
Abstract
Glasses in the ternary CaO–Fe2O3–P2O5 system were prepared and studied by means of density, differential scanning calorimetry, infrared, Raman and UV–Vis spectroscopies. The results showed that density and molar volume in the glass system decreased with increasing substitution of CaO for Fe2O3. The variation of glass transition temperature and thermal stability was strictly related to the nature of bonding in the vitreous network. Spectroscopic analysis showed that substitution of CaO for Fe2O3 induced an evolution of structural units from pyrophosphate to metaphosphate species indicating the polymerization of phosphate chains and the decrease of non-bridging oxygen concentrations. With increasing substitution of CaO for Fe2O3 The P–O–Ca linkage and (P–O− Ca2+ −O–P) chains participated in the glass network by replacing P–O–Fe bonds. The absorption band of the P–O–Ca stretching mode in the glasses with high CaO content (≥32 mol%) was assigned at around 1084 cm−1. The absorption edge would fall in the region between 332 and 420 nm which are the absorption bands of Fe3+ ions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Phosphate glasses have been proposed as good candidates for vitrifying high level nuclear wastes (HLW) due to their lower melting temperatures, higher waste loading and waste compositional flexibility [1, 2]. However, poor chemical durability of phosphate glasses and high crystallization tendency of their melts led to the exclusion of phosphate glasses from the pool of potential host matrices in the early 1970s [1–3]. Phosphate glasses are also important for their various interesting properties [4–8].
Several studies have shown that chemical durability of the glasses can be improved by the addition of one or more oxides such as CaO, TiO2, PbO, ZnO, CuO, Al2O3 and Fe2O3 [2, 9–12]. Among these additions, low cost and environmental friendly Fe2O3 enhances chemical durability dramatically. The binary iron phosphate glass with an approximate composition of 40Fe2O3–60P2O5 (mol%) forms more hydration resistant P–O–Fe bonds by replacing more easily hydrated P=O bonds and P–O–P bonds, leading to better chemical durability [2, 3]. CaO is another oxide which is usually added to phosphate glasses to improve chemical durability. The presence of CaO, acting as a network modifier, imparts wider glass forming region and lower viscosity [9, 12, 13]. Donald et al. [14] have indicated that calcium phosphate has appeared to be a viable starting material for the immobilization of a variety of actinide-, fluoride- and chloride-containing wastes.
Metcalfe et al. [9] have measured that dissolution rates of the ternary CaO–Fe2O3–P2O5 glass gravimetrically on monolithic samples for HLW waste. The results indicate that the glasses have more favorable properties on corrosion resistance than the binary iron phosphate glass (40Fe2O3–60P2O5, mol%). However, a little systematic work has been conducted to identify the change of the structural units in such glasses and correlate these changes with the non-bridging oxygens (NBOs) and bridging oxygens (BOs) in the glass network. Phosphate glasses can be prepared with a range of structures, from a cross-linked network of Q3 tetrahedra (vitreous P2O5) to polymer-like metaphosphate chains of Q2 tetrahedra to ‘invert’ glasses based on small pyro- (Q1) and orthophosphate (Q0) anions, depending on the [O]/[P] ratio as set by glass composition [11, 15].
In the present paper, the structural modifications in calcium–iron phosphate glasses obtained by replacing of Fe2O3 with CaO have been investigated by means of density, differential scanning calorimetry (DSC), infrared, Raman and UV–Vis spectroscopies.
2 Experimental details
CaO–Fe2O3–P2O5 glasses were investigated for the composition xCaO–(40–x)Fe2O3–60P2O5 for x = 0–40 mol% CaO. Glasses were prepared using CaO, Fe2O3 and NH4H2PO4 as the starting materials. The accurately weighed batches were introduced in alumina crucibles. In order to prevent the excess boiling and consequent spillage, water and ammonia in ammonium phosphate monobasic were removed initially by preheating it at 220 °C for about 2 h and then the temperature was raised to 1200 °C (heating rate was 10 °C min−1), and samples were melted at 1200 °C for 3 h. The melts were then quenched to room temperature in order to produce vitreous samples. Samples for property measurements were ground to give fine powder.
