods
Materials
All chemicals used here were purchased from Sigma-Aldrich: MCC (< 50 µm (Avicel-101)), sodium hydroxide (NaOH, 99%), graphite powder (< 20 µm), potassium permanganate (KMnO4), phosphoric acid (H3PO4), sulfuric acid (H2SO4, 98%), hydrogen peroxide (H2O2), potassium bromide (KBr), hydrochloric acid (HCl, 37%), acetic acid (CH3COOH, 99%), dialysis membrane, and all natural products were purchased from a local supermarket. The water used throughout the experiments was purified with a Milli-Q system from Millipore Co.
Preparation of Nanocrystalline Cellulose (NCC)
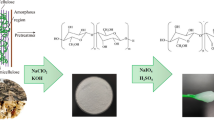
Two different acid hydrolysis processes (I and II, Scheme 1) have been applied to extract NCC from MCC. Extraction of NCC from alkali and bleached almond shells (AS) was performed using sulfuric acid hydrolysis according to protocols I and II. For the treated peanut shells, cotton and Alfa, NCC was obtained using only protocol II.

Acidic hydrolysis processes adopted to extract NCC from MCC
Protocol I
5 g of the treated AS were directly dispersed in 100 mL of 64% H2SO4 solution then heated at 44 °C under constant stirring. After 45 min of reaction, the mixture was diluted 10-fold with distilled water, to stop the hydrolysis process, then centrifuged four times at 4000 rpm for 15 min to remove the excess of acid. After that, the suspension was dialyzed against ultra-pure water using cellulose Sep T membrane (MWCO 12,000–14,000 Daltons) until reaching a constant pH. The obtained suspension was homogenized using an ultra-turrax T18 homogenizer (IKA) for 30 s, finally sonicated for 30 min and freeze-dried overnight to remove water.
Protocols II
a second hydrolysis protocol was adopted to isolate cellulose nanocrystals (CNCs) from alkali and bleached (treated) AS, peanut shells, cotton and Alfa fibers. It could be summarized as follows: 54 mL of sulfuric acid (98%) was added drop wise for 60 min, to 5 g of treated materials dispersed in 50 mL of distilled water. The suspension was then heated at 44 °C under magnetic stirring for 20 min. This hydrolysis process was stopped by adding cold distilled water. The excess of sulfuric acid was removed by centrifugation at 4000 rpm for 15 min, followed by dialysis against ultra-pure water using cellulose Sep T membrane (MWCO 12,000–14,000 Daltons) until reaching a constant pH. The obtained suspension was then homogenized using an ultra-turrax T18 homogenizer (IKA) for 30 s, finally sonicated for 30 min and freeze-dried overnight to remove water.
Preparation of Graphene Oxide (GO)
Graphene oxide (GO) was prepared from graphite powder as follows [35]: graphite powder (3 g) and KMnO4 (18 g) were added to a mixture of concentrated sulfuric and phosphoric acids (H2SO4/H3PO4) (9/1) and heated at 50 °C under constant magnetic stirring for 12 h. The mixture was then transferred in a beaker containing glacial Milli-Q-water and 2 mL of H2O2 (30%). The resulting precipitate was collected by centrifugation at 6000 rpm, washed successively with different solvents (HCl (37%), Milli-Q water until neutral pH, ethanol and ether) and finally dried in an oven at 50 °C.
In-Situ Preparation of Reduced Graphene Oxide/Nanocrystalline Cellulose (RGO/NCC) Composite
The composite RGO/NCC was prepared as follows: 20 mg of cellulose nanocrystals were added to 20 mL of an aqueous solution of GO (1 mg/mL). The mixture was kept under stirring for 72 h at 100 °C. The resulting black precipitate was separated from the aqueous supernatant by centrifugation at (5000 rpm for 15 min) then dried overnight at 50 °C.
Characterizations
Fourier Transform Infrared (FTIR) Spectroscopy
FTIR spectra of all samples were recorded in the transmittance mode in the range of 4000–400 cm− 1 using a Nicolet Avatar 360 spectrophotometer in attenuated total reflectance (ATR) mode. All the spectra were collected at 4 cm− 1 resolution and an accumulation of 200 scans.
X-Ray Diffraction (XRD) Analysis
X-ray diffraction (XRD) patterns were obtained using a BRUKER D8 ADVANCE powder diffractometer equipped with a copper anticathode (λCuKα = 1.5406 Å). Data was collected in the 2θ range varying from 5° to 40° at an increment step of 0.02°.
13C Cp-Mas NMR Spectroscopy
The solid state 13C CP-MAS NMR spectra were recorded with an AVANCE III Bruker NMR spectrometer equipped with a 7.049 T superconducting magnet, corresponding to a carbon resonance frequency of 75.422 MHz, using MAS spinning rates of 4–8 kHz. Samples were packed in MAS 4 mm diameter zirconia rotors. Chemical shifts were estimated relative to tetramethylsilane, which was used as external standard reference. All the spectra were collected for 3 h, which corresponds to 2000 scans.
Scanning Electron Microscopy (SEM)
SEM micrographs were obtained using an ULTRA 55 field emission scanning electron microscope (Zeiss, Auriga). The acquisitions were carried out at room temperature applying a voltage of 2.00 kV. The free working distance (FWD) varied from 5.9 to 30 mm. The samples were thoroughly grinded then deposited on aluminum substrates covered with carbon films. The samples were then coated with gold using a vacuum sputter-coater.
