
Abstract
Epidemiologically Parkinson’s disease (PD) is associated with chronic ingestion or inhalation of environmental toxins leading to the development of motor symptoms. Though neurotoxin-based animal models played a major role in understanding diverse pathogenesis, they failed to identify the risk assessment due to uncommon route of toxin exposure. Towards this, the available neurotoxin-based intranasal (i.n.) PD models targeting olfactory bulb (OB) have demonstrated the dopaminergic (DAergic) neurodegeneration in both OB and substantia nigra (SN). Despite that, the studies detecting the alpha-synuclein (α-syn) accumulation in OB and its progression to other brain regions due to inhalation of environmental toxins are still lacking. Herein, we developed oil in water microemulsion of rotenone administered intranasally to the mice at a dose which is not detectable in blood, brain, and olfactory bulb by LCMS method. Our data reveals that 9 weeks of rotenone exposure did not induce olfactory and motor dysfunction. Conversely, after 16 weeks of washout period, rotenone treated mice showed both olfactory and motor impairment, along with α-syn accumulation in the OB and striatum without glial cell activation and loss of dopaminergic neurons. The results depict the progressive nature of the developed model and highlight the role of α-syn in PD like pathology or symptoms. Together, our findings suggest the adverse consequences of early exposure to the environmental toxins on the olfactory system for a shorter period with relevance to the development of synucleinopathy or Parkinson’s disease in its later stage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by loss of dopaminergic (DAergic) neurons in substantia nigra pars compacta (SNc) (Poewe et al. 2017; GBD 2016 Parkinson’s Disease Collaborators 2018). It is marked by development of olfactory dysfunction or constipation as early non-motor symptoms followed by motor symptoms (Fullard et al. 2017). The main factor involved in pathogenesis of PD is the development of alpha-synuclein (α-syn) aggregates and subsequent neurodegeneration in selectively vulnerable neurons of peripheral and enteric nervous system (ENS). Once formed, the aggregates appear to be capable of propagating transneuronally involving different brain regions such as olfactory bulb (OB), anterior olfactory nucleus (AON), locus coeruleus, amygdala, dorsal motor nucleus of vagus (DMNV), cortex, striatum, and substantia nigra (SN) (Rey et al. 2018). Albeit the exact mechanism behind development of PD is not clear, however, a plethora of research reports indicate the contribution of inhalation or ingestion of environmental toxins in pervasive spreading and influencing peripheral parts of nervous system in the pathogenesis of PD. Based on the recognizable sequence of Lewy body lesions in postmortem brain biopsy of PD patients, Braak hypothesized that PD pathology originates either from ENS or OB due to exposure to environmental toxins and then spreads to other brain regions (Braak et al. 2003; Braak and Del Tredici 2017). This has sparked great interest in development of several animal models based on exposure to environmental toxins like MPTP, Paraquat, and rotenone that can provide key understanding of disease pathogenesis. Since these neurotoxins were administered through oral or intraperitoneal route avoiding natural route of exposure to toxins, they did not provide accurate information on risk from environmental toxin exposure. Very few studies are reported in which toxins were given through natural routes like ingestion or inhalation to induce PD (Pan-Montojo et al. 2010; Rojo et al. 2007; Sasajima et al. 2015, 2017; Souza et al. 2018). Rojo A and colleagues found that i.n. administration of rotenone or paraquat did not induce PD symptoms in mice or rats, whereas MPTP treatment induced motor symptoms along with DAergic neuronal loss in striatum (Rojo et al. 2007). While other study found olfactory dysfunction along with DAergic neuronal loss in OB even at lower dose of rotenone for 7 days given intranasally (Sasajima et al. 2015). However, the animal studies which gave rotenone intranasally did not report any changes in α-syn levels neither in OB nor in SNc (Sasajima et al. 2015, 2017).
Importantly severe α-syn accumulation has been observed in OB years before the development of motor symptoms in PD (Braak and Del Tredici 2017). Clinical studies with PD patients reported that α-syn tends to transfer from one neuron to another and it may behave like a prion protein (Longhena et al. 2017) and thus supported the Braak’s hypothesis. Furthermore, Desplats et al. showed inclusion formation and neurodegeneration through interneuronal transport of α-syn in both in vivo as well as in vitro studies (Desplats et al. 2009). In this direction, Rey et al. developed one slow progressive mouse model of PD in which they injected very small amount of oligomeric and fibrillar form of human α-syn via stereotaxic apparatus in OB of wild-type mice and found that injected α-syn formed pathological aggregates spread transneuronally to over 40 other brain regions and subregions, over the period of 12 months. They also demonstrated that induced α-synucleinopathy triggered neuronal loss in OB at early stage (Rey et al. 2013, 2016).
Based on the above studies, our aim was to induce α-syn accumulation in OB and check the progression of α-syn pathology from OB to other brain regions using rotenone which might be more naturalistic way of exposure to environmental toxins. For this purpose, we successfully developed mucoadhesive and nasoretentive microemulsion (ME) of rotenone. Based on the LC–MS results obtained from pilot study for rotenone dose determination, we administered a very low dose of rotenone to C57BL/6 mice for chronic period followed by long wash out period of rotenone administration to check progressive nature of the disease model. We investigated both motor and non-motor symptoms of PD, DAergic neurodegeneration, and neuroinflammation to examine development of α-syn pathology in OB and its progression to other brain regions.
Materials and Methods
Chemicals
Rotenone, acrylamide, sodium dodecyl sulfate (SDS), Tween-20, formaldehyde, 3,3′-diaminobenzidine (DAB), glucose oxidase, and D-glucose were obtained from Sigma-Aldrich. Ammonium persulfate (APS), tetramethyl-ethylenediamine (TEMED), sodium chloride, sodium lauryl sulfate (SLS), bis-acrylamide, sodium deoxycholate, Triton-X 100, phenylmethyl sulfonyl fluoride (PMSF), glycerol, and carboxymethylcellulose (CMC) were purchased from Hi-Media Laboratories Pvt. Ltd. β-Mercaptoethanol was purchased from Alfa Aesar.
Potassium chloride, hydrochloric acid, sodium hydroxide, and chloroform were procured from Fisher Scientific. Polyethylene glycol-400 was purchased from Merck. Tris–HCl was purchased from Thermo Fisher. Protein ladder and PVDF membrane were procured from Bio-rad. HRP conjugated Enhanced Chemiluminescent Substrate Reagent kit was obtained from Invitrogen. BCA (bicinchoninic acid) reagent kit was bought from Thermo Scientific. Primary antibodies (GAPDH, α-syn, phosphorylated α-syn (psyn)) and secondary antibodies were purchased from Abcam. Primary antibody glial fibrillary acidic protein (GFAP) (AB5804), tyrosine hydroxylase (TH) (AB152), and phosphorylated TH (pTH) (AB5935) were procured from Sigma-Aldrich. For immunohistochemical staining, the Vectastain ABC kit was procured from Vector Laboratories.