The density (ρ) of each glass was measured at room temperature using the Archimedes method with water as an immersing liquid. The sample weights varied between 3 and 4 g, and the measured densities were reproducible within 0.03 g cm−3. The molar volume (V m ) was calculated using the relation \( V_{m} = \sum (x_{i} M_{i} )/\rho \), where x i is the molar fraction and M i is the total molecular weight of the component. The composition of the prepared glass was used for the calculation of V m . The glass transition temperature (T g ) and first crystallization temperature (T r ) were measured on DSC by utilizing a SDT Q600 instrument (TA, USA) in a flowing air atmosphere at a heating rate of 20 °C min−1. The temperature was scanned over a range from room temperature to 800 °C and the estimated error in T g and T r were ±2 °C.
X-ray diffraction (XRD) analysis was performed on samples employing a X-ray diffractometer (PANalytical X’Pert PRO, The Netherlands). The 2θ scans were made between 5° and 80° with step width of 0.03° and utilized Cu Kα radiation (λ = 1.5405 Å). The infrared spectra of the samples were measured from 400 to 2000 cm−1 using a Spectrum One FT–IR spectrometer (Perkin Elmer, USA) and the KBr standard pellet method. Glass pellets were prepared by mixing about 2 mg powder with 200 mg dried KBr powder and compressing the resulting mixture in an evacuated die. The accuracy of this technique was estimated to be ±0.5 cm−1. Raman spectra at 400–1600 cm−1 were collected from glass powders using the InVia Raman Microscope (Renishaw, UK) at room temperature. The Raman spectra were excited by 514.5 nm light from an argon ion laser. The spectral resolution was about 1–2 cm−1 and the wavenumber accuracy was 0.2 cm−1. Six multiple measurements per sample were done to check for the potential micron-range heterogeneity and for the effects of sample orientation.
The diffuse reflectance spectra of the powdered glass samples were recorded at room temperature in the 200–800 nm range up to a resolution of 0.3 nm using a UV–Vis near-infrared spectrometer (UV-3150, Shimadzu, Tokyo, Japan) with the ISR-3100 integrating sphere attachment.
In the limiting case of an infinitely thick sample, thickness and sample holder has no influence on the value of reflectance (R). In this case, the Kubelka–Munk equation at any wavelength becomes [16, 17]:
F(R∞) is the so-called remission or Kubelka–Munk function, where \( R\infty = R_{sample} /R_{standard} \). The following relational expression proposed by Tauc et al. [15, 18] was used
where h is Planck’s constant, ν is frequency of vibration, α is absorption coefficient, E g is band gap and C 1 is proportional constant.
The acquired diffuse reflectance spectrum was converted to Kubelka–Munk function. Thus, the vertical axis was converted to quantity F(R∞), which was proportional to the absorption coefficient. The α in the Tauc equation is substituted with F(R∞). Thus, in the actual experiment, the relational expression becomes [16]:
Therefore, obtaining F(R∞) from Eq. (1) and plotting the [F(R∞)hν] 2 against hν, the band gap E g of a powder sample can be extracted easily.
3 Results and discussion
3.1 XRD, density and thermal properties
The X-ray diffraction patterns of the xCaO–(40–x)Fe2O3–60P2O5 glasses system show no sharp peaks ensuring amorphous nature of the samples. All prepared glass samples are homogeneous, and their glassy state has been checked by X-ray diffraction. The obtained compositional range corresponding to glass forming regions in the ternary CaO–Fe2O3–P2O5 system was described by Brow et al. [19].