Thermogravimetric Analysis (TGA)
The thermal stability of the as-synthesized samples was examined using a Thermo-Microbalance TG 209 F3 Tarsus®, from 25 to 800 °C with a heating rate of 25 °C min− 1 under nitrogen atmosphere.
UV–Vis Measurements
The UV–Vis absorption spectra were recorded using a Perkin Elmer Lambda UV–Vis 950 spectrophotometer in quartz cuvettes with an optical path of 10 mm. The wavelength range is 200–800 nm.
X-Ray Photoelectron Spectroscopy (XPS)
The near-surface composition of NCC/RGO was studied by X-ray photoelectron spectroscopy (XPS) using an ADES 400 angular-resolved photoelectron spectrometer (VG Scientific, U.K.) equipped with a twin anode X-ray source with the standard Al/Mg anodes and a hemispherical analyzer. The XPS spectra were recorded using MgKα source operated at a power of 200 W at constant pass energy of 100 or 20 eV. The peak areas were determined following the Shirley inelastic background subtraction method. Static sample charging of the spectra was corrected with respect to the C1s peak at 285.0 eV.
Results and Discussion
The main objective of this work was to study the evolution of the characteristic (structure, microstructure, shape and size, cristallinity,…) of extracted NCC as a function of the origin of the precursor and the hydrolysis conditions. NCC was thus prepared from different natural precursors (AS, peanut shells, cotton, Alfa fibers) using two distinct hydrolysis protocols (I and II). The most attractive results were summarized in Table 1. The reducing properties of the cellulose nanocrystals were exploited to prepare reduced graphene oxide (RGO/NCC) composite, to show the very important role of cellulose nanocrystals for green reduction of graphene oxide to a stable composite.
Yield of Cellulose
Different amounts of MCC were obtained after alkaline and bleaching treatment, depending on the precursor used (Table 1). The highest amount of cellulose was obtained from PS with a yield reaching 70%.
Scanning Electronic Microscopy (SEM)
Scanning electron micrographs of cotton microfibers, treated AS and PS as well as those of their corresponding materials obtained after acid hydrolysis according to protocol I or II were reported in Fig. 1. The SEM micrographs of the treated cotton and AS show microfibers with approximately 10–20 µm in diameter and a length reaching 80 µm. The surface of these microfibers (MCCC and MCCAS), displays nanorods held together, which is quite different depending on the precursor material. The acid hydrolysis of the MCCC and MCCAS permits the total dissolution of the amorphous regions, leading to individual rod-like nanorods with a morphology resembling to that present in the surface of the microfibers. The geometries and dimensions of nanocrystalline cellulose, obtained after hydrolysis, seem to highly depend on the hydrolysis conditions as well as on the origin of cellulose microfibrils. The very smooth surface of the nanorods extracted from AS (NRAS) indicates the total removal of the amorphous parts and is a real specific characteristic of the as-extracted samples. The nanorods (NR) extracted from MCCAS, hydrolyzed according to protocol I or II (NRAS−I and NRAS−II), exhibit the same morphologies, but different dimensions: the average diameter of these NRs is about 20 nm (Table 1). Their lengths reach 600 nm for NRAS−I, slightly shorter (about 200 nm) for those obtained according to protocol II (sample NRAS−II). Acidic hydrolysis of MCCC using the protocol II leads to nanorods with similar dimensions as the NRAS−II, but with a different morphology.

Scanning electron micrographs of microcrystalline cellulose (MCC) obtained from different precursors and the corresponding nanocrystalline cellulose (NCC) obtained by acid hydrolysis of MCC according to protocol I or II
For the PS, the first treatment (Protocol I) leads to micrometric particles (MCCPS) with an undefined morphology. The hydrolysis of these MCCPS samples using the protocol II allows the disintegration of the nanorods present at the surface of the micrometric particles under our experimental conditions. The crystallinity of the NCC obtained here using protocol I or II is relatively important (up to 75% for cellulose obtained from AS) (Table 1) [36].
FTIR Study
The FTIR spectra of the novel precursors (AS and PS), those of their alkali and bleached products (MCCAS and MCCPS), and their corresponding nanocrystalline samples (NRAS−I, NRAS−II and NRPS−II) obtained according to protocol I or protocol II are displayed in Fig. 2. All spectra exhibit the same bands in the 3700–3020 cm− 1 region, around 2911 and at 1060 cm− 1 characteristic of the stretching vibrations of the O–H, C–H and C–O of the carbohydrates, respectively. The intensity of these absorption bands increased considerably after alkali and hydrolysis treatments, indicating increase in the percentage of cellulosic components.