Animals
Three-month-old C57BL/6 mice were procured from Zydus Research Center, Ahmedabad, after being approved by IAEC (Approval number: IAEC/2019/02). Animals were housed in optimum temperature and moisture conditions suitable for them under 12 h light/dark cycle and provided with food and water ad libitum. Each and every experiment was performed according to guidelines of Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India.
Preparation and Characterization of Rotenone ME
In order to prepare the rotenone loaded ME, we first performed solubility study to select the oil, surfactant, and co-surfactant in which rotenone has the highest solubility (for details, refer supplementary Sect. S1.1b) (Shah et al. 2015). After selecting the components of ME, initial optimization of the different surfactant-co-surfactant mixtures (1:1, 2:1, and 3:1) was done by plotting pseudo ternary phase diagrams (refer supplementary Sect. S1.1c) (Patel et al. 2018). Following the selection of proportion of different constituents of ME, further optimization was done to achieve the desired globule size range. Later, mucoadhesive polymer concentration was optimized by adding different concentrations of polymer to the optimized ME to achieve greater retention time in the nasal mucosa (refer S1.1d). Characterization of the rotenone loaded ME was done using different techniques like diffusion light scattering (DLS), zeta sizer (Nano-ZS90, Malvern, Worcestershire, UK), UV meter, pH meter, anton par rheometer, texture analyzer) for determining average globule size, % transmittance, pH, viscosity, and mucoadhesive strength of the formulation, respectively (Thakkar et al. 2014; Shah et al. 2016) (for details, refer supplementary Sect. S1.2).
I.n. Dose Determination Using LC–MS
Using LC–MS, we determined the dose of rotenone that was not detectable in the blood, olfactory bulb, and in the brain. In brief, C57BL/6 mice were divided into three rotenone dose groups and administered with 15 µL volume (divided equally into both nostrils, i.e., 7.5 µL each nostril) containing a dose of 0.05 (n = 6), 0.1 (n = 6), and 0.35 mg/kg (n = 6) rotenone ME for 1 week. On 7th day, 200–250 µL of blood was collected from retro-orbital plexus at 1 and 2 h from different animals after rotenone administration and pooled in the same tube. The blood was allowed to stand for 30 min, and serum samples were isolated by centrifugation at 3000 rpm for 10 min. Animals were then sacrificed, and the brain along with OB was isolated from mice of all groups and stored in − 80 °C refrigerator for further analysis. Rotenone was extracted from blood serum, OB and brain homogenates using methanol by protein precipitation method and samples were analyzed using LC-QTOF-MS instrument, central instrumental facility, NIPER-Ahmedabad. In brief, initially 1 mg/mL rotenone stock solution was prepared in MS grade methanol. It was then diluted to 10, 20, 80, 200, 400, 800, and 1000 ng/mL as the standard solutions to prepare calibration curve. The detection threshold was computed by adding various concentrations of rotenone to control plasma before extraction, where detection limit was found to be 10 ng/mL.
LC–MS Method
The LC–MS analysis was performed on Agilent’s 1290 ultra-high-performance liquid chromatography system (UHPLC; Agilent technologies, USA) coupled to electrospray ionization-quadrupole-time of flight mass analyzer (ESI-Q-TOF; Agilent technologies, USA) of 6545 series. Initially, LC–MS parameters were optimized for rotenone in positive mode scan. The chromatographic separation was achieved on Poroshell 120 SB C-18 (3 × 100 mm, 1.8 μm, Agilent Infinity Lab, USA) using mobile phase consisted of methanol (solvent A) and 0.1% formic acid (solvent B). The gradient program was set as follows: 0 min, 70% B; 1 min, 70% B; 3 min, 30% B; 5.5 min, 5% B; 7.9 min, 5% B; 9 min, 70% B; and 10 min, 70% B. The autosampler temperature, flow rate, and the injection volume were set at 15 °C, 0.5 mL min−1 and 5 μL, respectively. The MS conditions of analyte were optimized on tuning mode. The optimized ESI source parameters were as follows: 160 V fragmentor voltage, 3000 V capillary voltage, 65 V skimmer, and 1000 V nozzle voltage. Nitrogen was used as drying gas at a flow rate of 10 L min−1 at 350 °C.
Study plan
Animals were randomized into two groups: control (n = 7) and rotenone ME treated (n = 9). In rotenone group, each mouse was laid on its back and administered with mucoadhesive ME of 15 µL volume (divided equally into both nostrils, i.e., 7.5 µL each nostril) containing a dose of 0.1 mg/kg rotenone intranasally using micropipette for 9 weeks (5 days/week). Control group mice were treated with the plain ME without rotenone for the same time period. Then, animals were given a washout period of 16 weeks followed by their sacrifice by cervical dislocation at the end of 25th week. Hereafter, OB and striatum were isolated from one hemisphere of mice brains kept on ice and stored at − 80 °C for western blotting, and another hemisphere was kept in 4% paraformaldehyde (PFA) solution in phosphate-buffered saline (PBS, pH 7.4) at 4 °C for immunohistochemical studies (refer Experimental design figure).
Behavioral tests
Olfactory and motor dysfunction tests were performed every 2 weeks till 9 weeks of rotenone administration and then on 25th week of the study.
-
(a)
Butyric acid avoidance test
This test was performed using Y maze as reported by Sasajima et al. with little modifications. Two filter papers soaked with 20 µL of water in petri dish were kept in two arms of the Y-maze, and animal was placed at the ending of the 3rd arm and allowed to explore the apparatus for 4 min. On the next day of the experiment, in one arm, water was replaced with same volume of butyric acid, and the duration that the animal spent in both the arms with petri plates was recorded for 4 min. Then, butyric acid avoidance rate was calculated as (time spent in arm with water × 100) / (time spent in arm with butyric acid + time spent in arm with water) (Sasajima et al. 2017).
-
(b)
Buried pellet test
Mice were fasted for 24 h with ad libitum supply of water and then acclimatized in behavioral room for 1 h before starting the test. Regular chow food pellet weighing 2 g was buried 8 cm beneath the bedding surface in one of the corners of the cage, and mouse was placed on bedding surface in another corner just opposite to the corner where pellet was hidden and time taken by mouse to touch or find the pellet was observed and reported as latency in minutes with a cut off time of 20 min (Machado et al. 2018).
-
(c)
Grip strength test
Grip strength test was performed to check the muscle strength of mice. Mouse was placed and moved on the grid with all of four paws, while holding its tail. The maximum force (in grams) applied by mice paws to hold the grid was recorded by grip strength meter. This was done in triplicate for each mouse at an interval of 30 min between each trial (Pan-Montojo et al. 2010).
