
Abstract
Aging plays a significant role in the progression of vascular diseases and vascular dysfunction. Activation of the ADP-ribosylation factor 6 and small GTPases by inflammatory signals may cause vascular permeability and endothelial leakage. Pro-inflammatory molecules have a significant effect on smooth muscle cells (SMC). The migration and proliferation of SMC can be promoted by tumor necrosis factor alpha (TNF-α). TNF-α can also increase oxidative stress in SMCs, which has been identified to persuade DNA damage resulting in apoptosis and cellular senescence. Peroxisome proliferator-activated receptor (PPAR) acts as a ligand-dependent transcription factor and a member of the nuclear receptor superfamily. They play key roles in a wide range of biological processes, including cell differentiation and proliferation, bone formation, cell metabolism, tissue remodeling, insulin sensitivity, and eicosanoid signaling. The PPARγ activation regulates inflammatory responses, which can exert protective effects in the vasculature. In addition, loss of function of PPARγ enhances cardiovascular events and atherosclerosis in the vascular endothelium. This appraisal, therefore, discusses the critical linkage of PPARγ in the inflammatory process and highlights a crucial defensive role for endothelial PPARγ in vascular dysfunction and disease, as well as therapy for vascular aging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammation is regarded as the critical biological response of body tissues to detrimental stimuli, including damaged cells, irritants, and/or pathogens (Hossain et al. 2016). In addition, inflammation is also part of a protective response involving molecular mediators, immune cells, and blood vessels (Ferrero-Miliani et al. 2007). The functions of inflammation include the initiation of tissue repair, clearing necrotic cells and tissues injured by the inflammatory process and the original insult and elimination of the preliminary cause of cell injury (Rispens et al. 2012). Rheumatoid arthritis, asthma, inflammatory bowel disease (IBD), psoriasis, multiple sclerosis, and atherosclerosis are the commonly observed inflammatory diseases. Over recent decades, it has been noticed that there is a significant rise in the prevalence of atopic illness counting asthma (Bloomfield et al. 2006). It is believed that diseases including ulcerative colitis and Crohn’s disease are prevalent only in Western societies. However, in Asia, the prevalence and incidence of these illnesses are rising (Jacobsen et al. 2006; Goh and Xiao 2009; Konkel 2016; Ng 2016). Individuals with IBD are at greater risk of developing colorectal carcinoma (Carter et al. 2009). Interestingly, lower incidence of cancer was observed in the geographical regions where diets are high in n-3 polyunsaturated fatty acids (PUFAs) (Caygill and Hill 1995). It has been recommended that various protective effects of dietary n-3 PUFAs might be because of their peroxisome proliferator-activated receptor (PPAR) activating and anti-inflammatory properties (Allred et al. 2008).
The PPARs are a member of the subclass of the nuclear receptor superfamily (Delerive et al. 1999). Cytoplasm is the primary site where classical hormone-activated receptors, including estrogen receptors, are commonly found. Furthermore, after binding of the activating ligand, they translocate to the nucleus. In contrast, PPARs are located in the nucleus bound to deoxyribonucleic acid (DNA) response elements (Glass and Rosenfeld 2000). Muscle, liver, smooth muscle cells (SMCs), vascular endothelial cells, bone, macrophages, heart, and adipose tissue are the main sites where PPARγ is extensively expressed (Sigmund 2010). Treatment with therapeutic PPARγ agonists including thiazolidinediones (TZDs) can lower blood pressure and ameliorate glucose control in individuals with type 2 diabetes (Sharma and Staels 2007). Furthermore, PPARγ agonists may also protect these individuals from the progression of atherosclerosis (Hodis et al. 2006; Mazzone et al. 2006; Nissen et al. 2008). It has been observed that TZDs can exhibit protective vascular and metabolic effects, which is compatible with genetic affirmation, suggesting that hypertension and severe insulin resistance are likely to be developed in the individuals who carry dominant-negative PPARγ mutations (Barroso et al. 1999).
The vasculature is largely affected by aging. The properties of SMCs and endothelial cells (ECs) are severely affected by vascular aging (i.e., cellular extrinsic and intrinsic changes), which can eventually result in poor vascular function and can further lead to the advancement of vascular disease (Mistriotis and Andreadis 2017). Blood vessels are found to be constituted by the SMCs and ECs. In addition, ECs have thromboresistant properties, line the lumen of a vessel, are found to be permeable to humoral factors and chemicals, and also offer a dynamic barrier to the blood cells (Yazdani et al. 2010). Moreover, via the translation of mechanical forces applied through the pulsatile blood flow into biological signals, endothelium can also act as a mechanosensor (Chiu and Chien 2011).
Increased reactive oxygen species (ROS) production is a vital element of vascular aging (Oeseburg et al. 2010; Seals et al. 2011). Moreover, uncoupled endothelial nitric oxide synthase (eNOS), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and mitochondria are the sources of aging-linked increased ROS production in the endothelium (Seals et al. 2011; Touyz and Briones 2011). Irrespective of the source, increased ROS have been found to be connected with the functional alterations in the endothelium along with senescence, apoptosis, and EC inflammation (Oeseburg et al. 2010; Touyz and Briones 2011). Vascular cell senescence, DNA damage, environmental factors (Csiszar 2009), genetic diseases (Olive et al. 2010), atherosclerosis, and hypertension are the noteworthy factors that can play a part in the proinflammatory phenotype of aged vessels. Furthermore, these factors can alter homeostasis and eventually result in the deterioration of the vasculature. Milk fat globule-EGF factor VIII (MFG-E8), intercellular adhesion molecule (ICAM), interleukin-6 (IL-6), interferon gamma (IFNγ), interleukin 1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), matrix metalloproteinase 2 (MMP2), angiotensin II (Ang II), and calpain-1 are the pro-inflammatory signals that can increase with age-related diseases and aging (Belmin et al. 1995; Donato et al. 2008; Lesniewski et al. 2011; Wang et al. 2014b). Therefore, the aim of this review is to discuss the mechanisms through which endothelial PPARγ controls inflammatory responses and protects against vascular dysfunction connected to aging.
PPARγ — a Family of Nuclear Hormone Receptors
PPARs are members of nuclear hormone receptors and there are three isoforms of PPARs, including PPARα, PPARβ, and PPARγ (Tyagi et al. 2011). Inhibition of inflammatory signaling through nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), promotion of adipogenesis, glucose metabolism, and fatty acid synthesis and storage are the biological roles of PPARγ (Corona and Duchen 2016). There is a growing interest in recent years regarding the role of PPARγ in inflammation. Dendritic cells (DCs), lymphocytes, platelets, and numerous immune cells (i.e., including macrophages/monocytes) are the common sites of PPARγ expression (Padilla et al. 2002; Akbiyik et al. 2004; Asada et al. 2004). Usually, PPARγ occurs as a heterodimer complexed with retinoid X receptor alpha (RXRα), whereby these two molecules are normally bound to corepressors. The corepressor molecules are dislocated by ligand stimulation; furthermore, coactivators and ligand can form an active complex, binding to PPARγ response elements (PPRE). On the other hand, PPARγ can bind with NF-κB target genes upon ligand stimulation, which ultimately suppress NF-κB target genes (Marcu et al. 2010).