FTIR spectra of almond shells (a), treated almond shells (AS) (b), treated peanuts shells (PS) (c), almond nanorods (NRAS−I) (d), almond nanorods (NRAS−II) (e), and peanut nanorods (NRPS−II) (f)
The absorption bands observed at 1515 and 1246 cm− 1 in the FTIR spectra of the precursor materials correspond to the C=C stretching of aromatic rings of lignin and –COO vibrations of acetyl groups in hemicellulose, respectively [36, 37]. The smaller band appearing at 1739 cm− 1 in the untreated materials can be assigned to the carbonyl (C=O) of acetone groups in hemicellulose and lignin. These three bands (1246, 1515 and 1739 cm− 1) were no longer present in the FTIR spectra of the treated materials, which indicates that the alkali, subsequent bleaching and acid treatments adopted permitted the complete removal of lignin and polysaccharides, such as pectin, xylan and hemicelluloses [37, 38]. Furthermore, all bands observed on the spectra of the white products (MCCAS and MCCPS) obtained after alkali and bleaching treatment of the precursors and the samples (NRAS−I, NRAS−II, NRPS−II), obtained after acid hydrolysis according to protocol I or protocol II, correspond to those of pure cellulose (Table 2). No significant difference was observed between the spectra of MCCAS, MCCPS and NRAS−I, which confirms that the cellulose molecular structure remains unchanged after hydrolysis according to protocol I. For the FTIR spectra of hydrolyzed AS according to protocol II, the bands observed at 1436, 1370 and 1327 cm− 1 on the spectra of NRAS−I and NRPS−I, were shifted to lower values by 2–4 cm− 1 on the spectra of NRAS−II (Table 2), which is in good agreement with a change in the cellulose structure by modifying the hydrolysis process. A small change in the peak position could also be noted for bands in the 3500–2000 cm− 1 range, which is highly sensitive to the changes of intra-and inter-molecular hydrogen bonds related to the transformation from cellulose I to cellulose II [39].
X-Ray Diffraction Patterns
The X-ray diffraction patterns of the alkali and treated AS and PS as well as those of their corresponding hydrolyzed samples according to protocol I or II are depicted in Fig. 3. The X-ray diffractograms of MCCAS, MCCPS and NRAS−I (Fig. 3a) show diffraction profiles typical of monolithic cellulose type I (also referred to as native cellulose) with three well defined crystalline peaks at around 2θ = 15.8°, 22.5° and 34.6° and no doublet for the peak at 22.5° [40, 41]. Thus, the alkali and bleaching treatments as well as the acid hydrolysis according to protocol I adopted seem to not alter the crystalline structure of the native cellulose.

X-ray diffractograms of: a almondshells (AS) treated almond shells (MCCAS) and almond shells nanorods P-I(NRAS−I); b peanut shells nanorods P-II (NRPS−II), almond shells nanorods P-II (NRAS−II), nanorods of cotton P-II (NRC−II)and nanorods of Alfa P-II(NCCAF−II)
The presence of four well resolved peaks at 2θ = 12.15°, 20°, 21.69° and 34.75° in the diffractogram of the crystalline nanorods (NRAS−II), obtained by hydrolyzing the AS microfibrils according to protocol II (Fig. 3b), clearly indicates predominance of cellulose with structure II [39]. Possibly, the dropwise as well as the time of addition of sulfuric acid in protocol II, unlike protocol I, causes a change in the crystalline arrangement of cellulose chains by regeneration of cellulose in water following its progressive dissolution in the 64% sulfuric acid solution: the original parallel-chains of cellulose I change to anti-parallel ones in cellulose II [42]. To further confirm this hypothesis, we have applied the hydrolysis protocol II to two other conventional precursors, cotton and Alfa fibers. The NCC (NRC−II and NCCAF−II) obtained also adopts the structure II (Fig. 3b). These results illustrate the effect of the hydrolysis process on the structure of the extracted nanocellulose that either retains the initial profile of the sample or transforms in to another cellulose polymorph.
For the PS, the peaks observed on the diffractogram of the nanocrystalline cellulose, obtained by hydrolyzing MCCPS according to protocol II, are also characteristic of cellulose II polymorph (Fig. 3b). However, the two peaks at around 22° are not well defined, which supports the hypothesis of unfinished hydrolysis process as was deduced from the SEM observations. So, the time of the hydrolysis process sufficient for almond shells, cotton and Alfa fibers to dissolve all amorphous regions, should be prolonged in the case of PS, which raises the relation between the origin of cellulose and the results of the hydrolysis process.
13C Cp-Mas NMR Spectroscopy
The crystalline structure of the as-elaborated samples was further verified by 13C CP-MAS NMR spectroscopy (Fig. 4). The signal patterns of the samples MCCAS and MCCPS, analyzed on the basis of previous reports [43, 44], are typical of cellulose I polymorph. The peaks observed on these spectra could be assigned as follows: the large peaks observed at 105, 89 and 63 ppm are due to C1, C4 and C6 cellulose ordered carbons, respectively. Those appearing at 84 and 61 ppm correspond respectively to C4 and C6 disordered carbons. The cluster at 71–73 ppm is attributed to the C2, C3 and C5 carbons. For the sample NRAS−II, its NMR spectrum is characteristic of pure cellulose II polymorph. It highly differs from that of cellulose I because of the difference between the chains conformations of cellulose type I and type II [39]. The peaks at 81 and 61 ppm disappear completely, indicating that the hydrolysis process adopted here permits the total removal of amorphous cellulose parts from cellulose I or fibrils. For the crystalline signal, peaks become thinner and well resolved. Peaks corresponding to C1, C4 and C6 were split into two.

13C CPMAS NMR spectra of the different treated materials and their corresponding nanocrystalline cellulose obtained by acid hydrolysis
For the MCCAS hydrolyzed according to protocol I (sample NRAS−I), despite that the XRD diffractogram is characteristic of pure cellulose I structure, signals corresponding to cellulose II were detected on its NMR spectrum. Thus, the protocol I adopted leads not to pure cellulose I, but to a mixture of cellulose polymorphs I and II. For the peanut shells, the presence of amorphous regions is evidenced on its NMR spectrum, confirming that the protocol II adopted here does not permit the total disintegration of the crystalline regions for this sample, which corroborates the conclusions based on SEM and XRD observations.