-
(d)
Round beam walk test
Beam walk test was used to measure impairment in hind limbs. This test measures foot-slips and time taken to cross the beam. Mice were firstly habituated in the test room for 1 h in their home cage and then for 15 min in the test cage before starting the test. Animals were then trained to walk on large diameter (25 mm) and medium diameter (15 mm) 120-cm-long wooden beams placed horizontally 1.5 feet above the floor surface. Mice were placed on one open end of the beam and permitted to walk to their home cage placed on another end of the beam. After training, this step was repeated with smallest diameter (10 mm) beam, and video covering the hind limb was recorded. Beams were cleaned with 1% acetic acid amidst the experiments. Time taken to cross the beam and number of hind limb slips were counted (Rabl et al. 2017).
-
(e)
Y maze test
Y maze test was used to measure the short-term memory impairment in mice. Animals were placed one by one in y-shaped runway at the end of one arm so that they can move freely through the maze for 7 minutes. Maze was wiped with 1% acetic acid before placing the next mouse in Y maze. One arm entry was considered, when the mice entered any arm of the maze with all of its four paws. Total number of entries and linear entries was counted for 7 minutes. Spontaneous alternation was defined as the consecutive entries into all the three arms in overlapping triplet sets. Percent alternation was calculated by the formula: % alternation=Linear entries/total entries*100.
Immunoblotting
Striatum and OB samples were thawed, and tissue lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer constituting Tris–HCl, NaCl, SDS, sodium deoxycholate, and 1% Triton X-100 mixed with PMSF (protease inhibitor). Lysate samples were centrifuged at 12,000 rpm for 7 min, and supernatant was collected and stored for SDS-PAGE. Protein concentration was calculated using BCA method. Thirty µg of protein was separated on 15% and 12% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membrane using Bio-rad trans-blot assembly. Membrane was then blocked using 3% BSA and incubated with primary antibodies of α-syn (rabbit polyclonal antibody (pAb), ab212184, 1:1000), psyn (rabbit monoclonal antibody (mAb), ab51253, 1:1000), and GAPDH (mouse mAb, ab8245, 1:10,000), overnight at 4 °C with gentle shaking. Similarly, we also performed blotting for neuroinflammatory marker (GFAP (rabbit pAb, AB5804, 1:1000)) and neurodegenerative marker (TH (rabbit pAb, AB152, 1:10,000)) and pTH (AB5935, 1:1000). Blots were then incubated with HRP-conjugated secondary antibody (rabbit pAb, AB6721, 1:10,000; mouse pAb, AB6789, 1:10,000), followed by chemiluminescent detection. Then, blot analysis was done with the help of ImageJ software (NIH, USA) (Parkhe et al. 2020).
Immunohistochemistry (IHC) staining
Hemispheres of one side kept in 4% PFA overnight were transferred to 15% sucrose (sucrose solution in 0.1 M phosphate buffer (PB) pH 7.4) at 4 °C for 24 h followed by 30% sucrose solution incubation till their submersion. After that, these hemispheres were processed for cryostat sectioning. Forty µm free-floating coronal sections of SN of control and rotenone mice were taken using cryostat (Thermo Fisher Scientific) at three different levels (− 3.52 mm, − 3.16 mm, − 2.80 mm) in 24-well culture plates and processed for IHC-DAB staining for TH. IHC was done using Vectastain ABC kit as per protocol mentioned by Khairnar et al. (2010) with minor changes (Khairnar et al. 2010). Sections were incubated in 1% hydrogen peroxide solution in 0.1% Triton-X-100 for 10 min and then blocked with 5% normal goat serum in 0.1% Triton-X-100 in PBS. Thereafter, sections were incubated with anti-tyrosine hydroxylase antibody (rabbit pAb, AB152, 1:1000) overnight at 4 °C. Next day, PBS washing was done followed by secondary biotinylated antibody (for one and half hour) and avidin–biotin-peroxidase (Diluted ABC solution, Vector Laboratories) for 1 h in dark at room temperature with intermediate three PBS washings of 10 min each. The peroxidase reaction was developed with DAB substrate in presence of glucose and ammonium chloride dissolved in 0.1 M PB for 5 min followed by glucose oxidase incubation for 8–15 min. Then sections were taken on slides, dried overnight, and dehydrated in ethanol gradient solutions. Mounting was done using dibutylphthalate polystyrene xylene (DPX), and then images were taken using Leica DMi1 inverted microscope.
Quantitative Analysis of DAB Staining
The number of DAergic neurons was determined as previously described (Kuhn et al. 2003). Briefly, we manually counted TH‑positive cells under bright‑field illumination in all the three levels (− 3.52 mm, − 3.16 mm, − 2.80 mm) of SNc using a 20 × objective and calculated mean count of these levels. DAergic neurons of ventral tegmental area were excluded during counting, and neurons were only counted if they contained a nucleus that was surrounded by cytoplasm. Cell counting was done by the authors blind to the experimental groups and reported as percentage of the control group.
Immunofluorescence Staining and Analysis
40 µm free-floating coronal sections of SN at three different levels (− 3.52 mm, − 3.16 mm, − 2.80 mm) of control, and rotenone mice brains were processed for immunofluorescence staining for TH and α-syn. The sections were firstly rinsed three times with 0.1 M PB followed by blocking with protein block (ab64226) for 20 min. The sections were then incubated overnight at 4 °C with anti-tyrosine hydroxylase (rabbit pAb, AB152, 1:500) and anti-α-syn (mouse mAb, AB1903, 1:500) antibody diluted in antibody diluent (ab64211). The sections were then rinsed thrice in PB mixed with 0.025% triton-x and incubated with goat pAb secondary to mouse, Alexa-Fluor 488 (ab150113) (1:1000) and goat pAb secondary to rabbit, and Alexa-Fluor 647 (ab150079) (1:1000) for 1 h at room temperature. The sections were again rinsed with PB mixed with triton-x, and the nuclei were stained with DAPI (Sigma-Aldrich, USA). Images were captured at 10 × magnification under confocal scanning laser microscope (Leica TCS SP8 Microsytem). Mean fluorescence from α-syn was measured for all the three levels in SNc using ImageJ software (version 1.42, NIH, USA) (Farrand et al. 2020). Background measures were also taken for each section and subtracted from the mean fluorescence values, and the corrected values were presented as percentage of control.
Statistical Analysis
All the data were expressed as mean ± SEM. Two-sided Student’s t test was used for comparison between control and rotenone groups. Values were considered statistically significant if p < 0.05. GraphPad Prism, version 5.01, GraphPad Inc. software was used for statistical analysis.