The density, mole volume and DSC parameters of the studied glasses are given in Table 1. The density of glasses decreases from 3.06 to 2.57 g/cm3 and the molar volume decreases slightly when Fe2O3 is replaced by CaO. Density of glasses decreases with increasing CaO substitution, which is due to the smaller atomic weight of calcium (40 g/mol) compared to iron (56 g/mol). It is well known that the change of molar volume is associated with the change of the glass structure. The decrease in molar volume is ascribed to a decrease in the number of non-bridging oxygens [18]. This decrease in molar volume indicates that the phosphate glass network becomes more compact.
For the glass system, the glass transition temperature (T g ) and the first crystallization peak (T r ) increase for glasses containing ≤24 mol% CaO, while T g and T r decrease for glasses containing ≥24 mol% CaO (Table 1). The variation of glass transition temperature is strictly related to the nature of bonding in the vitreous network. The difference between the glass transition temperature (T g ) and the onset crystallization temperature (T r ), \( \varDelta T = T_{r} - T_{g} \), has been frequently used as a accurate estimate of glass thermal stability [20]. ΔT values in the glasses increase as Fe2O3 is replaced by CaO (Table 1). It indicates that glass thermal stability improves with increasing substitution of CaO for Fe2O3.
3.2 Infrared spectroscopy
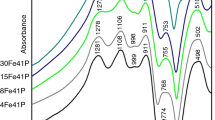
FT-IR spectra together with vibration frequency and their identifications for the xCaO–(40–x)Fe2O3–60P2O5 glasses system are shown in Fig. 1. As Fe2O3 is replaced by CaO, the most notable change is that the band at ~1268 cm−1 appears for glass containing 24 mol% CaO and shifts to the high frequency band at ~1304 cm−1 with increasing substitution of CaO. The most intense peak in the spectra has an obvious red shift from 1111 cm−1 up to 1070 cm−1. However, there is a slight increase in frequency and decrease in intensity beyond 24 mol% CaO. In addition, the band at 517 cm−1 shifts to the low frequency band at 506 cm−1, and a weak band at ~491 cm−1 appears. The assignment of the IR absorption bands for the studied 40CaO–60P2O5 glasses is similar to that reported in the literature for different alkali phosphate glasses [1, 21].

IR spectra of different composition of xCaO–(40–x)Fe2O3–60P2O5 glasses. Different peaks are shown by arrows
The IR spectra of the parent 40Fe2O3–60P2O5 glass in the glasses system reveal five bands at ~1617, ~1111, ~941, ~759 and ~517 cm−1, respectively. The band at ~1617 cm−1 reflected water-bending mode [2, 20]. The band at ~1111 cm−1 is assigned to the asymmetric stretch of (PO3)2− group in Q1 units [22], while the absorption bands at ~941 and ~517 cm−1 correspond to the symmetric modes of P–O–P bonds in the Q1 units [22] and fundamental frequencies (PO4)3− of Q0 units [2, 20], respectively.
As Fe2O3 is replaced by CaO, the absorption bands in the region 900–1300 cm−1 for the glasses system become wider and the the new band at ~1268 cm−1 appears for glass containing ≥24 mol% CaO, which is assigned to the asymmetric stretching mode of the (PO2)− group in Q2 units [20, 22]. It may be due to the formation of the (P–O− Ca2+ −O–P) ionic cross-links between two different phosphate chains [12, 23]. The bond strengthens the crosslinking of the glass network, with consequent increases in Tg. The new band at ~1304 cm−1 is assigned to the asymmetric stretching vibration of P=O bonds [22]. It is concluded that calcium ions act as a glass modifier and occupy the positions between P–O–P layers, leading to the increase of the P=O bonds. Similar behavior in Fe2O3–PbO–P2O5 glasses has been observed by Doweidar et al. [24].