Thermogravimetric Analysis (TGA)
The thermogravimetric curves corresponding to microcrystalline and NCC of different precursors are displayed in Fig. 5. The initial weight loss observed on the most of the curves between 30 and 100 °C can be attributed to the evaporation of adsorbed moisture and corresponds to about 1.9% mass loss. The effective thermal degradation process of the microfibers (MCCAS and MCCPS) occurs in one step and begins at about 240 °C with a total weight loss of about 76%. A significant change in the degradation temperature and profile was observed after acid hydrolysis: the NCC degradation starts at around 200 °C, and occurs in two steps: the first step of degradation could be attributed to the decomposition of the highly sulfated regions. The second degradation step could be due to the breakdown of the crystalline regions, which have not been attacked by sulfuric acid. The lower degradation temperature of NCC compared to microcrystalline one could be related to the smaller dimension of its nanorods as well as the treatment with sulfuric acid, in accordance with previous reports [45, 46], leading to a notable decrease in the thermal stability of cellulose. It can be noticed that the thermal behavior of the NCC, obtained by hydrolyzing the MCCAS and MCCPS according to protocols I and II, respectively, is similar to that of NRC−II, resulting from cotton hydrolysis using protocol II. For the NRAS−II, despite the nanorods constituting this sample are of the smallest dimensions than those of NRAS−I, its decomposition occurs at relatively higher temperature as the other nanocrystalline samples. These findings indicate that NCC prepared from AS using the hydrolysis process II are well adapted to be used as reinforcement for polymer composites since they resist to thermal decomposition until temperatures of approximately 240 °C. Similar conclusions were also found by Jeong and collaborators [47, 48].

Thermogravimetric curves corresponding to as-extracted microcrystalline and nanocrystalline cellulose
Reduced Graphene Oxide–Nanocrystalline Cellulose (RGO–NCC) Composite
The GO reduction involves the elimination of oxygen-containing groups like epoxy, carbonyl and carboxyl groups from GO, leading to partial restoration of the graphenic network [49]. A color change is adopted as a direct signature of GO reduction. GO and RGO have been used in nanocomposite materials [49, 50], polymer composite materials [49], energy storage [50], biomedical applications [51,52,53], catalysis [54,55,56], and as a surfactant [57] with some overlaps between these fields.
In the present study, we investigated the reduction of GO using the as-prepared NCC at 100 °C for the preparation of reduced graphene oxide/nanocrystalline cellulose (RGO–NCC) composite. The first signal of success of the reduction is the change in the color of the GO suspension from brown to black. To further confirm the reduction process additional characterizations based on UV–Vis, Raman, SEM, FTIR, XPS and XRD were performed.
UV–Vis Spectroscopic Study
The UV–Vis absorbance spectra (Fig. 6) were recorded for the as-prepared nanocrystalline cellulose (NCC), the exfoliated GO and the RGO. The NCC suspension shows no absorption band in the wavelength range between 200 and 800 nm. For the GO, an absorption peak corresponding to the π–π* transition of aromatic (C=C) bonds and a small shoulder attributed to the n–π* transition of carbonyl groups (C=O) appear at 227 and 302 nm, respectively. The red-shift of the π–π* absorption band from 227 to 260 nm for the RGO–NCC composite is in good agreement with a reduction of the GO. However, this shift is not as important as that observed when using a strong reducing agent (e.g. hydrazine), suggesting that the GO was only partially reduced, and its electronic conjugation level is still lower than that of perfect graphene sheets. In addition, it is clearly seen that the region (> 300 nm) was increased compared to that of the starting GO spectrum [58,59,60]. The color of the obtained solutions clearly confirmed the GO reduction; the photos of the different suspensions are added in the inset of Fig. 6.

UV–Vis absorption spectra obtained for exfoliated GO, NCC, RGO–NCC solutions (0.2 mg/mL); the inset corresponds to the photographs of NCC, GO and NCC/RGO suspensions in water
FTIR Spectroscopic Analysis
The FTIR spectrum of the GO is depicted in Fig. 7. The stretching vibrations observed at 3440 and 1745 cm− 1 could be assigned to hydroxyl (OH) and carbonyl (C=O) groups, respectively. The band at 1628 cm− 1 can be attributed to absorbed water molecules or to the (C=C) aromatic skeleton vibrations. The bands detected at 1190 and 1050 cm− 1 could be assigned to the stretching vibrations of the C–O epoxy and alkoxy, respectively. The band at 1120 cm− 1 can be attributed to the stretching vibration of phenolic hydroxyl group (C–OH). After the reaction between NCC and GO, the spectrum was quite different from the starting ones. The new band appearing at 1426 cm− 1 could be assigned to the tertiary C–O groups vibrations in RGO structure. The peaks at 1227 and 1060 cm− 1 were assigned respectively to C–O–C vibration and C–O stretching. The peaks at 1745 and 1628 cm− 1 were almost absent, which confirms the total removal of the carbonyl group of GO. The broad band detected at 1583 cm− 1, attributed to C=C stretching vibration, is related to the restoration of the π–π system by removing of oxygen containing groups.