Results
Formulation and Characterization of the Rotenone Loaded ME
Rotenone showed highest solubility in lauryl glycol (2.98 ± 0.010 mg/ml), Tween 80 (3.82 ± 0.002) mg/ml), and transcutol P (2.93 ± 0.015 mg/ml) as oil, surfactant, and co-surfactant, respectively (for details, refer supplementary Fig. 1 in Sect. S2.1). Phase diagram with surfactant: co-surfactant ratio 1:1 showed the maximum region of ME preparation (refer supplementary Fig. 2 in Sect. S2.2). The average globule size and polydispersity index (PDI) obtained was 154.80 ± 2.77 nm and 0.288 ± 0.075, respectively (refer Table I in supplementary Sect. 2). We obtained the required globule size, PDI and transparency with the optimized formula: 5% lauryl glycol, 37% Smix, and 58% water. We selected 0.5% w/v carbopol 974 P solution in water for mucoadhesive microemulsion (MME) preparation (refer Table II and Table IV in supplementary Sect. 2). pH of all the formulations was observed between 4.5 and 6.5 (refer Table IV in supplementary Sect. 2) that is pH of nasal mucosa. This indicates the safety of our formulation for i.n. administration. The percentage transmittance of rotenone MME was found to be 99.969 (refer Table IV in supplementary Sect. 2). Mucoadhesive strength of the rotenone ME, carbopol solution, and rotenone MME was found to be 66.27 ± 3.88 g, 132.58 ± 27.12 g, and 120.67 ± 6.43 g, respectively (refer Fig. 3 in supplementary Sect. 2). Viscosity was found to be 165.44 mPa.s and 276.16 mPa.s for rotenone ME and rotenone MME, respectively.
I.n. Dose Determination Using LC–MS
We got the linearity in LC–MS method displaying the calibration curve correlation factor of 0.99. We were unable to detect rotenone at 0.05 and 0.1 mg/kg dose in serum samples (Fig. 1a) and at 0.05, 0.1, and 0.35 mg/kg dose in OB (Fig. 1b) and brain samples (Fig. 1c). 0.1 mg/kg was the maximum dose which was not detectable in serum and brain tissue, which indicates that rotenone couldn’t reach serum and tissue, when 0.1 mg/kg microemulsion was administered intranasally for one week. Therefore, we selected 0.1 mg/kg dose of rotenone ME for further administrations in mice.

Experimental design. Diagrammatic illustration of the experimental procedure

LC–MS results. a, b, and c Standard (50 ng/ml) and chromatogram from blood serum, OB, and brain samples of 0.05, 0.1, and 0.35 mg/kg rotenone microemulsion treated (for 1 week) mice. d, e, and f Quantification of rotenone levels in serum, OB, and brain samples collected at 1 and 2 h after treatment with different doses on 7th day of treatment
Behavioral Alterations in Mice After i.n. Rotenone Administration
Intranasally Administered Rotenone Microemulsion Induced Olfactory Dysfunction
We found no significant difference between control and rotenone group mice in olfactory tests after 9 weeks of study (Fig. 2a, b). Rotenone administration for 9 weeks followed by washout period of 16 weeks induced hyposmia in rotenone-treated mice as we found a significant difference (p < 0.001) in latency to find the food pellet buried in bedding as compared to control animals in 25th week of study (Fig. 3a).

Effect of low-dose rotenone on behavioral parameters in 9th week of the study. a Time taken to find the hidden food pellet as measured by buried pellet test, b percent avoidance of butyric acid measured in Y-maze test, c percentage alternation in Y maze test, d force applied by mice paws in grip strength test, e and f number of slips and time to travel beam in RBWT in control and rotenone animals after 9 weeks of i.n. rot ME administration. Data was analyzed by unpaired t test and expressed as mean ± SEM (n = 5 in control group and n = 8 in rotenone group)

Effect of low dose rotenone on behavioral parameters in 25th week of the study. a Time taken to find the hidden food pellet as measured in buried pellet test, b avoidance of butyric acid measured in Y-maze test, c percentage alternation in Y maze test, d force applied by mice paws in grip strength test, e, f number of slips and time for traveling distance in RBWT in control and rotenone animals after 9 weeks of i.n. rot ME administration followed by washout period of 16 weeks. Data was analyzed by unpaired t test and expressed as mean ± SEM (n = 5 in control group and n = 8 in rotenone group), p*** ˂ 0.001 vs control, p** ˂ 0.01 vs control, p* ˂ 0.05 Vs control
There was also a significant decrease (p < 0.01) in butyric acid avoidance rate (%) in rotenone treated animals as compared to control animals (Fig. 3b).
Intranasally Administered Rotenone Microemulsion Induced Neuromuscular Dysfunction in Grip Strength Test
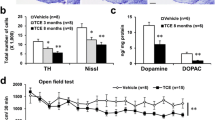
There was observed significant decrease (p < 0.05) in grip strength force in rotenone group animals as compared to control animals in 25th week of study (Fig. 3d), which was not there in 9th week (Fig. 2d).
Intranasally Administered Rotenone Microemulsion Induced Motor Impairment in Round Beam Walk Test (RBWT):
We could not see any motor impairment in RBWT in rotenone treated mice while compared to control mice in 9th week of study while in 25th week, there was significant difference (p < 0.05) in number of slips between control and rotenone animals (Fig. 2e, 3e). However, we didn’t find any significant difference in time taken by animals to cover a certain distance in RBWT in 9th week as well as 25th week of study (Figs. 2f and 3f).
Intranasally Administered Rotenone Microemulsion Didn’t Induce Memory Impairment in Y-Maze Test
There was no significant difference in percentage alternation in control and rotenone mice as observed in 9th week as well as in 25th week of study (Figs. 2c and 3c).
Effect of i.n. Rotenone on Different Protein Markers
Synucleinopathy
We observed significant increase in α-syn (p < 0.01) and psyn (p < 0.05) in OB of rotenone administered mice as compared to control group mice (Fig. 4d, e). Similarly, expression of these proteins was elevated in striatum (Fig. 5d, e) of rotenone treated mice as compared to control mice (p < 0.01 for both α-syn and psyn).

Rotenone induced α-syn aggregation and altered TH activity, but did not affect neurodegeneration and neuroinflammation in OB. Expression of TH, pTH, GFAP, Phospho α-syn (psyn) and α-syn in OB of control and rotenone animals after 9 weeks of i.n. rotenone ME administration followed by washout period of 16 weeks. Data was analyzed by unpaired t test and expressed as mean ± SEM (n = 3 in both groups), p** ˂ 0.01 vs control, p* ˂0.05 vs control

Rotenone induced α-syn aggregation, but did not affect TH activity, neurodegeneration and neuroinflammation in striatum. Expression of TH, pTH, GFAP, Phospho α-syn (psyn) and α-syn in striatum of control and rotenone animals after 9 weeks of i.n. rotenone ME administration followed by washout period of 16 weeks. Data was analyzed by unpaired t-test and expressed as mean ± SEM (n = 3 in both groups), p** ˂ 0.01 vs control
Immunofluorescent staining results indicated that there is no significant difference in α-syn immunoreactivity in TH+ neurons in SNc of rotenone mice as compared to control mice as can be seen by mean florescence intensity results (Fig. 6).