In addition, the new band at ~1070 cm−1 is assigned to the asymmetric stretch of (PO4)3− tetrahedra (P–O− ionic group) in Q0 units [25, 26]. Shih et al. [27] have shown that the absorption band near 1080 cm−1 in the IR spectra of CuO–Na2O–P2O5 glasses is associated with the stretching mode of the P–O–Cu linkage by the replacing P–O−. Therefore, we suggest that the band at ~1084 cm−1 in the 40CaO–60P2O5 glass may be associated with the P–O–Ca linkage, which may lead to the increase of frequency in the IR spectra of the glass system for CaO ≥ 24 mol%. The decrease of Tg in Table 1 can be due to the formation of the P–O–Ca linkage by the replacing P–O–Fe bonds with increasing substitution of CaO for Fe2O3. The low frequency band at ~491 cm−1 appears, which may be assigned to the harmonics of bending vibration of O=P–O linkages in Q3 units [28]. These changes can be attributed to the depolymerization of pyrophosphate chains when the O/P ratio decreases (Table 1).
3.3 Raman spectroscopy
Raman spectra of the studied glasses are shown in Fig. 2. In the Raman spectra of the glasses system, the prominent band at ~1092 cm−1 shifts markedly to the high frequency band at ~1173 cm−1 and the new band at ~1293 cm−1 appears with increasing substitution of CaO. In addition, the bands at ~767 and ~952 cm−1 shift gradually to the low frequency bands at ~705 and ~879 cm−1, respectively.

Raman spectra of different composition of xCaO–(40–x)Fe2O3–60P2O5 glasses
In Fig. 2, the Raman spectra of the 40Fe2O3–60P2O5 glass reveals three bands at ~1092, ~952 and ~767 cm−1, respectively. The most intense peak at ~1092 cm−1 in the Raman spectra is assigned to the symmetric stretching mode of (PO3)2− bonds in Q1 units [2]. With increasing substitution of CaO, the band shifts markedly to the high frequency band at ~1173 and ~1293 cm−1, which correspond to the asymmetric stretching mode of the (PO2)− group in Q2 units and the P=O symmetric stretching [12, 28], respectively. The shift of these bands indicates that the metaphosphate (Q2) species dominate the structure of the CaO–P2O5 glass and substitution of CaO for Fe2O3 depolymerizes the pyrophosphate groups to form the metaphosphate long chains or rings.
The bands at ~952 and ~767 cm−1 correspond to the asymmetric stretching mode of (PO4)3− in Q0 units [11] and the symmetric stretching mode of P–O–P bonds in Q1 units [2, 11], respectively. These bands shift to the lower frequency at ~879 and ~705 cm−1 with increasing CaO substitution. The two bands can correspond to the symmetric stretching mode of P–O–P bonds in Q2 units [11]. Similar bands have been observed in the Raman spectra from the metaphosphate glasses [10, 29]. Therefore, calcium oxide in the studied glasses acts as a network modifier and incorporates into the network with increasing CaO for Fe2O3 substitution.
Both variations in Raman and IR spectra demonstrates the concomitance of Q0, Q1 and Q2 species and the variation of dominant structural units from pyrophosphate to metaphosphate species, resulting in the increase of the average phosphate chain length and the decrease of non-bridging oxygen concentrations. The change of dominant structural units may lead to the increase of the thermal stability (ΔT). Since the field strength of the iron ion (Fe3+ and Fe2+) is higher than that of Ca2+, the lower Ca–O bond force constant leads to the shift of structural units.
3.4 UV–Vis spectroscopy
In order to get further into the structure of the investigated glasses, diffuse reflectance UV–Vis (DR UV–Vis) spectra of the studied glasses are presented in Fig. 3. The DR spectra of the glasses system shows that the reflectivity increases in the visible region as Fe2O3 is replaced by CaO in the glass composition. Parent iron phosphate glass is black, and the black coloration become shallower with increasing substitution of CaO for Fe2O3. Calcium phosphate glasses are colourless. It is expected to result in the increase of the reflectivity in the visible region.

Diffuse reflectance spectra of different composition of xCaO–(40–x)Fe2O3–60P2O5 glasses
The three absorption bands are observed in the parent 40Fe2O3–60P2O5 glasses at around 324 and 352 nm in the ultraviolet region and at around 540 nm in the visible region, and the three bands decrease in intensity or disappear with increasing CaO substitution. A new band at 235 nm in the 40CaO–60P2O5 glass appears.