FTIR spectra of GO, NCC and RGO–NCC
X-Ray Diffraction
The XRD diffractograms of the GO and RGO–NCC were recorded in the 2θ range from 5° to 60° (Fig. 8). The NCC formation was confirmed as in the first part of the study. The diffraction peak observed at 2θ = 10° on the XRD pattern of GO corresponds to the (002) plane. After chemical reduction of exfoliated GO with cellulose nanocrystals, this peak disappeared in the XRD pattern of RGO–NCC. The diffractogram was mainly dominated by the diffraction peaks of the NCC. This implies that NCC not only reduced GO to RGO but also forms a composite material with RGO [61,62,63,64].

XRD patterns of NCCI, exfoliated GO and the obtained RGO–NCC powder
Raman Spectroscopy
The Raman spectra of GO (Fig. 9) shows two main bands at 1582 and 1362 cm− 1 assigned to G and D bands in the graphitic structure, respectively. The G band is due to the symmetric E2g mode and the D band is arising from the A1g mode, which is induced by disorders and it is assigned to the defects introduced upon the oxidation process. The (ID/IG) ratio was 0.84 for GO and 0.90 for RGO–NCC. The significant results are the shift of the obtained peaks. The G band at 1582 cm− 1 in GO, shifted to 1592 cm− 1 for RGO–NCC. This shift is most likely due to successful reduction of GO to RGO using NCC as a reducing agent. The formation of new graphitic crystallites form is due to the restoration of the π-conjugated system by removing oxygen-containing functional groups [65, 66]. Another characteristic of single-layer graphene is the relatively strong Raman intensity of the 2D band with respect to the G-band. Usually, a Lorentzian peak for the 2D band of monolayer graphene sheets is observed at 2700 cm− 1, whereas this peak is broadened and shifted to a higher wave number in the case of multilayer graphene. We observed the 2D band at 2720 cm− 1, which is the same as the previously reported peak position for single-layer graphene. Thus, our sample could consist of single-layer graphene sheets.

Raman spectra of GO and RGO–NCC
XPS Spectroscopy
XPS analysis was performed to confirm the GO reduction to RGO and RGO–NCC formation upon heating a GO solution with NCC (Fig. 10). The high resolution XPS spectrum of C1s of NCC can be fitted with three components at 284.6, 286.3, and 288.3 eV due to (C–C/C–H), C–O and C=O groups (Fig. 10a). Figure 10b exhibits the C1s high resolution XPS spectrum of GO. It can be deconvoluted into three different components at 284.3, 286.6 and 288.2 eV due to Csp2, C–O, and C=O, respectively with a carbon to oxygen (C/O) ratio of 1.73. The C1s XPS spectrum of RGO–NCC consists of peaks at 284.7, 286.3, 287.7, 288.5, and 290.4 eV assigned to Csp2/C–C, C–O, O–C–O, C=O, and O=C–OH, respectively [67]. In our case, the use of C/O ratio to estimate the reduction degree of GO to RGO is not appropriate as NCC is mainly composed of carbon and oxygen and as such introduces oxygen and as such introduces oxygen and carbon signals in the XPS spectrum of RGO–NCC.

High resolution XPS spectra of the C1s of nanocellulose (a), graphene oxide (b), and RGO–NCC composite (c)
SEM Characterization
Figure 11 depicts the SEM images of RGO–NCC composite, deposited on a silicon wafer. The SEM image of RGO–NCC consists of randomly aggregated, thin, crumpled sheets closely connected with each other, forming a disordered solid. The SEM image indicates that the cellulose nanocrystals were totally covered with the graphene sheets.

SEM images of the prepared RGO–NCC composite at low and high magnifications
To conclude, even though there is no direct evidence for the reduction mechanism of GO by NCC, the reduction process is believed to be analogous to that reported for reducing sugars, previously described by Zhu et al. [32]. GO is an oxidant and can easily oxidize the various oxygen containing groups of NCC, while at the same time being reduced to RGO. The presence of various oxygen functional groups in both NCC and RGO is favourable for hydrogen bond formation between NCC and RGO and thus leads to the formation of stable RGO–NCC nanocomposite (Scheme 2). Additionally, these interactions can disrupt the π–π interactions between the RGO sheets and thus limit their aggregation.

A general scheme showing the partial reduction of graphene using cellulose nanocrystals
Conclusion
Pure MCC fibers were isolated from different precursors (almond shells, peanut shells, cotton and Alfa fibers) then hydrolyzed using sulfuric acid using two different protocols. The hydrolysis processes adopted, leading to nanocrystalline rods for some precursors and to incomplete hydrolysis process for the others, allowed confirming the effect of the cellulose precursor and the hydrolysis parameters on the structure and the morphology of the obtained cellulose samples. To the best of our knowledge, this is the first time that cellulose nanorods with structure II were directly produced from AS. Furthermore, the nanorods extracted from this origin present higher aspect ratio, smoother surface and superior thermal stability than those obtained from other conventional resources. Thus, due to these characteristics, the as-extracted NRs from the AS have a potential to be used as a reinforcement element in composite materials. Furthermore, the obtained nanocellulose was successfully applied as a reducing agent for the preparation of reduced graphene oxide–nanocellulose composite from graphene oxide. The results obtained in the present study represent a very food way to enhance the green chemistry then biomass valorization.