Rotenone did not alter α-syn expression in SNc. Colocalization of tyrosine hydroxylase and alpha-synuclein in the SNc of control and rotenone mice. Immunofluorescent staining of DAPI (A, E), tyrosine hydroxylase (TH) (B, F), and alpha-synuclein (α-syn) (C, G) in SN. Panels D and H represent merged images of A + B + C, and E + F + G, respectively. Scale bar is 300 μm. Student’s t test was applied. Data is expressed as mean ± SEM (n = 3)
Neuroinflammation
GFAP can be observed as neuroinflammatory marker, as the increased expression of GFAP indicates astroglial cell activation or reactive astrogliosis. Reactive astrocytes act as pro-inflammatory regulator and release a variety of molecular signals, which contribute to inflammatory state of central nervous system by activating the release of cytokines, chemokines, and various growth factors. We found no significant change in expression of GFAP in OB and striatum of control and rotenone animals (Figs. 4c and 5c), which shows that there is no astroglial cell activation after rotenone administration.
Neurodegeneration
To determine the DAergic neurodegeneration in OB and striatum, we checked the expression of TH (rate limiting enzyme in dopamine (DA) synthesis) and found no significant change in expression of TH in OB and striatum of rotenone animals as compared to control animals (Figs. 4a and 5a).
In PD, there is higher risk of death of DAergic neurons of SNc. We didn’t find any significant change in percentage of TH+ neurons in SN of rotenone mice as compared to control mice, as observed by DAB staining results (Fig. 7).

Rotenone treatment did not alter the number of TH+ neurons in SN. Representative images of TH+ neurons in the SN of control and rotenone mice at 10 × (scale bar 100 µm) and 20 × magnification (scale bar 50 µm). No significant difference was observed in number of TH+ neurons in the SN of control and rotenone animals; Student’s t test was applied. Data is expressed as mean ± SEM (n = 3)
Among the different phosphorylation sites of TH, Ser40 is the major regulatory site contributing to TH activity and DA synthesis in vivo. We also checked the pTH (phosphorylated at serine 40) expression in control and rotenone mice and found significant increase (p < 0.05) in OB (Fig. 4b) of rotenone animals as compared to control animals, while there was no significant difference in expression of pTH in striatum between control and rotenone mice (Fig. 5b).
Discussion
The present study suggests that low-level pesticide exposure in the OB of mice for short duration followed by long wash out period is enough to trigger the α-syn pathology and its progression to other brain regions even in the absence of continued exposure to pesticide. The study is in line with Braak’s hypothesis stating that α-syn pathology may originate from OB and later transfer to other brain regions in retrograde manner (Braak et al. 2003). Importantly, 9 weeks of low-dose i.n. administration of rotenone did not develop olfactory dysfunction and motor impairment, whereas after wash out period of around 16 weeks, we could observe both olfactory dysfunction and motor impairment suggesting progressive nature of the model. Surprisingly, we were able to see significant α-syn accumulation, but not neuroinflammation and neurodegeneration in the OB and striatum of rotenone-treated animals. Moreover, we did not find α-syn pathology and DAergic neurodegeneration in SN.
PD models developed based on neurotoxins though helped in understanding the mechanism behind development of PD, but these studies lack the natural way of exposure to toxins (Meredith and Rademacher 2011; Bové et al. 2005). In our studies, we developed i.n. oil in water microemulsion of rotenone which is the common way of exposure to environmental toxin in agrochemical use for example in fisheries (Rojo et al. 2007). We understand that the effects observed by i.n. administration of rotenone will not be similar as those by inhaled toxins which might go into the lungs as well as respiratory tract, but i.n. approach at least mimics one condition that is direct exposure to OB. Based on this, the aim of the present study was to induce local exposure of rotenone to OB and to study development and progression of α-syn pathology to other brain regions. Pan Montojo F and his colleagues developed intragastric rotenone mouse model targeting enteric neurons and induced α-syn accumulation selectively in ENS, which later progressed to the brain in a retrograde manner. For this, they optimized the rotenone dose and found that intragastric administration of rotenone at a dose of 5 mg/kg was not detectable in the blood and brain (Pan-Montojo et al. 2010). Similarly, in our studies, we targeted OB and given 0.1 mg/kg i.n. rotenone administration which was not detectable in the blood serum, OB, and brain.
Several animal models of PD targeting OB by i.n. administration of environmental toxin have been developed (Rojo et al. 2007; Sasajima et al. 2015, 2017). The studies reported changes in DAergic neurons along with olfactory and motor impairment. However, very few studies have reported the α-syn accumulation which is central to PD pathogenesis using environmental toxin (Voronkov et al. 2017). Towards this, the present study reported for the first time α-syn accumulation after low-dose rotenone exposure followed by 16 weeks of wash out period in the OB and striatum. We kept 16 weeks of wash out period based on our previous studies on intragastric rotenone model in which we gave rotenone for 8 weeks followed by 16 weeks of wash out time and scan the animals for diffusion kurtosis imaging (DKI) and found kurtosis changes in the SN and thalamus which were not present after 8 weeks of rotenone administration (Khairnar et al. 2021).
In the case of PD, intraneuronal accumulation of α-syn plays a central role among all other mechanisms. Of several post translational modifications of α-syn in PD, phosphorylation at serine 129 position is found to be critical in the pathogenesis of PD. Similarly, only a small fraction of α-syn (~ 4%) is phosphorylated in healthy brains, and substantial accumulation of psyn phosphorylated at 129 position (~ 90%) is observed in brains with Lewy pathology (Fields et al. 2019). Pre-clinical studies have reported that psyn increases the toxicity of α-syn by enhancing aggregation of α-syn (Oueslati 2016). In the present study, we found increase in level of total α-syn and psyn in OB and striatum after low-dose rotenone microemulsion given through intranasal route. Rotenone is reported to upregulate α-syn expression which is evident by the studies performed by Sala et al. 2013 on human neuroblastoma SH-SY5Y cells, in which they found rotenone induced de novo synthesis of α-syn (Sala et al. 2013). Similarly, Yang et al. 2018 also reported increase in α-syn expression and psyn levels in SN after oral administration of rotenone (Yang et al. 2018). In our previous study with intragastric rotenone mouse model, we observed progressive increase in α-syn expression in DMV, striatum, and SN (Khairnar et al. 2021). Rotenone is also known to induce phosphorylation of α-syn by decreasing the protein phosphatase 2A activity (PP2A). PP2A is reported to be involved in dephosphorylation of α-syn (Wang et al. 2016). From these reports, we can say that in our studies, increase in α-syn expression might be due to rotenone induced de novo synthesis and increase in psyn levels might be due to decrease in PP2A activity by rotenone.