For binding and activation of PPARγ, a number of PPARγ ligands have been found. Several prostaglandins such as prostaglandin D2 (PGD2) and its metabolite (i.e., 15-deoxy- D12,14-prostaglandin J2 [15d-PGJ2]), oleanolic acids, certain eicosanoids, and TZDs can act as PPARγ ligands (Croasdell et al. 2015). It has been found that potent anti-inflammatory effects can be achieved via ligand-dependent activation of PPARγ. Furthermore, several of these molecules have anti-inflammatory effects and these effects are not reliant on PPARγ (Youssef and Badr 2004). Though the extent to which these ligands exert their effects may vary in an independent way. For example, TZDs act in a heavily PPARγ-dependent manner, prostaglandins exhibit independent/mixed dependent activity, and ibuprofen acts largely independent of PPARγ. Most of these ligands have been studied to assess their PPARγ-independent effects (Kulkarni et al. 2012). Currently, PPARγ studies have started emphasizing on the anti-inflammatory effects and exploring the contribution of PPARγ and its ligands in the resolution of inflammation.
Ligand-Dependent PPARγ-Facilitated Gene Repression
It has been observed that PPARγ can affect gene expression either via direct promoter-binding and transcriptional modulation of target genes called transactivation or via indirect intervention with other transcription factor mechanisms leading in gene repression (Chinetti et al. 2003). Ligand-dependent gene repression is the principal way by which PPARγ exerts its anti-inflammatory effects. In addition, for ligand-dependent gene repression via PPARγ, there are several distinct mechanisms, all occurring by the indirect regulatory effects of ligand binding. The subsequent negative regulation of gene repression is known as transrepression if PPARγ activity is not facilitated by its binding to PPRE target DNA sequences. In this regard, ligand-dependent transrepression of inflammatory genes perhaps is the best characterized model. As described by Li et al. (2000b), in the absence of inflammatory signals, there is an inhibitory protein complex bound to the promoter of inflammatory genes (i.e., inducible nitric oxide synthase 2 [iNOS2]) that can keep these genes repressed.
There are few requirements that must need to be met for effective expression of NOS2 upon lipopolysaccharide (LPS)-triggered activation of toll-like receptor 4 (TLR4). Firstly, from the promoter region, the inhibitory complex must be separated. Only following this separation, the key immune regulator NF-κB can activate expression of NOS2 (Varga et al. 2011). The ubiquitin-proteasome system is an important pathway by which the displacement of the inhibitory complex normally takes place. A portion of the liganded PPARγ will be SUMOylated on lysine K365 in cells that receive concomitant LPS treatments and PPARγ ligand. The SUMOylated PPARγ will not be capable to bind its typical heterodimerization, retinoid X receptor (RXR). Rather, SUMOylated PPARγ will bind to the repressor complex, which is situated on the promoter of inflammatory genes (Varga et al. 2011). Blockage of ubiquitination and subsequent effective removal of the repressors will be achieved via binding of PPARγ to these repressor complexes. Consequently, even in the presence of active TLR4 signaling, ligand-bound PPARγ will maintain the repression on the promoter of inflammatory genes, as for example iNOS2. In addition, other forms of transrepression do also exist. NF-κB and activator protein 1 (AP-1) are the examples of other transcription factors to which PPARγ was found to bind directly (Delerive et al. 1999; Chung et al. 2000) and interfere with the DNA binding ability of these transactivators. On the other hand, it has been confirmed that the p38 mitogen-activated protein (MAP) kinase activity can be modulated through the ligand-activated PPARγ (Syrovets et al. 2002). Even though the previously mentioned processes can elucidate a subset of the anti-inflammatory effects of PPARγ, positive transcriptional regulation of inhibitory proteins play crucial roles in ligand-induced repression instead of transrepression of other transcription factors. In addition, the significance of in vivo and the pathway by which these suggested processes contribute to the decrease in gene expression are yet to be determined.
Phosphorylation can modulate the outcome of ligand-mediated PPARγ activation. In this regard, the phosphorylation of PPARγ2 at serine-112 by MAP kinases was reported. It was observed that the transcriptional activity of PPARγ was reduced by this phosphorylation (Hu et al. 1996). Perhaps, as observed by the increased PPARγ target gene expression, aP2 (i.e., FABP4), it was observed that, via the activity of MAP kinases, insulin mediated the ligand-dependent activation of PPARγ (Zhang et al. 1996). On the other hand, through epidermal growth factor (EGF) and platelet-derived growth factor (PDGF), PPARγ1 might also be phosphorylated at serine-82 (i.e., which correlates with serine-112 of PPARγ2) (Camp and Tafuri 1997). Instead, the transcriptional action of the ligand-bound PPARγ can be attenuated by this phosphorylation event. Choi et al. (2010) mentioned in their study that adipocyte impairment in the expression of several metabolism-related target genes took place due to cdk5 arbitrated phosphorylation at serine-273 of PPARγ2. Normalized gene expression and this phosphorylation were hindered by the treatment of adipocytes with the rosiglitazone (i.e., a PPARγ agonist).
PPARγ and Inflammation
Multiple sclerosis (MS) is a well-known autoimmune disease of the central nervous system (CNS). Genetic screening of individuals with MS, where the effect of PPARγ was apparently evidenced on inflammatory diseases. A number of genetic connections to immune genes have been identified for MS, particularly the receptors for IL-7 and IL-2 (Parnell and Booth 2017). In recent times, in the list of MS-linked genes, a PPARγ polymorphism has been introduced. In a study of 211 healthy age-matched controls and 116 patients, the Ala/Ala genotype of the PPAR Pro12Ala polymorphism was linked with a 10-year significant delay of the onset of disease in individuals with MS (Klotz et al. 2009b). Remarkably, through research on atherosclerosis, it has been found that Pro12Ala polymorphism mediated anti-inflammatory effects. The findings of this study of males with coronary artery disease suggested that the carriers of the Pro12Ala allele of PPARγ have less prevalent atherosclerosis and are markedly secured against 10-year vascular mortality and morbidity (Regieli et al. 2009).
The contribution of natural PPAR ligands in regulating inflammation was observed in the PPARγ knockout (KO) mice. Alternatively, in a seminal experiment, colitis was provoked in mice lacking in colonic PPARγ through two distinct mechanisms, either via oral dextran sodium sulfate treatment or through the transfer of CD4+CD45RBhi T-cells (Itzkowitz and Yio 2004). It is noteworthy that in the previous process colitis is facilitated by antigen-mediated activation of TH1-cell, whereas in the latter mentioned method, colitis is reliant on activation of macrophage and is triggered by chemical irritation of the mucosa. Interestingly, in both models, the colonic PPAR KO mice were not responsive, while the wild-type mice were responsive to dietary conjugated linoleic acid through improvement of colitis (Bassaganya-Riera et al. 2004).
Interestingly, the anti-inflammatory properties of PPARγ are found to be firmly related to its anti-diabetic effects (Pascual et al. 2007). Instead, to resist a challenge with Leishmania major, mice with a macrophage-specific deletion of PPARγ were studied for their capability. Remarkably, less susceptibility to Leishmania spp. infection was noticed with mice lacking macrophage PPAR. Furthermore, as anticipated, the same animals exhibited susceptibility to diabetic illness (Odegaard et al. 2007). Reduced ability for the skeletal muscle to metabolize sugars and fatty acids was caused by the selective KO of macrophage PPARγ. Furthermore, the expression of co-activator proteins and transcription factors, which are essential for mitochondrial biogenesis, is also markedly lessened. Therefore, macrophage PPAR activation not only has significant contributions to the metabolic status of the whole mouse but also persuades the differentiation of macrophages into the non-inflammatory, M2 type. In contrast, a related exhibition of the significant physiological effect of selective KO of the PPARγ gene, via selectively eliminating the gene from ECs, caused entire trunk hair loss in the offspring of the KO mice (Martin 2010). On the other hand, targeted removal of PPARγ in mice caused genesis of toxic milk having higher levels of inflammatory lipids (Wan et al. 2007). Generation of these oxidized lipids took place because of a marked surplus of lipoxygenase activity, usually repressed by the endothelial PPARγ gene. In the skin of the suckling mice, inflammation and succeeding alopecia were caused due to the buildup of the inflammatory lipid mediators. Alopecia was found to be caused by skin inflammation. This inflammation was cured by the use of topical aspirin and this treatment averted the hair loss (Wan et al. 2007). Therefore, it is well understood from these findings that systemic inflammation is the usual physiological response, controlled by PPARγ under usual circumstances.