Iron exists in the 40Fe2O3–60P2O5 glass as Fe2+ ions (occupies usually octahedral positions and plays the network modifier role) and Fe3+ ions (occupies both tetrahedral and octahedral positions with the network former role) [30]. Some authors [31, 32] have shown that the bands centered in the 325–450 nm region are due to the presence of the Fe3+ ions, the UV–Vis bands associated with the Fe2+ ions are located in the ultraviolet region due to the iron–oxygen charge transfer and in the visible range in the 450–550 nm region. The absorption band at around 352 nm is due to the d–d transitions of the Fe3+ ions. The d–d transitions of the Fe3+ ions decrease with increasing CaO content. The weak absorption bands at 324 nm and 540 nm is due to a strong Fe–O charge transfer derived to the Fe2+ and Fe3+ ions and the d–d transitions of the Fe2+ ions in the near infrared region, respectively [31, 33]. Then, the new band centered at 235 nm would be due to the charge transfer transition of Ca–O in the 40CaO–60P2O5 glass (Fig. 3) [34]. The increase of CaO content for Fe2O3 substitution produces a shift of the fundamental absorption edge from 420 nm to a shorter wavelength at about 332 nm, indicating the decrease of disorder degree of the glass system. The region of absorption edge is the absorption bands of Fe3+ ions.
Kubelka–Munk transformed reflectance spectra of the studied glasses are shown in Fig. 4. From the extrapolation of the linear portion of these spectra, the values of the band gap (E g ) have been determined. The value of the band gap in the glasses system is found to decrease with increasing CaO content (Table 1). The modifications in the band gap energy can be understood in terms of the variation in non-bridging oxygen concentrations in the glass network. Some authors [35, 36] have attributed the increase of the band gap energy to the decrease of the amount of NBOs. The results are in agreement with Raman spectra and density data. The non-bridging orbitals have higher energies than the bonding orbitals [37]. The non-bridging oxygen ions contribute to the valence band maximum. Accordingly, addition of CaO causes a decrease in non-bridging oxygen concentration, which increases the band gap energy.

Kubelka–Munk transformed reflectance spectra of different composition of xCaO–(40–x)Fe2O3–60P2O5 glasses
4 Conclusions
The ternary glasses in the system xCaO–(40–x)Fe2O3–60P2O5 with 0–40 mol% CaO have been obtained. As Fe2O3 is replaced by CaO in the glass system, density and molar volume of glasses decrease. It indicates that the phosphate glass network becomes more compact. Infrared and Raman spectra indicate that substitution of CaO for Fe2O3 creates the P–O–Ca linkage and (P–O− Ca2+ −O–P) chains, resulting in the variation of dominant structural units from pyrophosphate to metaphosphate species. The absorption band is assigned the P–O–Ca stretching mode in the glasses with high CaO content at around 1084 cm−1. UV–Vis spectra show that the absorption edge falls in the region between 332 and 420 nm in the absorption bands of Fe3+ ions. The increase of the band gap energy with increasing CaO substitution is attributed to the decrease of non-bridging oxygen concentration. The variation of these bonds with increasing CaO substitution leads to the change of Tg and thermal stability.