References
Henrique, M.A., Silvério, H.A., Flauzino Neto, W.P., Pasquini, D.: Valorization of an agro-industrial waste, mango seed, by the extraction and characterization of its cellulose nanocrystals. J. Environ. Manag. 121, 202–209 (2013)
Habibi, Y., Lucia, L.A., Rojas, O.: Cellulose nanocrystals: chemistry, self-assembly, and applications. Chem. Rev. 11, 3479–3500 (2010)
Lam, E., Male, K.B., Chong, J.H., Leung, A.C., Luong, J.H. Applications of functionalized and nanoparticle-modified nanocrystalline cellulose. Trends Biotechnol. 30, 283–290 (2012)
Lavoine, N., Desloges, I., Dufresne, A., Bras, J.: Microfibrillated cellulose—its barrier properties and applications in cellulosic materials: a review. Carbohyd. Polym. 90, 735–764 (2012)
Siqueira, G., Bras, J., Dufresne, A.: Cellulosic bionanocomposites: a review of preparation, properties and applications. Polymers. 2, 728–765 (2010)
Dos Santos, M.R., Wilson Pires, F.N., Ferreira Martins, S.H.A., Dantas, D.N.O., Pasquini, D.: Cellulose nanocrystals from pineapple leaf, a new approach for the reuse of this agro-waste. Ind. Corps Products 50, 707–714 (2013)
Flauzino, N.W.P., Silvério, H.A., Dantas, N.O., Pasquini, D.: Extraction and characterization of cellulose nanocrystals from agro-industrial residue—Soy hulls. Ind. Corps Products 42, 480–488 (2012)
Rafieiana, F., Shahedi, M., Keramat, J., Simonsen, J.: Mechanical, thermal and barrier properties of nano-biocomposite based on gluten and carboxylated cellulose nanocrystals. Ind. Corps Products 53, 282–288 (2014)
Moon, R.J., Martini, A., Nnain, J., Simonsen, J., Jeff, Y.: Cellulose nanomaterials review: structure, properties and nanocomposites. Chem. Soc. Rev. 40, 3941–3994 (2012)
Wang, Y., Li, Y., Tang, L., Lu, J., Li, J.: Application of graphene-modified electrode for selective detection of dopamine. Electrochem. Commun. 11, 889–892 (2011)
Xiao, X., Miller, P.R., Narayan, R.J., Brozik, S.M., Wheeler, D.R., Brener, I., Wang, J., Burckel, D.B., Polsky, R.: Simultaneous detection of dopamine, ascorbic acid and uric acid at lithographically-defined 3D graphene electrodes. Electroanalysis. 26, 52–56 (2014)
Wang Q., Li M., Szunerits S., Boukherroub R.: Environmentally friendly reduction of graphene oxide using tyrosine for nonenzymatic amperometric H2O2 detection. Electroanalysis 26, 156–163 (2014)
Xiao, H., Xiaoying, Q., Freddy, B., Hua, Z.: Graphene-based composites. Chem. Soc. Rev. 41, 666–686 (2012)
Teixeira, S., Ferreira, N.S., Conlan, R.S., Guy, O.J., Sales, M.G.F.: Chitosan/AuNPs modified graphene electrochemical sensor for label-free human chorionic gonadotropin detection. Electroanalysis. 26, 2591–2598 (2014)
Kuila, T., Bose, S., Khanra, P., Mishra, A.K., Kim, N.H., Lee, J.H.: Recent advances in graphene-based biosensors. Biosensens. Bioelectron. 12, 4637–4648 (2011)
Wang, Y., Li, Y., Tang, L., Lu, J., Li, J.: Application of graphene-modified electrode for selective detection of dopamine. Electrochem. Commun. 11, 889–892 (2009)
Qi, W., Li, M., Szunerits, S., Boukherroub, R.: Environmentally friendly reduction of graphene oxide using tyrosine for nonenzymatic amperometric H2O2 detection. Electroanalysis. 26, 156–163 (2014)
Yiming, Z., Xun, Y., Yuan, W., Yi, Ch: One-pot photochemical synthesis of graphene composites uniformly deposited with silver nanoparticles and their high catalytic activity towards the reduction of 2-nitroaniline. J. Mater. Chem. 22, 7245 (2012)
Dubin, S., Gilje, S., Wang, K., Tung, V.C., Cha, K., Hall, A.S., Farrar, J., Varshneya, R., Yang, Y., Kaner, R.B.: A one-step, solvothermal reduction method for producing reduced graphene oxide dispersions in organic solvents. ACS Nano. 4, 3845 (2012)
Paredes, J.I., Villar-Rodil, S., Fern_andez-Merino, M.J., Guardia, L., Martínez-Alonso, A., Tascon, J.M.: Environmentally friendly approaches toward the mass production of processable graphene from graphite oxide. J. Mater. Chem. 21, 298 (2011)
Chen, Y., Zhang, X., Yu, P., Ma, Y.: Stable dispersions of graphene and highly conducting graphene films: a new approach to creating colloids of graphene monolayers. Chem. Commun. 30, 4527–4529 (2009)
Li, H., Pang, S., Feng, X., Mullen, K., Bubeck: Handbook of carbon nano materials C. Chem. Commun. 46, 6243 (2010)
Liang, Y., Wu, D., Feng, X., Mullen, K.: Dispersion of graphene sheets in organic solvent supported by ionic interactions. Adv. Mater. 21, 1679 (2009)