Molecular Mechanisms of PPARγ in Anti-inflammatory Processes
Previous studies recommended that the anti-inflammatory effects of PPARγ can be exhibited by the number of molecular mechanisms (Fig. 1 and Fig. 2) (Varga et al. 2011). As stated earlier, transactivation and transrepression are the ways of gene regulation. It has been found that PPAR can bind and sequester other distinct transcriptional regulators or transcription factors, during transrepression. Regulatory circuits controlled by the related transcription factors are weakened, as a final outcome of this sequestration. In fact, when activated by ligand binding, PPARγ was reported to transrepress other transcription factors (Ricote and Glass 2007).

Mechanisms of ligand-dependent genetic regulation by PPARγ cause displacement of corepressors and recruit of co-activators to promoters comprising PPRE and subsequently activate gene expression to exert anti-inflammatory effects. L, Ligand; CA, Coactivator; CR, Corepressor; RXR, Retinoid X receptor; PPRE; Peroxisome proliferator-activated receptor gamma response element

Mechanisms of ligand-dependent genetic regulation by PPARγ binds with NF-κB to repress NF-κB target genes and exert anti-inflammatory effects. L, Ligand; RXR, Retinoid X receptor; NF-κB RE, Nuclear factor kappa light chain enhancer of activated B cells response element
It is not well known how the molecular mechanisms of PPARγ action are translated into anti-inflammatory effects. Possibly, PPARγ controls the expression of genes (i.e., via transrepression, either indirectly or directly) that have direct contributions in inflammation (Brunmeir and Xu 2018). Instead, by altering lipid metabolism, it is also likely that PPARs indirectly modulate inflammatory processes (Varga et al. 2011). In fact, PPARs would directly alter the intracellular and extracellular pool of lipid molecules and this modified lipid environment would trigger secondary regulatory processes in the body (Varga et al. 2011). A similar process was reported in human dendritic cells (DCs), where activation of PPARγ can lead to the formation of retinoic acid (Szatmari et al. 2006). In the well-fed state, PPARγ is activated and can regulate the production of fatty acids and associated lipids (Szatmari et al. 2006). Like PPARγ, it has been found that the other PPAR subtypes (i.e., PPARβ/δ) also modify lipid metabolism in markedly discrete ways and produce different categories of lipid molecules, which possess a similar anti-inflammatory contribution in diseases. Consequently, the regulatory circuits of PPARs in inflammation and metabolism are partially diverse. It is also possible that the noticed anti-inflammatory effects of PPARs are not completely arbitrated by their ability to modify entire homeostasis of body lipid. In addition, direct molecular regulatory processes are moderately responsible for their anti-inflammatory effects.
PPARγ and Inflammatory Diseases
Impact of PPARγ in Animal Studies
In several mouse models, the contribution of PPARγ in IBD was studied. From these studies, generally, it was found that PPARγ activity protected from IBD (Annese et al. 2012). In both resident macrophages of the colonic mucosa and epithelial cells, PPARγ is usually expressed. Henceforth, in both cell types, disease progression can be potentially improved by the PPARγ activity. In heterozygous PPARγ mice, the chemically stimulated inflammation model was used (Desreumaux et al. 2001); on the other hand, in colonic epithelium-specific mice (Adachi et al. 2006), T cell–specific or macrophage-specific (Shah et al. 2007) PPARγ knockout (Hontecillas and Bassaganya-Riera 2007), while in wild-type mice, the treatment of PPARγ ligand was subjected to modulate inflammation (Su et al. 1999). An augmented susceptibility to disease was observed with PPARγ deficiency, in all of the abovementioned animal models. On the other hand, even without the presence of an exogenously administered PPARγ ligand, animals lacking PPARγ exhibited more serious disease symptoms. These findings recommended that either an endogenous ligand with a powerful PPARγ agonist activity was generated in these animals, or instead, unliganded PPARγ had an action that saved mice from the disease. Furthermore, these findings raised the chance to improve human IBD by using PPARγ agonists.
In order to study the contribution of PPARγ in the modulation of inflammation, an animal model of brain inflammation known as experimental autoimmune encephalomyelitis (EAE) can be used. It has been noticed that EAE in rodents coexist with demyelination. Due to the previously mentioned phenomenon, for acute disseminated encephalomyelitis (ADEM) and human MS, EAE can be used as a suitable model. To improve experimentally stimulated EAE, studies have confirmed that the TZD drugs (i.e., ciglitazone and troglitazone) and the endogenous ligand 15d-PGJ2 were found to be effective (Niino et al. 2001; Natarajan and Bright 2002). It was also found that a reduction in TH1 cell differentiation and IL-12 production lead to a reduction in disease duration and severity. In accordance with the abovementioned findings, in the same EAE model, PPARγ-deficient heterozygous mice were found to develop an aggravated disease than the wild-type littermates (Natarajan et al. 2003). Recently, a different study recommended that, through the decrease in TH17 T cell differentiation, activation of PPARγ inhibited CNS inflammation in the EAE model (Klotz et al. 2009a, b). In wild-type mice, the previously mentioned study also confirmed both the harmful actions of the CD4 T cell–specific PPARγ deletion and the beneficial effects of the PPARγ agonist treatment.
Dysregulation of lipid metabolism is responsible for atherosclerosis and metabolic syndrome that play a role in the inflammatory state. Since PPARγ has anti-inflammatory effects and regulates lipid metabolism, thus it is a reasonable target in associated studies that dissect inflammatory and lipid metabolism resulting in components of the diseases. PPARγ is vastly expressed in macrophage foam cells of atherosclerotic lesions. In a study, Li et al. (2000a, b) reported that PPARγ-specific agonist, rosiglitazone, inhibited the development of atherosclerosis in low-density-lipoprotein (LDL) receptor KO male mice. In another study, Collins et al. (2001) investigated the doubt regarding whether the beneficial effects of PPARγ was facilitated by the changed phenotype of cells involved in the lesions or by the changed systemic lipid homeostasis. This study confirmed that the beneficial effects of PPARγ on insulin sensitivity and its anti-atherogenic effects were uncoupled. The findings of Collins et al. (2001) recommend that PPARγ improved atherosclerosis by affecting cells locally. Moreover, it was also revealed that the consequence of atherosclerosis was deteriorated by the bone marrow transplantation of PPARγ−/− cells, this finding firmly signifying a contribution of PPARγ in macrophages involved in lesion formation (Chawla et al. 2001).