Extensive studies on human brain samples and pre-clinical studies with rodents support the involvement of neuroinflammation either in the degeneration of DAergic neurons or progression of PD pathology. Mc Geer and his colleagues in 1988 reported the first study with microglial cell activation in SN of PD patients (Hirsch and Hunot 2009). Pre-clinical studies with neurotoxins such as 6-OHDA, MPTP, and rotenone have shown the involvement of glial cells such as astroglia and microglia along with lymphocyte infiltration (Zhou et al. 2007; Cicchetti et al. 2002; Lofrumento et al. 2011). In the present study, we analyzed the expression of GFAP in OB and striatum and did not find significant astroglial cell activation in both the regions. In vitro studies with primary microglial culture from C57BL/6 mice suggested that rotenone is not involved in direct activation of microglia or neuroinflammatory process. Therefore, in vivo studies with rotenone showing neuroinflammation might be a cause of DAergic neuronal damage or due to factors secreted by neurons (Klintworth et al. 2009). This might be the reason we may not have seen the glial cell activation in OB and striatum as we found α-syn accumulation but not degeneration of DAergic neurons in the OB and striatum.
One of the main pathological hallmarks of PD is degeneration of DAergic neurons and decrease in DA levels in the striatum which causes akinesia, tremor, and rigidity in PD patients (Dickson 2012). In the present study, we did not find any alteration in TH levels in the OB and striatum which is a classical marker for DAergic neurons. In contrast, studies in which rotenone was given systemically or through nasal route showed significant DAergic cell loss and decrease in DA level in the striatum (Sasajima et al. 2015, 2017; Souza et al. 2018; Alam and Schmidt 2002). The discrepancy of DAergic neuronal loss between ours and others (Sasajima et al. 2015, 2017) might be due to the low dose of rotenone, the timing of sacrifice of animals, or due to age. When we performed similar pilot studies with 1-year-old mice, we found significant α-syn accumulation and degeneration of DAergic neurons in the OB and striatum suggesting the role of aging factors in α-syn induced DAergic neurodegeneration (DATA NOT SHOWN). Similarly, our previous studies with TNWT-61 mice (Line 61 transgenic mouse model) showed α-syn accumulation from 3 months of age without any degeneration of DAergic neurons, while at the age of 14 months, the animals showed DAergic neuronal loss suggesting the role of aging factors in DAergic neuronal loss (Khairnar et al. 2016, 2017).
Surprisingly, we found significant motor impairment in rotenone-treated mice without any neurodegeneration in the mid brain, suggesting α-syn accumulation is enough to induce motor deficits. Moreover, there are studies with transgenic mice overexpressing α-syn in the nigrostriatal system which reported the motor impairment without DAergic neuronal loss (Chesselet et al. 2012; Lee et al. 2012). Interestingly when we detected the levels of pTH at serine 40 position, we found that it is significantly decreased in rotenone-treated animals in OB. The reason behind it might be due to increase in α-syn levels, as it is reported that α-syn overexpression increases PP2A activity which will reduce phosphorylation of TH at serine 40 position (Peng et al. 2005). So, decrease in pTH level might have lowered the DA in the OB, which may have led to olfactory dysfunction in rotenone treated mice. While it is still not clear why presence of α-syn accumulation induced motor impairment without alteration in the pTH and TH levels in the striatum. Similar to our previous studies, TNWT-61 mice showed motor impairments due to α-syn accumulation without the DAergic neuronal loss (Khairnar et al. 2016, 2017).
However, in the similar model after MPTP administration, the TNWT-61 mice showed enhanced sensitivity towards DAergic neurodegeneration suggesting α-syn accumulation might have altered the nigrostriatal functioning (Chesselet et al. 2012). In our studies too, the α-syn accumulation might have altered the nigrostriatal functioning or might have decreased the DA level leading to motor impairment. The studies with transgenic mice overexpressing α-syn after DA agonist administration showed worsening of motor deficit due to excessive DA in the regions without loss of DA (Fleming et al. 2006). However, this is the limitation of the present study as we did not perform the studies with DA agonist and due to sample scarcity could not able to determine the DA levels in the OB and striatum. Further studies are warranted in this direction.
The above results suggest the progressive nature of the developed model as mice received rotenone only for 9 weeks, a time point at which there was no olfactory dysfunction and motor impairment. However, when the same animals were kept for 16 weeks without rotenone treatment, they showed both olfactory dysfunction and motor impairment that might be due to α-syn accumulation in OB and striatum. As these mice did not show any DAergic neurodegeneration in OB and striatum but showed the progression of α-syn pathology, we may say that this model is more suitable to study synucleinopathy rather than PD. Also, with this study, we can’t say that the pathology first developed in OB and later progressed to nigrostriatal pathway as we performed the studies only at one time point. Further longitudinal studies are warranted in this direction. In conclusion, our findings highlight the adverse consequences of early exposure to the environmental toxins on the olfactory system for a shorter period with relevance to the development of synucleinopathy or Parkinson’s disease in its later stage.