References
R O Omrani, S Krimi, J J Videau, I Khattech, A E Jazouli and M Jemal J. Non-Cryst. Solids 389 66 (2014)
B Qian, X F Liang, S Y Yang, S He and L Gao J. Mol. Struct. 1027 31 (2012)
M Karabulut, G K Marasinghe, C S Ray, D E Day, O Ozturk and G D Waddill J. Non-Cryst. Solids 249 106 (1999)
S Kabi and A Ghosh Solid State Ion. 262 778 (2014)
S Shaw and A Ghosh EPL Europhys. Lett. 100 66003 (2012)
S Shaw and A Ghosh J. Phys. Chem. C 116 24255 (2012)
S Kabi and S Ghosh Solid State Ion. 187 39 (2011)
A Pan and A Ghosh J. Chem. Phys. 112 1503 (2000)
P A Bingham and R J Hand Mater. Res. Bull. 43 1679 (2008)
X Y Li, H M Yang, X L Song and Y Wu J. Non-Cryst. Solids 379 208 (2013)
R K Brow, D R Tallant, S T Myers and C C Phifer J. Non-Cryst. Solids 191 45 (1995)
A M B Silva, R N Correia, J M M Oliveira and M H V Fernandes J. Eur. Ceram. Soc. 30 1253 (2010)
Z Wu, C S Ray and P Hrma J. Non-Cryst. Solids 241 1 (1998)
I W Donald, B L Metcalfe, S K Fong, L A Gerrard, D M Strachan and R D Scheele J. Nucl. Mater. 361 78 (2007)
R K Brow J. Non-Cryst. Solids 263 1 (2000)
J Zheng, Z Q Liu, X Liu, X Yan, D D Li and W Chu J. Alloys Compd. 509 3771 (2011)
F Yakuphanoglu J. Alloys Compd. 507 184 (2010)
M Rada, E Culea, S Rada, A Bot, N Aldea and V Rednic J. Non-Cryst. Solids 358 3129 (2012)
R K Brow, D R Tallant, W L Warren, A McIntyre and D E Day Phys. Chem. Glasses 38 300 (1997)
B Qian, X F Liang, C L Wang and S Y Yang J. Nucl. Mater. 443 140 (2013)
Y M Moustafa and K El-Egili, J. Non-Cryst. Solids 240 144 (1998)
B Qian, S Y Yang, X F Liang, Y M Lai, L Gao and G F Yin J. Mol. Struct. 1011 153 (2012)
B C Bunker, G W Arnold and J A Wilder J. Non-Cryst. Solids 64 291 (1984)
H Doweidar, Y M Moustafa, K El-Egili and I. Abbas Vib. Spectrosc. 37 91 (2005)
P Pascuta, G Borodi, A Popa, V Dan and E Culea Mater. Chem. Phys. 123 767 (2010)
L Baia, M Baia, W Kiefer, J Popp and S Simon Chem. Phys. 327 63 (2006)
P Y Shih, J Y Ding and S Y Lee Mater. Chem. Phys. 80 391 (2003)
H J Li, X F Liang, C L Wang, H J Yu, Z Li and S Y Yang J. Mol. Struct. 1067 154 (2014)
J J Hudgens, R K Brow, D R Tallant and S W Martin J. Non-Cryst. Solids 223 21 (1998)
X Yu, D E Day, G J Long and R K Brow J. Non-Cryst. Solids 215 21 (1997)
S Rada, A Dehelean, M Stan, R Chelcea and E Culea J. Alloys Compd. 509 147 (2011)
L Rus et al. J. Non-Cryst. Solids 402 111 (2014)
S M Abo-Naf, M S El-Amiry and A A Abdel-Khalek Opt. Mater. 30 900 (2008)
H Tanaka, T Okumiya, S Ueda, Y Taketani and M Murakami Mater. Res. Bull. 44 328 (2009)
S Rada, M Rada and E Culea J. Non-Cryst. Solids 357 62 (2011)
S Rada et al. Electrochim. Acta 109 82 (2013)
M Rada, E Culea, S Rada, A Bot, N Aldea and V Rednic J. Non-Cryst. Solids 358 3129 (2012)
Acknowledgments
This work was supported by the Science Foundation of Southwest University of Science and Technology (11zx7157), the Scientific Research Fund of SiChuan Provincial Education Department (14ZA0105, 14ZD1122) and Postgraduate Innovation Fund Project by Southwest University of Science and Technology (14ycx018).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, H.J., Liang, X.F., Yu, H.J. et al. Studies of structure of calcium–iron phosphate glasses by infrared, Raman and UV–Vis spectroscopies. Indian J Phys 90, 693–698 (2016). https://doi.org/10.1007/s12648-015-0794-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12648-015-0794-5