Unpredictably, in metabolic studies, insulin sensitivity was found to be regulated by macrophage PPARγ (Hevener et al. 2007; Odegaard et al. 2007). Both human medical practice and murine studies have confirmed the fact that ligand activation of PPARγ can improve the sensitivity of insulin. Initially, it was presumed that, in one of the main target tissues of lipid and glucose metabolism (i.e., for example, liver, muscle, or fat), beneficial effects of ligand activation were facilitated by PPARγ. Remarkably, insulin resistance was found to be caused by the loss of PPARγ in hematopoietic cells. In the mouse, it has been proposed that inflammation is a constituent present in developing insulin resistance; furthermore, this inflammation was exacerbated by the macrophage PPARγ deletion. Nevertheless, conserved glucose tolerance in high-fat-fed C57BL/6 mice transplanted with LXRα−/− (liver X receptor alpha), PPARβ/δ−/− or PPARγ−/− hematopoietic cells was also noticed in a similar study (Marathe et al. 2009). Indeed, adipose tissue was found to be the major site of the insulin-sensitizing action of ligand-activated PPARγ (Sugii et al. 2009). Due to these opposing findings, to elucidate the contribution of macrophage PPARγ in the development of insulin resistance, further studies are required.
Impact of PPARγ in Human Studies
Single nucleotide polymorphisms (SNPs) and mutations of human PPARγ are linked with inflammatory and metabolic diseases. Conversely, in 85 distinct individuals with severe insulin resistance, all the coding exons of PPARγ 1 and 2 were examined by Barroso et al. (1999). Whereas, in three patients, they found two heterozygous missense mutations of PPARγ ligand-binding domain (i.e., V290M and P467L). Hypertension and type 2 diabetes mellitus were observed in all 3 patients, which further signifies the substantial contribution of PPARγ in the adjustment of blood pressure, glucose homeostasis, and sensitivity of insulin. In a study conducted by Agostini et al. (2006), it stated that in lipodystrophic insulin–resistance patients, extra PPARγ mutations were observed. These formerly mentioned mutations made PPARγ to act in a dominant-negative fashion and also made unable to bind DNA. It is known that the modulation of lipid and glucose metabolism is considered as the canonical PPARγ function. Henceforth, there is a strong possibility that, in these patients, these phenotypes were produced because of the weakened metabolism, and increased inflammation in metabolic disease was not contributing due to the loss of PPARγ.
A number of SNPs of PPARγ were recognized, among them Pro12Ala is the most frequently studied SNP. It has been firmly found that the metabolic status of patients is influenced by Pro12Ala (Deeb et al. 1998). Moreover, growing evidence propose that inflammatory diseases are also influenced by SNPs of PPARγ. Oh et al. (2009) in their study exhibited that the development of asthma was associated with the combination of the major allele of the non-synonymous Pro12Ala SNP with the major allele of the synonymous His449His polymorphism. In the same study, they also found that when compared to healthy controls, the occurrence of the major Pro12Ala allele in combination with the minor His449His allele was markedly lesser in individuals with asthma (Oh et al. 2009).
Interestingly, Penyige et al. (2010) exhibited in their study that in chronic obstructive pulmonary disease (COPD), major allele of the His447His polymorphisms had a safeguarding role. Furthermore, they also noticed that minor His447His allele was found to be linked with the development of COPD (Penyige et al. 2010). Moreover, in the central Chinese but not Dutch IBD population, the link of the minor allele of the same SNP was observed (Shrestha et al. 2010). It needs to be considered that genetic variations in PPARγ have only a minor effect on the expression level/function of the receptor, while these variations can have a statistically significant effect on a number of chronic inflammatory conditions. In the near future, similar research in other inflammatory diseases is estimated to arise.
PPARγ and Proinflammatory Cytokines
In many studies, it was noticed that the expression of proinflammatory cytokines is affected by PPARγ. Similarly, the expression of TNF-α has also been found to be significantly affected by the PPARγ. In terms of regulation of immune cell function, TNF-α plays an important role. Furthermore, TNF-α can stimulate other cytokines, promote apoptosis, and also can induce fever (D’Elia et al. 2013). TNF-α also plays a role as a neutrophil chemoattractant and macrophage. Although normally this cytokine has significant contribution in bacterial killing, overexpression can cause poor health effects (i.e., rapid weight loss) and promotion of chronic inflammation. It is likely that in a feedback loop or compensatory mechanism, TNF-α can increase protein PPARγ expression and mRNA in human neutrophils (Reddy et al. 2008). In association with this elevated expression, TNF-α expression can be significantly reduced by the PPARγ ligands, notably TZDs (Sharma et al. 2009). A reduction in the expression of TNF-α was observed in LPS-exposed and pioglitazone-treated guinea pigs and mice. Certainly, both rosiglitazone and pioglitazone decreased TNF-α expression in a broad variety of inflammatory models, including spinal trauma, gastric injury, colitis, reperfusion/ischemia, and sepsis models (Cuzzocrea et al. 2003; Villegas et al. 2004; Akahori et al. 2007; Zhang et al. 2010; Celinski et al. 2011; Gao et al. 2015). Interestingly, these observed effects were not dependent on the route of administration. Furthermore, in mouse livers, the oral delivery of pioglitazone to reduce TNF-α is specifically remarkable from a therapeutic viewpoint (Collino et al. 2010). With the decrease in TNF-α expression blocked by PPARγ antagonists, these effects were mostly found to be PPARγ-dependent (Cuzzocrea et al. 2003; Zhang et al. 2010). Other PPARγ ligands, counting oleanolic acid and 15d-PGJ2, can also reduce the expression of TNF-α (Zingarelli et al. 2003; Genovese et al. 2008; Reddy et al. 2013).
Some ILs, together with TNF-α, can be generated in response to inflammatory stimuli. IL-6 is considered as a mediator of fever and also as an element of the acute inflammatory response. Local production of IL-6 in the lung, colon, liver, and intestine was found to be decreased by ciglitazone, rosiglitazone, and pioglitazone, respectively (Zingarelli et al. 2003; Celinski et al. 2011; Gao et al. 2015). In a similar manner, bacteria-triggered upsurges in IL-6 can be reduced by the PGD2 microspheres and 15d-PGJ2 (Zingarelli et al. 2003; Pereira et al. 2015). On the other hand, for neutrophil trafficking, IL-8 is known as an important chemokine. Generally, expression of IL-8 can take place in a group of cell types (i.e., macrophages) and this expression can be triggered by a range of inflammatory stimuli.
IL-8 production, induction of leukotrienes, and tobacco smoke–triggered decreases in PPARγ can be prevented by rosiglitazone (Yin et al. 2014). Stimulation of acute inflammation can be promoted by the IL-1β; on the other hand, it can be lessened by pioglitazone, 15d-PGJ2, and rosiglitazone in a PPARγ-dependent manner (Cuzzocrea et al. 2003; Akahori et al. 2007; Pisanu et al. 2014). Since the expression of NF-κB has found to be decreased by PPARγ, the wide range of effects of PPARγ ligands on proinflammatory cytokines perhaps due to effects on the NF-κB pathway (Cuzzocrea et al. 2003; Villegas et al. 2004; Sánchez-Hidalgo et al. 2005; Celinski et al. 2011).
Debatably, pro-resolving and anti-inflammatory cytokines are also produced by PPARγ. Alternatively, in both anti- and pro-inflammatory conditions, IL-10 is produced (Jaudszus et al. 2013). Macrophages can produce IL-10 to arbitrate pro-resolving effects. Rosiglitazone can induce the production of IL-10 in the experimental models for Parkinson’s disease (PD) and colitis (Celinski et al. 2011; Pisanu et al. 2014). However, IL-10 expression was found to be reduced by 15d-PGJ2 and pioglitazone in a septic lung (Zingarelli et al. 2003; Celinski et al. 2011). Further studies are required to elucidate whether these variations are because of the differences in the time points selected, PPARγ ligand-specific signaling, in inflammatory stimuli. In models of PD, the expression of another pro-resolving cytokine, TGFβ, was found to be induced by rosiglitazone in microglia (Pisanu et al. 2014). Rather than suppression of all cytokine signaling, this change in the production of cytokine associates that PPARγ is not merely acting in an anti-inflammatory manner, nonetheless also in a pro-resolving manner.