Change history
26 April 2022
A Correction to this paper has been published: https://doi.org/10.1007/s12640-022-00497-4
Abbreviations
- α-syn:
-
Alpha-synuclein
- AON:
-
Anterior olfactory nucleus
- APS:
-
Ammonium persulphate
- BCA :
-
Bicinchoninic acid
- BSA :
-
Bovine serum albumin
- CMC :
-
Carboxymethylcellulose
- CPCSEA :
-
Committee for the Purpose of Control and Supervision of Experiments on animals
- DAB:
-
3,3′-Diaminobenzidine
- DA:
-
Dopamine
- DAergic:
-
Dopaminergic
- DKI:
-
Diffusion kurtosis imaging
- DLS:
-
Diffusion light scattering
- DMNV:
-
Dorsal motor nucleus of vagus
- DPX:
-
Dibutylphthalate polystyrene xylene
- ENS:
-
Enteric nervous system
- ESI-Q-TOF :
-
Electrospray ionization-quadrupole-time of flight mass analyzer
- GFAP:
-
Glial fibrillary acidic protein
- HPLC:
-
High-performance liquid chromatography
- IAEC:
-
Institutional animal ethics committee
- IHC :
-
Immunohistochemistry
- i.n.:
-
Intranasal
- LC-MS:
-
Liquid chromatography–mass spectrometry
- mAb:
-
Monoclonal antibody
- ME :
-
Microemulsion
- MME:
-
Mucoadhesive microemulsion
- OB:
-
Olfactory bulb
- OD :
-
Optical density
- pAb:
-
Polyclonal antibody
- PB:
-
Phosphate buffer
- PBS:
-
Phosphate-buffered saline
- PD:
-
Parkinson’s disease
- PDI:
-
Polydispersity index
- PFA:
-
Paraformaldehyde
- PMSF :
-
Phenylmethyl sulfonyl fluoride
- PP2A:
-
Protein phosphatase 2A
- pTH :
-
Phosphorylated tyrosine hydroxylase
- psyn:
-
Phosphorylated alpha-synuclein
- PVDF:
-
Polyvinylidene fluoride
- RIPA buffer:
-
Radioimmunoprecipitation assay buffer
- RBWT:
-
Round beam walk test
- SDS :
-
Sodium dodecyl sulfate
- SLS :
-
Sodium lauryl sulfate
- SN:
-
Substantia nigra
- SNc:
-
Substantia nigra pars compacta
- TNWT-61:
-
Line 61 transgenic mouse model
- TEMED :
-
Tetramethyl-ethylenediamine
- TH :
-
Tyrosine hydroxylase
- UHPLC :
-
Ultra-high-performance liquid chromatography system
References
Alam M, Schmidt WJ (2002) Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res 136(1):317–324. https://doi.org/10.1016/s0166-4328(02)00180-8
Bové J, Prou D, Perier C, Przedborski S (2005) Toxin-induced models of Parkinson’s disease. NeuroRx 2(3):484–494. https://doi.org/10.1602/neurorx.2.3.484
Braak H, Del Tredici K (2017) Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis 7(s1):S71–S85. https://doi.org/10.3233/JPD-179001
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211. https://doi.org/10.1016/s0197-4580(02)00065-9
Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR (2012) A progressive mouse model of Parkinson’s disease: the Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9(2):297–314. https://doi.org/10.1007/s13311-012-0104-2
Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O (2002) Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci 15(6):991–998. https://doi.org/10.1046/j.1460-9568.2002.01938.x
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106(31):13010–13015. https://doi.org/10.1073/pnas.0903691106.Erratum.In:ProcNatlAcadSciUSA.106(41):17606
Dickson DW (2012) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2(8):a009258. https://doi.org/10.1101/cshperspect.a009258
Farrand AQ, Verner RS, Mcguire RM, Helke KL, Hinson VK, Boger HA (2020) Differential effects of vagus nerve stimulation paradigms guide clinical development for Parkinson’s disease. Brain Stimul 13(5):1323–1332. https://doi.org/10.1016/j.brs.2020.06.078
Fields CR, Vergniory NB, Martins RW (2019) Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease 12:299. https://doi.org/10.3389/fnmol.2019.00299
Fleming SM, Salcedo J, Hutson CB, Rockenstein E, Masliah E, Levine MS, Chesselet M-F (2006) Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-synuclein. Neuroscience 142(4):1245–1253. https://doi.org/10.1016/j.neuroscience.2006.07.005
Fullard ME, Morley JF, Duda JE (2017) Olfactory dysfunction as an early biomarker in Parkinson’s disease. Neurosci Bull 33(5):515–525. https://doi.org/10.1007/s12264-017-0170-x
GBD 2016 Parkinson’s Disease Collaborators (2018) Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17(11):939–953. https://doi.org/10.1016/S1474-4422(18)30295-3
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8(4):382–397. https://doi.org/10.1016/S1474-4422(09)70062-6
Khairnar A, Plumitallo A, Frau L, Schintu N, Morelli M (2010) Caffeine enhances astroglia and microglia reactivity induced by 3,4-methylenedioxymethamphetamine (‘ecstasy’) in mouse brain. Neurotox Res 17(4):435–439. https://doi.org/10.1007/s12640-009-9125-y
Khairnar A, Ruda-Kucerova J, Arab A, Hadjistyllis C, Minsterova AS, Shang Q, Chovsepian A, Drazanova E, Szabo N, Starcuk ZS Jr, Rektorova I, Pan-Montojo F (2021) Diffusion kurtosis imaging detects the time-dependent progress of pathological changes in the oral rotenone mouse model of Parkinson’s disease. J Neurochem 158(3):779–797. https://doi.org/10.1111/jnc.15449
Khairnar A, Ruda-Kucerova J, Drazanova E, Szabo N, Latta P, Arab A, Hutter-Paier B, Havas D, Windisch M, Sulcova A, Starcuk Z Jr, Kiraly A, Rektorova I (2016) Late-stage α-synuclein accumulation in TNWT-61 mouse model of Parkinson’s disease detected by diffusion kurtosis imaging. J Neurochem 136(6):1259–1269. https://doi.org/10.1111/jnc.13500
Khairnar A, Ruda-Kucerova J, Szabo N, Drazanova E, Arab A, Hutter-Paier B, Neddens J, Latta P Jr, Starcuk Z, Rektorova I (2017) Early and progressive microstructural brain changes in mice overexpressing human α-synuclein detected by diffusion kurtosis imaging. Brain Behav Immun 61:197–208. https://doi.org/10.1016/j.bbi.2016.11.027
Klintworth H, Garden G, Xia Z (2009) Rotenone and paraquat do not directly activate microglia or induce inflammatory cytokine release. Neurosci Lett 462(1):1–5. https://doi.org/10.1016/j.neulet.2009.06.065
Kuhn K, Wellen J, Link N, Maskri L, Lubbert H, Stichel CC (2003) The mouse MPTP model: gene expression changes in dopaminergic neurons. Eur J Neurosci 17(1):1–12. https://doi.org/10.1046/j.1460-9568.2003.02408.x
Lee Y, Dawson VL, Dawson TM (2012) Animal models of Parkinson’s disease: vertebrate genetics. Cold Spring Harb Perspect Med 2(10):a009324. https://doi.org/10.1101/cshperspect.a009324
Lofrumento DD, Saponaro C, Cianciulli A, Nuccio FD, Mitolo V, Nicolardi G, Panaro MA (2011) MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. NeuroImmunoModulation 18(2):79–88. https://doi.org/10.1159/000320027. Epub 2010 Oct 13
Longhena F, Faustini G, Missale C, Pizzi M, Spano P, Bellucci A (2017) The contribution of α-synuclein spreading to Parkinson’s disease synaptopathy. Neural Plast 2017:5012129. https://doi.org/10.1155/2017/5012129