Ai, K., Liu, Y., Lu, L., Cheng, X., Huo, L.: Facile synthesis of a Ag nanoparticle/polyoxometalate/carbon nanotube tri-component hybrid and its activity in the electrocatalysis of oxygen reduction. J. Mater. Chem. 21, 14917–14924 (2011)
Pham, V.H., Cuong, T.V., Hur, S.H., Oh, E., Kim, E.J., Shin, E.W., Chung, J.S.: Chemical functionalization of graphene sheets by solvothermal reduction of a graphene oxide suspension in N-methyl-2-pyrrolidone. J. Mater. Chem. 21, 3371–3377 (2011)
Lin, Z., Yao, Y., Li, Z., Liu, Y., Li, Z., Wong, C.-P.: J. Phys. Chem. C. 114, 14819 (2010)
Gao, J., Liu, F., Liu, Y., Ma, N., Wang, Z., Zhang, X.: Environment-friendly method to produce graphene that employs vitamin C and amino acid. J. Chem. Mater. 22, 2213–2218 (2010)
Dua, V., Surwade, S.P., Ammu, S., Agnihotra, S.R., Jain, S.Roberts, K.E., Park, S., Ruoff, R.S., Manohar, S.K.: All-organic vapor sensor using inkjet-printed reduced graphene oxide. Angew. Chem. Int. Ed. 49, 2154–2157 (2010)
Fang, M., Long, J., Zhao, W., Wang, L., Chen, G.: American chemical society ph-responsive chitosan-mediated graphene dispersions. Langmuir. 26(22), 16771–16774 (2010)
Zhang, J., Yang, H., Shen, G., Cheng, P., Zhang, J., Guo, S.: Reduction of graphene oxide via L-ascorbic acid. Chem. Commun. 46, 1112–1114 (2010)
Fernandez-Merino, M.J., Guardia, L., Paredes, J.I., Villar-Rodil, S., Solís-Fern_andez, P., Martínez-Alonso, A., Tascon, J.M.D., Vitamin, C.: Is an ideal substitute for hydrazine in the reduction of graphene oxide suspensions. J. Phys. Chem. C. 114, 6426–6432 (2010)
Zhu, C., Guo, S., Fang, Y., Dong, S.: Reducing sugar: new functional molecules for the green synthesis of graphene nanosheets. ACS Nano. 4, 2429–2437 (2010)
Liu, J., Fu, S., Yuan, B., Li, Y., Deng, Z.: Toward a universal “adhesive nanosheet” for the assembly of multiple nanoparticles based on a protein-induced reduction/decoration of graphene oxide. J. Am. Chem. Soc. 132, 7279–7281 (2010)
Wang, Y., Shi, Z.X., Yin, J.: Facile synthesis of soluble graphene via a green reduction of graphene oxide in tea solution and its biocomposites. ACS Appl. Mater. Interfaces. 3, 1127–1133 (2011)
Hummers, W.S. Jr., Offeman, R.E.: Preparation of graphitic oxide. J. Am. Chem. Soc. 80(6), 1339 (1958)
George, J., Sabapathi, S.N.: Cellulose nanocrystals: synthesis, functional properties, and applications. Nanotechnol. Sci. Appl. 8, 45–54 (2015)
Nurain, J., Ahmad, I., Dufresne, A.: Extraction, preparation and characterization of cellulose fibres and nanocrystals from rice husk. Ind. Corps Products 37, 93–99 (2012)
Ping, L., Hsieh, Y.L.: preparation and properties of cellulose nanocrystals/rods, spheres and network. Carbohydr. Polym. 82, 329–336 (2010)
Oh, S.Y., Yoo, D. I., Shin, Y., Seo, G.: FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohyd. Res. 340, 417–428 (2005)
Sebe, G., Ham-Pichavant, F., Ibarboure, E., Akissi, L., Chantal, K., Tingaut, Ph.: Supramolecular structure characterization of cellulose II nanowhiskers produced by acid hydrolysis of cellulose I substrates. Biomacromol. 13, 570–578 (2012)
Borysiak, S., Doc zekalska, B.: Research into the mercerization process of beech wood using the waxs method fibres. Text East Eur. 16, 101–103 (2008)
Idström, A., Brelid, H., Nydén, M., Nordstierna, L.: CP/MAS 13C NMR study of pulp hornification using nanocrystalline cellulose as a model system. Carbohyd. Polym. 92, 881–884 (2013)
Ishikawa, A., Okano, T., Sugiyama, J.: Fine structure and tensile properties of ramie fibres in the crystalline form of cellulose I, II, IIII and IVI. J. Polym. 38, 463–468 (1997)
Brinchia, L., Kenny, J.M., Cotana, F., Fortunati, E.: Production of nanocrystalline cellulose from lignocellulosic biomass: technology and applications. Carbohyd. Polym. 94, 154–169 (2013)
Haiping, Y., Rong, Y., Hanping, Ch., Dong H. L, Chuguang. Z., Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel, 86, 1781–1788 (2007)
Zhang, X., Yang, W., Blasiak, W.: Kinetics study on thermal dissociation of levoglucosan during cellulose pyrolysis. Fuel, 109, 476–483 (2013)
Chang, C.-P., Wang, I.-C., Hung, K.-J., Perng, Y.-S.: Preparation and characterization of nanocrystalline cellulose by acid hydrolysis of cotton linter. Taiwan J. For. Sci. 25(3), 231–244 (2010)