Anti-inflammatory Effects of PPARγ Ligands
PPARγ ligands can be categorized into a range of groups, including reversible/covalent, exogenous/endogenous, or synthetic/natural. Although for the treatment of diabetes, PPARγ agonists particularly TZDs are normally used to treat diabetes; nevertheless, their anti-inflammatory effects have also been explored (Wang et al. 2014a, b). Interestingly, PPARγ protein was recognized in the macrophages, monocytes, and antigen presenting cells. Suppression of the inflammatory cytokine production by these cells was noticed with the use of synthetic PPARγ agonists such as rosiglitazone, troglitazone, and pioglitazone (Jiang et al. 1998; Ricote et al. 1998). Afterwards, PPARγ was detected in DCs which are highly differentiated and potent, specialized antigen-presenting cells. Furthermore, DC secretion of IL12 (i.e., a potent TH1-type inflammatory cytokine) was found to be decreased by the same thiazolidinedione compounds (Faveeuw et al. 2000; Gosset et al. 2001).
TZDs were also found to lessen the production of TH1-cell but not TH2-cell-recruiting chemokines (Gosset et al. 2001). The contribution of rosiglitazone through PPAR as a general repressor of a wide variety of interferon and LPS-target genes was demonstrated by an mRNA profiling study with mouse macrophages. Collectively, these findings strongly suggest that PPARγ exerts anti-inflammatory effects via its capability to subdue production of TH1-type cytokine in DCs and macrophages (Welch et al. 2003). Rosiglitazone as a PPARγ activator was found to be beneficial to treat ulcerative colitis in the clinical trials (Lewis et al. 2008; Liang and Ouyang 2008).
Role of PPARγ Ligands in Animal Studies
The PPARγ agonists have been studied in animal models of inflammation. Atherosclerosis, renal disease, and production of autoantibodies were reduced by rosiglitazone, in a mouse model of systemic lupus erythematosus (Aprahamian et al. 2009). On the other hand, in a mouse model of renal fibrosis, inflammation and renal scarring were reduced by the use of PPARγ agonist, troglitazone (Kawai et al. 2009). Rosiglitazone inhibited the expression of TNF-α and the cytokine markers of inflammation (i.e., notably IL-1β) and also weakened the activity of myeloperoxidase in a rat model of postoperative brain inflammation (Hyong et al. 2008). However, the crossing of the blood-brain barrier (BBB) by rosiglitazone is poor, which may have an impact on its beneficial effects. Probably, the effects of rosiglitazone are arbitrated outside the brain, possibly in the brain vasculature.
By activation of PPARγ, rosiglitazone barred gut colonization in a mouse model of gastrointestinal candidiasis (Coste et al. 2008). It has been noticed that elevated mannose-receptor expression on mucosal macrophages mediated the previously mentioned effect. It was shown by means of a rat model of pancreatitis that troglitazone and the PPAR agonists, including 15d-PGJ2, both can reduce inflammation of the pancreas, which was detected by low levels of the transforming growth factor beta 1 (TGF-β1) and inflammatory cytokines IL-6 and by lessened pancreatic weight (Yu et al. 2008). In the IBD treatment, 5-aminosalicylic acid (5-ASA) is most commonly used (Williams et al. 2011). In a PPAR+/− mouse model of colitis, the anti-inflammatory effects of 5-ASA have been found to be reliant on the activation of PPAR (Rousseaux et al. 2005).
Role of PPARγ Ligands in Human Studies
The systemic anti-inflammatory activity of PPARγ agonist has been confirmed by preclinical and clinical trials. During exposure of the cells to the inflammatory molecules, IL-1β and LPS, the nutritional PUFAs, PPAR ligands, eicosapentaenoic acid, docosahexaenoic acid, and linolenic acid were administered to treat the three different cells such as human enterocyte cell line, Caco-2 and human DCs (Martin 2010). PUFAs enhanced PPARγ expression while reduced the release of pro-inflammatory cytokines IL-6 and IL-8 (Marion-Letellier et al. 2008). Interestingly, this anti-inflammatory activity was blocked by GW 9662, a synthetic PPARγ antagonist, which revealed that the anti-inflammatory activity of PUFAs was PPARγ-dependent. Recently, linseed oil suggestively decreased the TNF-α expression but increased muscle mass in pigs (Huang et al. 2008). The main components (i.e., linoleic and linolenic acid) of linseed oil are PPAR agonists. This study also showed that the concentration of PPARγ in the muscle and spleen depressingly associated with serum TNF-α.
The mechanism of PPARγ activation has been confirmed by its X-ray crystallographic investigations with bound ligands. Based on crystal structures, the ligand-binding pocket can accommodate two fatty acids concurrently. In fact, by a Michael addition mechanism, oxo-fatty acid metabolites (Itoh et al. 2008) and the anti-inflammatory 15d-PGJ2 (Waku et al. 2009) attached covalently to the sulfhydryl group at Cys285, present in the binding domain of ligand. The ligands, those that have properties for covalent binding, mainly serve as PPARγ activators.
Vascular Endothelial Dysfunction and Aging
Studies point out that in humans, aging is responsible for vascular endothelial dysfunction in the absence of clinical cardiovascular disease (CVD) and foremost risk factors for CVD (Seals et al. 2014). The impairment in the endothelium-dependent dilation (EDD) decreased the function for fibrinolysis, but elevated leukocyte adhesion and/or other indicators of endothelial dysfunction have been detected in older as compared to younger adult humans, in addition to rodents and primates those are non-human in nature (Brandes et al. 2005; Yamamoto et al. 2005; Ungvari et al. 2010).
Endothelial dysfunction happens during aging as shown in Fig. 3 (Rodríguez-Mañas et al. 2009; Faraci 2011; Martens and Seals 2016). While the damage of endothelial function increases with aging, the temporal pattern is contingent on the species and the definite vessel (Brown et al. 2007; Modrick et al. 2009; Rodríguez-Mañas et al. 2009). De Silva et al. (2018) found a slight change in endothelial role in carotid arteries at the ages considered, results dependable with earlier investigations that used mice at about the same age in non-transgenic mice (Didion et al. 2006; Modrick et al. 2012). In the carotid artery and aorta of control mice, endothelial dysfunction becomes a manifest as the animals rise even older (Brown et al. 2007; Fleenor et al. 2012). Considerable impairment of endothelium-dependent vasodilation existed in old E-V290M mice. All of the variations were specific for endothelium because responses to an endothelium-independent vasodilator were not suggestively changed in old E-V290M mice.

Aging triggers vascular endothelial dysfunction and causes a shifting of vasodilation, anticoagulation, antiproliferation and anti-inflammatory state to vasoconstriction, procoagulation, proliferation, and pro-inflammatory state
PPARγ Activation in the Vasculature
It was primarily supposed that PPARγ has a function within adipocytes, but it is now clear that PPARγ can be active in numerous cell types (Ketsawatsomkron et al. 2010). PPARγ is expressed in endothelium such as in the carotid artery (Marx et al. 1999; Ketsawatsomkron et al. 2010). In numerous portions of the circulation, systemic TZD treatment affects the vascular structure, as well as it has promising effects on vascular permeability and vasomotor tone in the disease models (Ketsawatsomkron et al. 2010; Faraci 2011). As per research findings, PPARγ activation has defensive properties on vascular cells (Ketsawatsomkron et al. 2010; Angulo et al. 2012). Patients with dominant undesirable mutations in the domain for ligand binding of PPARγ (i.e., V290M or P467L) show early onset of hypertension and type 2 diabetes (Barroso et al. 1999). Using mice expressing these same mutations provides a selective genetic tactic to study the influence of PPARγ, avoid off-target properties of TZDs, and gain insight into PPARγ-mediated potential driven by ligands that are endogenous. Mice expressing the murine equal to the P467L mutation in all cells share characteristics of human disease such as irregular fat distribution, variations in circulating metabolites, and increased blood pressure (Barroso et al. 1999; Tsai et al. 2004; Pendse et al. 2012). In contrast, upon targeting these genetic manipulations to ECs, there are no substantial variations in body weight, distribution of fat, arterial pressure, the glucose level in plasma, or cholesterol (Beyer et al. 2008; Pelham et al. 2013; Hu et al. 2016).