Machado CF, Reis-Silva TM, Lyra CS, Felicio LF, Malnic B (2018) Buried food-seeking test for the assessment of olfactory detection in mice. Bio Protoc 8:e2897
Meredith GE, Rademacher DJ (2011) MPTP mouse models of Parkinson’s disease: an update. J Parkinsons Dis 1(1):19–33. https://doi.org/10.3233/JPD-2011-11023
Oueslati A (2016) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: what have we learned in the last decade? J Parkinsons Dis 6(1):39–51. https://doi.org/10.3233/JPD-160779
Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, Jackson S, Gille G, Spillantini MG, Reichmann H, Funk RH (2010) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5(1):e8762. https://doi.org/10.1371/journal.pone.0008762
Parkhe A, Parekh P, Nalla LV, Sharma N, Sharma M, Gadepalli A, Kate A, Khairnar A (2020) Protective effect of alpha mangostin on rotenone induced toxicity in rat model of Parkinson’s disease. Neurosci Lett 716:134652. https://doi.org/10.1016/j.neulet.2019.134652
Patel MR, Hirani SN, Patel RB (2018) Microemulsion for nasal delivery of asenapine maleate in treatment of schizophrenia: formulation considerations. J Pharm Investig 48:301–312. https://doi.org/10.1007/s40005-017-0318-8
Peng X, Tehranian R, Dietrich P, Stefanis L, Perez RG (2005) Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J Cell Sci 118(Pt 15):3523–3530. https://doi.org/10.1242/jcs.02481
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE (2017) Parkinson Disease Nat Rev Dis Primers 3:17013. https://doi.org/10.1038/nrdp.2017.13
Rabl R, Breitschaedel C, Flunkert S, Duller S, Amschl D, Neddens J, Niederkofler V, Rockenstein E, Masliah E, Roemer H, Hutter-Paier B (2017) Early start of progressive motor deficits in line 61 α-synuclein transgenic mice. BMC Neurosci 18(1):22. https://doi.org/10.1186/s12868-017-0341-8
Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, Lee VM, Brundin P (2018) Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol 135(1):65–83. https://doi.org/10.1007/s00401-017-1792-9
Rey NL, Petit GH, Bousset L, Melki R, Brundin P (2013) Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126(4):555–573. https://doi.org/10.1007/s00401-013-1160-3
Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016) Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213(9):1759–1778. https://doi.org/10.1084/jem.20160368
Rojo AI, Cavada C, de Sagarra MR (2007) Cuadrado A (2007) Chronic inhalation of rotenone or paraquat does not induce Parkinson’s disease symptoms in mice or rats. Exp Neurol 208(1):120–126. https://doi.org/10.1016/j.expneurol.2007.07.022
Sala G, Arosio A, Stefanoni G, Melchionda L, Riva C, Marinig D, Brighina L, Ferrarese C (2013) Rotenone upregulates alpha-synuclein and myocyte enhancer factor 2D independently from lysosomal degradation inhibition. Biomed Res Int 2013:846725. https://doi.org/10.1155/2013/846725
Sasajima H, Miyazono S, Noguchi T, Kashiwayanagi M (2015) Intranasal administration of rotenone in mice attenuated olfactory functions through the lesion of dopaminergic neurons in the olfactory bulb. Neurotoxicology 51:106–115. https://doi.org/10.1016/j.neuro.2015.10.006
Sasajima H, Miyazono S, Noguchi T, Kashiwayanagi M (2017) Intranasal administration of rotenone to mice induces dopaminergic neurite degeneration of dopaminergic Neurons in the substantia nigra. Biol Pharm Bull 40(1):108–112. https://doi.org/10.1248/bpb.b16-00654
Shah B, Khunt D, Misra M, Padh H (2016) Non-invasive intranasal delivery of quetiapine fumarate loaded microemulsion for brain targeting: formulation, physicochemical and pharmacokinetic consideration. Eur J Pharm Sci 91:196–207. https://doi.org/10.1016/j.ejps.2016.05.008
Shah BM, Misra M, Shishoo CJ, Padh H (2015) Nose to brain microemulsion-based drug delivery system of rivastigmine: formulation and ex-vivo characterization. Drug Deliv 22(7):918–930. https://doi.org/10.3109/10717544.2013.878857
Souza MF, Freire MAM, Medeiros KAAL, Lins LCRF, Bispo JMM, Gois AM, Leal PC, Engelberth RCGJ, Ribeiro AM, Silva RH, Marchioro M, Santos JR (2018) Deltamethrin Intranasal administration induces memory, emotional and tyrosine hydroxylase immunoreactivity alterations in rats. Brain Res Bull 142:297–303. https://doi.org/10.1016/j.brainresbull.2018.08.007
Thakkar HP, Patel AA, Chauhan NP (2014) Formulation and optimization of mucoadhesive microemulsion containing mirtazapine for intranasal delivery. Chron Young Sci 5:25–32. https://doi.org/10.4103/2229-5186.129333
Voronkov DN, Kutukova KA, Ivanov MV, Khudoerkov RM (2017) Immunomorphological Changes in the Olfactory Bulbs of Rats after intranasal Administration of Rotenone. Bull Exp Biol Med 164(2):203–206. https://doi.org/10.1007/s10517-017-3958-9
Wang Y, Liu J, Chen M, Du T, Duan C, Gao G, Yang H (2016) The novel mechanism of rotenone-induced α-synuclein phosphorylation via reduced protein phosphatase 2A activity. Int J Biochem Cell Biol 75:34–44. https://doi.org/10.1016/j.biocel.2016.03.007
Yang X, Qian Y, Xu S, Song Y, Xiao Q (2018) Longitudinal analysis of fecal microbiome and pathologic processes in a rotenone induced mice model of parkinson’s disease. Front Aging Neurosci 9:441. https://doi.org/10.3389/fnagi.2017.00441
Zhou F, Wu J, Sun X, Yao H, Ding J, Hu G (2007) Iptakalim alleviates rotenone-induced degeneration of dopaminergic neurons through inhibiting microglia-mediated neuroinflammation. Neuropsychopharmacology 32(12):2570–2580. https://doi.org/10.1038/sj.npp.1301381
Funding
This supplement was supported by the seed fund of the National Institute of Pharmaceutical Education and Research (NIPER), Ahmedabad.
Author information
Authors and Affiliations
Contributions
We declare that all authors made fundamental contributions to the manuscript. MS and AMK designed the study. MS, JK, SR, and NS conducted experiments. MS, JK, SR, DK, and AMK analyzed the data. MS and AMK participated in the interpretation and writing of the manuscript. DK and AMK advised the experiments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
IAEC approval number: IAEC/19/02.
Consent to Participate
Not applicable.
Consent for Publication
Each author has provided his/her consent for this publication.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sharma, M., Kaur, J., Rakshe, S. et al. Intranasal Exposure to Low-Dose Rotenone Induced Alpha-Synuclein Accumulation and Parkinson’s Like Symptoms Without Loss of Dopaminergic Neurons. Neurotox Res 40, 215–229 (2022). https://doi.org/10.1007/s12640-021-00436-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-021-00436-9