Jeong, H.D., Yoon, C.R., Lee, J.H., Band, D.S.: Preparation and characterization of cellulose nano-whiskers extracted from microcrystalline cellulose by acid hydrolysis. Elastom. Compos. 45, 51–57 (2010)
Huang, X., Qi, X., Boey, F., Zhang, H.: Graphene-based composites. Chem. Soc. Rev. 41, 666 (2012)
Light cap, I., Kamat, P.V.: Graphitic design: prospects of graphene-based nanocomposites for solar energy conversion, storage, and sensing. Acc. Chem. Res. 46, 2235 (2013)
Sun, X., Liu, Z., Welsher, K., Robinson, J.T., Goodwin, A., Zaric, S., Dai, H.: Nano-graphene oxide for cellular imaging and drug delivery. Nano. Res. 1, 203 (2008)
Chung, C., Kim, Y.-K., Shin, D., Ryoo, S.-R., Hong, B.H., Min, D.-H.: Biomedical applications of graphene and graphene oxide. Acc. Chem. Res. 46, 2211 (2013)
Wang, Y., Li, Z., Wang, J., Li, J., Lin, Y.: Graphene and graphene oxide: bio functionalization and applications in biotechnology. Trends Biotechnol. 29, 205 (2011)
Pyun, J.: Graphene oxide as catalyst: application of carbon materials beyond nanotechnology. Angew. Chem. Int. Ed. 50, 46 (2011)
Yeh, T.-F., Syu, J.-M., Cheng, C., Chang, T.-H., Teng, H.: Graphite oxide as a photocatalyst for hydrogen production from water. Adv. Func. Mater. 20, 2255 (2010)
Dreyer, D.R., Jia, H.-P., Bielawski, C.W.: Graphene oxide: a convenient carbocatalyst for facilitating oxidation and hydration reactions. Angew. Chem. Int. Ed. 49, 6813 (2010)
Kim, J., Cote, L.J., Franklin Kim, F., Yuan, W., Shull, K.R., Huang, J.: Graphene oxide sheets at interfaces. J. Am. Chem. Soc. 132, 8180 (2010)
Dohyung, K., Seung Jae, Y., Yern Seung, K., Haesol, J., Chong Rae P.: Simple and cost-effective reduction of graphite oxide by sulfuric acid. Carbon 50, 3229–3232 (2012)
Mousumi, M., Krishanu, Ch., Kajari, K., Saibal, G., Dipali, B.: Reduced graphene oxide-polyaniline composites—synthesis, characterization and optimization for thermoelectric applications. Diamond Relat. Mater. 37, 74–79 (2013)
Sheng, Y., Wenbo, Y., Dazhen, H., Caifeng, Ch, Hao, L., Xiaojing, Y.: A facile green strategy for rapid reduction of graphene oxide by metallic zinc. RSC Adv. 2, 8827–8832 (2012)
Zhu, C., Guo, S., Fang, Y., Dong, S.: Reducing sugar: new functional molecules for the green synthesis of graphene nanosheets. ACS Nano. 4(4), 2429–2437 (2010)
Zhang, S., Shao, Y., Liao, H., Engelhard, M.H., Yin, G., Lin, Y.: Polyelectrolyte-induced reduction of exfoliated graphite oxide: a facile route to synthesis of soluble graphene nanosheets. ACS Nano. 5, 1785–1791 (2011)
Hassan, H.M.A., Abdelsayed, V., Khder, A., AbouZeid, K.M., Terner, J., El-Shall, M.S., Al-Resayes, S.I., El-Azhary, A.A.: Microwave synthesis of graphene sheets supporting metal nanocrystals in aqueous and organic media. J. Mater. Chem. 19, 3832–3837 (2009)
Mc Allister, M.J., Li, J.-L., Adamson, D.H., Schniepp, H.C., Abdala, A.A., Liu, J., Herrera-Alonso M. et al.: Single sheet functionalized graphene by oxidation and thermal expansion of graphite. Chem. Mater. 19, 4396–4404 (2007)
Yang, S., Yue, W., Huang, D., Chen, C., Lin, H., Yang, X.: A facile green strategy for rapid reduction of graphene oxide by metallic zinc. RSC Adv. 2, 8827–8832 (2012)
Mousumi, M., Krishanu, Ch, Kajari, K., Saibal, G., Banerjee, D.: Reduction of graphene oxide through a green and metal-free approach using formic acid. Diamond Relat Mater. 37, 74–79 (2013)
Petit, C., Seredych, M., Bandosz, T.J.: Revisiting the chemistry of graphite oxides and its effect on ammonia adsorption. J. Mater. Chem. 19, 9176–9185 (2009)
Acknowledgements
This work was partly supported by The Tunisian Ministry of Higher Education and Scientific Research and the Portuguese Science and Technology Foundation through projects FCT/5964/27/5/2013/S, PTDC/FIS/NAN/0117/2014 and by FEDER funds through the COMPETE 2020 Program and National Funds through FCT - Portuguese Foundation for Science and Technology under the project UID/CTM/50025/2013/S and by the Portuguese Nuclear Magnetic Resonance Network (PTNMR).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khili, F., Borges, J., Almeida, P.L. et al. Extraction of Cellulose Nanocrystals with Structure I and II and Their Applications for Reduction of Graphene Oxide and Nanocomposite Elaboration. Waste Biomass Valor 10, 1913–1927 (2019). https://doi.org/10.1007/s12649-018-0202-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12649-018-0202-4