The contribution of PPARγ in aging has been suggested by a few studies. For example, a global reduction of its expression reduces the lifespan of mice (Argmann et al. 2009). Treating the mesenteric arteries with a PPARγ agonist enhanced endothelial function in aging (Angulo et al. 2012). Aging unmasked endothelial dysfunction in mice expressing the P465L mutation in PPARγ in all cells has been confirmed (Modrick et al. 2012). Such data make evidence for the idea that PPARγ defends the vasculature but not supported insight into the cell types or mechanisms which are involved. Concerning a study described a direct indication that vascular endothelium is a crucial cell type where PPARγ typically acts to prevent mechanisms that help oxidative stress and vascular aging (De Silva et al. 2018). Together, the results designate that the protective role of PPARγ in aging, being functionally significant in the absence of any pharmacological influence.
Oxidative Stress and Vascular Aging
Vascular aging is known as an outcome of ordinary fatigue and physical stress that might be interpreted for the main physical variations observed in aging such as stiffening (i.e., by transmission of stress to the more inflexible collagenous constituent of the arterial wall) and dilation (i.e., after rupturing of load-bearing material) (Xu et al. 2017). However, aging-connected variations on vascular functioning have been measured a set up for diseases of the cardiovascular system (CVS), but alterations on cardiac and central role could sluggish down or quicken this set point. Therefore, it is critical to know how aging and other pathophysiological conditions disturb the interaction between the arterial system and heart. In a study, Wojciechowska et al. (2012) stated on the variations with age in central and peripheral systolic blood pressure based on collected data from arbitrarily employed European and Chinese participant’s subsidiary a vicious circle between age-related arduousness, cardiovascular difficulties, and elevated systolic blood pressure. Chantler and Lakatta (2012) defined the perception of arterial-ventricular coupling and suggested crucial evidence on how aging, in the presence and absence of cardiovascular disease, disturbs the coupling both at resting and exercising conditions and its pathophysiological manifestations.
Vascular aging has been mainly connected to senescence of the vascular endothelium (El Assar et al. 2012). Cau et al. (2012) described that the influence of NOS isoform changes on aging-connected vascular irregularities, addressing the possible inhibition by few agents that modify the NOS expression/activity. In another study, Blanco and Bernabéu (2012) recapitulated data that supports the relationship between the splicing factor SRSF1 and endothelial cell senescence and recommend the availability of a common genetic program concerning alternate splicing of a cluster of genes, which donates to a senescent condition in the vessels. Nevertheless, aging-connected damage to the endothelium may not only be a consequence of the EC dysfunctioning but also as a consequence of impaired maintenance repair systems caused by endothelial progenitor cell. As stated by Williamson et al. (2012), a weakening of endogenous endothelial progenitor cell function in aging may terminate in a reduced capability for neovascularization and/or decreased re-endothelialization of vascular lesions, helping the progress, development, and clinical sequelae of disease in the CVS.
ROS and oxidative stress play a crucial role in age-dependent changes within the vasculature. For instance, both pharmacological and genetic methods designate that oxidative stress triggers damage of endothelium-mediated vasodilation, a crucial factor of vascular aging (Brown et al. 2007; Mayhan et al. 2008; Modrick et al. 2009; Faraci 2011). Indeed, interactions between nitric oxide (NO) and superoxide cause loss of NO-dependent signaling, which is the mechanistic source for this dysfunction (Didion et al. 2006; Eisenach et al. 2014; Toth et al. 2014).
Activation of phosphatidylinositol 3-kinase (PI3K)/Akt, PDGF, and extracellular signal-regulated kinase (ERK) 1/2 can be triggered by oxidative stress, which can result in enhanced transcription of PPARs as a protective mechanism. On the other hand, oxidized lipids can cause activation of PPARs and transcription. Moreover, 5′-AMP–activated protein kinase (AMPK) can be increased by oxidative stress which, in turn, can cause phosphorylation of PPAR proteins and further suppress the transcription of PPARs (Fig. 4) (Kim 2013). However, the activity against oxidative stress has been described following the systemic treatment with TZDs or genetic interference with PPARγ in all cells, but the cellular sites, which mediate these effects, have not been identified and are not easy to predict. For instance, genetic interference with PPARγ in smooth muscle has profound effects on vascular function without any apparent effect by oxidative stress (Pelham et al. 2013).

Oxidative stress triggers copious signaling pathways that affect PPARγ transcription. ERK1/2, Extracellular signal-regulated kinase 1/2; PDGF, Platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; Akt, Protein kinase B; AMPK, AMP-activated protein kinase
NADPH oxidase is a major source of superoxide in vascular cells including endothelium (Park et al. 2007; Rodríguez-Mañas et al. 2009; Faraci 2011). According to the various evidence on cell lines, this enzyme complex acts a crucial role in vascular anomalies in models focusing on aging and in arteries of elderly people (Park et al. 2007; Mayhan et al. 2008; Rodríguez-Mañas et al. 2009). In a study, a superoxide scavenger or an NADPH oxidase inhibitor intensely returned endothelial activity to typical levels in old E-V290M mice that supports oxidative stress as a vital contributor to this vascular anomaly (De Silva et al. 2018). Besides the oxidative stress, additional processes can possibly interrelate with ROS and thus subsidize the changes with aging (Faraci 2011). While initiation of a cyclooxygenase (COX)-mediated mechanism is one such progression (Faraci 2011), the study of De Silva et al. (2018) did not provide such a prospect in old E-V290M mice. These results in mice are dependable with data found in aged humans where COX inhibition did not affect damaged endothelium (Eisenach et al. 2014). Rather it appears that the NO-component of the vasodilator is the crucial mechanism affected (Eisenach et al. 2014).
ROCK and Vascular Aging
Aging has been considered as the utmost risk aspect for vascular disease and consequent clinical difficulties (Rothwell et al. 2005; Wang et al. 2014a, b) but comparatively little is known about the vascular aging biology (Faraci 2011; Rodríguez-Mañas et al. 2009; Wang et al. 2014a, b). Endothelial dysfunction is an autonomous interpreter of vascular events in investigational models and humans. It has an essential role in the initiation and progression of vascular disease (Emdin et al. 2018; Faraci 2011; Martens and Seals 2016; Wang et al. 2014a, b). However, the loss of endothelial health is serious in relation to vascular disease, but investigations using endothelial specific models or manipulation along with aging are rare. In the models utilizing human and animals, (Rothwell et al. 2005) low-grade inflammation and oxidative stress are common features of vascular disease throughout aging (Park et al. 2007; Mayhan et al. 2008; Rodríguez-Mañas et al. 2009; Wang et al. 2014b; Martens and Seals 2016).
Rho-kinase (ROCK) is a serine/threonine kinase and effector of RhoA, a small GTPase triggered by different stimuli including ROS (Sawada and Liao 2014). Both ROCK with RhoA exerts significant activity in the vasculature, but their influence is cell-specific and differs regionally (Sawada and Liao 2014). ROCK is the main basis of vascular tone in vascular muscle due to the properties of calcium sensitivity and the directing myosin light chain (Sawada and Liao 2014). The different interaction between ROCK and eNOS/NO-mediated signaling has been defined (Sawada and Liao 2014). ROCK prevents eNOS expression and activity (i.e., via possessions on mRNA stability and phosphorylation of protein) in ECs, but similarly, Y27632 hinders ROCK isoforms efficiently (De Silva et al. 2016).
Rao et al. (2017) described that in glomerular endothelial cells, RhoA/ROCK-dependent pathway improves inflammatory infiltration and adhesion caused by advanced glycation end products (AGEs). According to the study, AGEs elevated MCP-1 and ICAM-1 through the RhoA/ROCK pathway in the ECs of the glomerulus. On the other hand, RhoA/ROCK pathway inhibition ameliorated albuminuria, urinary albumin-creatinine ratio, and enhanced adhesion and inflammatory cell infiltration in the glomerulus.
As part of the effort to assess mechanisms in old E-V290M mice, De Silva et al. (2018) assessed the role of ROCK. It has been found that endothelial dysfunction in old E-V290M mice was inverted by this inhibitor. Though the data recommend the idea that ROCK has a significant contribution to vascular aging, it does not show which isoform was involved. However, recent experiments recommend that ROCK-2 has a role in endothelial dysfunction caused by Ang II (De Silva et al. 2016). As per De Silva et al. (2018), PPARγ in ECs generally prevents the activation of RhoA/ROCK, thus supporting the endothelium-dependent vasodilation. PPARγ-induced chronic inhibition of ROCK is possibly important because the action of ROCK is definitely connected to cardiovascular actions including stroke (Kajikawa et al. 2014).
Endothelial PPARγ in Vascular Protection
Vascular protection is the vascular endothelial growth factor (VEGF)–triggered endothelial function improvement that facilitates the prevention of proliferation of vascular SMC, enhancement of endothelial cell survival, prevention of thrombosis, and effects against inflammation. By inducing VEGF, the syntheses of NO and prostacyclin are both possible to be crucial mediators of VEGF-mediated vascular defense (Zachary et al. 2000).
In order to attain insight into the cell-specific effect of PPARγ, De Silva et al. (2018) addressed the proposition that genetic interference with endothelial PPARγ would augment age-induced vascular dysfunction. In their study, they used carotid arteries from both adult and old mice with endothelial-specific expression of a human dominant negative mutation in PPARγ regulated by the vascular cadherin promoter (i.e., E-V290M), accompanied by age-matched, nontransgenic littermates. Endothelial function in old E-V290M mice was not changed by a COX inhibitor, but it was restored to typical function by a superoxide scavenger, an inhibitor of NADPH oxidase, or inhibiting the ROCK. Vascular expression of IL-6, Nox-2, and CDKN2A (i.e., a marker of senescence) was elevated in old E-V290M mice compared with controls. The study concluded that age-related vascular dysfunction, inflammation, and senescence are enhanced following the interference with endothelial PPARγ through the mechanisms concerning oxidative stress and ROCK.
Mukohda et al. (2016) stated that endothelial PPAR-γ exerts vascular defense against oxidative stress by IL-1β induction in transgenic mice with endothelium-specific overexpression of either dominant negative PPAR-γ (i.e., E-V290M) or wild-type. In the aorta of NF-κB-LUC transgenic mice, IL-1β improved luciferase activity and phospho-p65, but dose-dependently reduced IκB-α. In addition, IL-1β decreased endothelial-dependent relaxation by acetylcholine (ACh) in a dose-dependent manner. The sensitivity was partly improved by pretreatment with the PPARγ agonist, rosiglitazone or in either wild-type. On the other hand, in the aorta of E-V290M mice, IL-1β–induced endothelial dysfunction was deteriorated. However, IL-1β elevated the expressions of NF-κB target genes. However, endothelial dysfunction caused by IL-1β was not treated by the inhibitor NF-κB p65. A superoxide dismutase (SOD) mimetic, tempol, partly restored ACh receptiveness in the IL-1β–treated aorta. Particularly, tempol only modestly enhanced protection in the E-WT aorta. Nonetheless, tempol had an improved defensive effect as compared to the aorta from nontransgenic mice in the E-V290M aorta. These aforementioned findings signifying that PPAR-γ–mediated defense may be due to the involvement of antioxidant effects. Therefore, IL-1β–induced ROS and reduced the ratio between phospho-endothelial nitric oxide synthase (i.e., Ser1177) and eNOS in the nontransgenic aorta. In the aorta with E-V290M, these activities were totally eliminated in the aorta with endothelial overexpression of WT PPAR-γ and were found deteriorated even in the absence of IL-1β. Regarding the study conclusion, PPAR-γ defends against IL-1β–mediated endothelial dysfunction via decreasing the oxidative stress signaling.
Vascular thrombosis is known as a major impediment of diseases of the CVS and is strongly accompanying with risk influences including hypertension, metabolic syndrome, atherosclerosis, and diabetes that are identified to be moderated by PPARγ (Roach et al. 2013). However, it is not well understood how PPARγ affects thrombotic vulnerability. Following the cultured ECs studies, it was found that the activation of PPARγ may defend against thrombosis by decreasing the pro-inflammatory cell adhesion molecule expression, suppressing the initiation of the transcription factor, NF-κB, and elevated production of endothelial NO (Jackson et al. 1999; Wang et al. 2002; Calnek et al. 2003; Sasaki et al. 2005; Ricote and Glass 2007). In contrast, agonists for PPARγ have been known to excite an elevated synthesis of procoagulant microparticles from macrophages and monocytes (Neri et al. 2012). In addition, findings suggest that may help to elucidate the paradoxical elevate in risk of myocardial infarction observed in individuals with diabetes managed by certain TZDs (Nissen and Wolski 2007). Therefore, via activities on numerous target cells, PPARγ agonists might show contrasting properties on thrombotic vulnerability. Evidently, a good understanding regarding the tissue-specific activities of PPARγ in modulating antithrombotic ability is anticipated (Duan et al. 2008).
A study of Jin et al. (2015) employing a transgenic mouse model expressing a PPARγ mutant (i.e., E-V290M) selectively in endothelium, endothelial PPARγ defends against vascular thrombosis. In E-V290M mice, the time required for occlusive thrombosis of the carotid artery was reduced as compared to the non-transgenic littermates after either chemical-induced injury or photochemical injury. In aortic endothelial cells in E-V290M mice, gene set enrichment analysis reported a significant elevation of NF-κB target genes, such as P-selectin. Furthermore, P-selectin in plasma and carotid artery mRNA was significantly raised in E-V290M mice. In E-V290M mice, P-selectin–dependent leukocytes rolling on mesenteric venules were elevated as compared to non-transgenic mice. The reduced time to arterial occlusion in mice (E-V290M) was upturned by treatment of P-selectin preventing antibodies or neutrophil-depleting antibodies prior to injury caused by photochemical. Endothelial PPARγ defends against thrombosis which comprises downregulation of expression of P-selectin and lessened P-selectin–dependent leukocyte-endothelial communications.
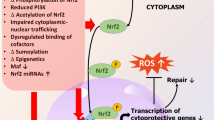
The close relationship between aging and/or inflammation and redox-regulated transcription factors has been revealed in various studies (Kim et al. 2002), but the properties of age on the changes in PPARs are yet to be explored. In order to clarify the changes in PPARs throughout aging and their variation at the molecular level, Sung et al. (2004) examined how age and calorie restriction affect PPARs in kidneys attained from Fischer 344 rats. Following the study, it was found that a nuclear protein, the mRNA level, and DNA binding action of PPARs decrease in an age-dependent manner, whereas calorie restriction blunted the reduction. In addition, the results were confirmed in distinct trails in which rats were injected with LPS, with the outcome of enhanced vulnerability to inflammation (Sung et al. 2004). According to these aforementioned findings, the investigators decided that the changed expression of PPARs perhaps due to an elevation of oxidative stress with age and calorie restriction averts these reduce through its action against oxidative stress.
Conclusion
Age-induced endothelial dysfunction is considered to be mediated by the inflammatory and oxidative-dependent mechanisms. PPARγ plays crucial roles in all phases of inflammation. During aging, suppression of the increased inflammatory processes is considered to take place partly because of the prevention of the age-related decline in PPARs. Following the intervention with endothelial PPARγ, age-related senescence, inflammation, and vascular dysfunction were attenuated. Interestingly, systemic inflammation is suppressed by the activation of PPARγ in healthy individuals. Furthermore, endothelial PPARγ has implications for therapeutic approaches for vascular aging, as well as vascular pathophysiology. Molecular insights into the calorie restriction’s capability to prevent decreases in PPARs and the modulation of PPARs throughout aging is obligatory to provide a better understanding of the impact of PPARγ in the mechanisms underlying anti-inflammatory action of calorie restriction and the role in the aging process.
Change history
27 May 2019
The original article unfortunately contains minor errors in Figs. 1 and 2. The correct Fig. 1 is shown below. The correct Fig. 2 is shown below.
Abbreviations
- PPARγ:
-
peroxisome proliferator-activated receptor gamma
- ROCK:
-
rho-kinase
- ROS:
-
reactive oxygen species
- eNOS:
-
endothelial nitric oxide synthase
- PUFAs:
-
polyunsaturated fatty acids
- TZDs:
-
thiazolidinediones
- IBD:
-
inflammatory bowel disease.
References
Adachi M, Kurotani R, Morimura K et al (2006) Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut 55(8):1104–1113. https://doi.org/10.1136/gut.2005.081745
Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, Rajanayagam O, Semple R, Luan J', Bath L, Zalin A, Labib M, Kumar S, Simpson H, Blom D, Marais D, Schwabe J, Barroso I, Trembath R, Wareham N, Nagy L, Gurnell M, O'Rahilly S, Chatterjee K (2006) Non-DNA binding, dominant-negative, human PPARγ mutations cause lipodystrophic insulin resistance. Cell Metab 4(4):303–311. https://doi.org/10.1016/j.cmet.2006.09.003
Akahori T, Sho M, Hamada K, Suzaki Y, Kuzumoto Y, Nomi T, Nakamura S, Enomoto K, Kanehiro H, Nakajima Y (2007) Importance of peroxisome proliferator-activated receptor-γ in hepatic ischemia/reperfusion injury in mice. J Hepatol 47(6):784–792. https://doi.org/10.1016/j.jhep.2007.07.030
Akbiyik F, Ray DM, Gettings KF, Blumberg N, Francis CW, Phipps RP (2004) Human bone marrow megakaryocytes and platelets express PPARγ, and PPARγ agonists blunt platelet release of CD40 ligand and thromboxanes. Blood 104(5):1361–1368. https://doi.org/10.1182/blood-2004-03-0926
Allred CD, Talbert DR, Southard RC, Wang X, Kilgore MW (2008) PPARgamma1 as a molecular target of eicosapentaenoic acid in human colon cancer (HT-29) cells. J Nutr 138(2):250–256
Angulo J, Vallejo S, El Assar M et al (2012) Age-related differences in the effects of α and γ peroxisome proliferator-activated receptor subtype agonists on endothelial vasodilation in human microvessels. Exp Gerontol 47(9):734–740. https://doi.org/10.1016/j.exger.2012.06.014
Annese V, Rogai F, Settesoldi A, Bagnoli S (2012) PPAR γ in inflammatory bowel disease. PPAR Res 2012:1–9
Aprahamian T, Bonegio RG, Richez C, Yasuda K, Chiang LK, Sato K, Walsh K, Rifkin IR (2009) The peroxisome proliferator-activated receptor gamma agonist rosiglitazone ameliorates murine lupus by induction of adiponectin. J Immunol 182(1):340–346
Argmann C, Dobrin R, Heikkinen S, Auburtin A, Pouilly L, Cock TA, Koutnikova H, Zhu J, Schadt EE, Auwerx J (2009) Pparγ2 is a key driver of longevity in the mouse. PLoS Genet 5(12):e1000752. https://doi.org/10.1371/journal.pgen.1000752
Asada K, Sasaki S, Suda T, Chida K, Nakamura H (2004) Antiinflammatory roles of peroxisome proliferator–activated receptor γ in human alveolar macrophages. Am J Respir Crit Care Med 169:195–200
Barroso I, Gurnell M, Crowley VE et al (1999) Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402(6764):880–883. https://doi.org/10.1038/47254
Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui Y, Hennighausen L, Gonzalez F, Rohrer J, Benninghoff AU, Hontecillas R (2004) Activation of PPAR γ and δ by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 127(3):777–791. https://doi.org/10.1053/j.gastro.2004.06.049
Belmin J, Bernard C, Corman B, Merval R, Esposito B, Tedgui A (1995) Increased production of tumor necrosis factor and interleukin-6 by arterial wall of aged rats. Am J Physiol Circ Physiol 268:H2288–H2293. https://doi.org/10.1152/ajpheart.1995.268.6.H2288
Beyer AM, De Lange WJ, Halabi CM et al (2008) Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res 103(6):654–661. https://doi.org/10.1161/CIRCRESAHA.108.176339
Blanco FJ, Bernabéu C (2012) The splicing factor SRSF1 as a marker for endothelial senescence. Front Physiol 3(54):1–6
Bloomfield SF, Stanwell-Smith R, Crevel RWR, Pickup J (2006) Too clean, or not too clean: the hygiene hypothesis and home hygiene. Clin Exp Allergy 36(4):402–425
Brandes RP, Fleming I, Busse R (2005) Endothelial aging. Cardiovasc Res 66(2):286–294
Brown KA, Didion SP, Andresen JJ, Faraci FM (2007) Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol 27(9):1941–1946. https://doi.org/10.1161/ATVBAHA.107.146852
Brunmeir R, Xu F (2018) Functional regulation of PPARs through post-translational modifications. Int J Mol Sci 19(6).
Calnek DS, Mazzella L, Roser S, Roman J, Hart CM (2003) Peroxisome proliferator-activated receptor gamma ligands increase release of nitric oxide from endothelial cells. Arterioscler Thromb Vasc Biol 23(1):52–57. https://doi.org/10.1161/01.ATV.0000044461.01844.C9
Camp HS, Tafuri SR (1997) Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem 272(16):10811–10816. https://doi.org/10.1074/JBC.272.16.10811
Carter AB, Misyak SA, Hontecillas R, Bassaganya-Riera J (2009) Dietary modulation of inflammation-induced colorectal cancer through PPARγ. PPAR Res 2009:498352–498359. https://doi.org/10.1155/2009/498352
Cau SBA, Carneiro FS, Tostes RC (2012) Differential modulation of nitric oxide synthases in aging: therapeutic opportunities. Front Physiol 3(218):1–11. https://doi.org/10.3389/fphys.2012.00218
Caygill CP, Hill MJ (1995) Fish, n-3 fatty acids and human colorectal and breast cancer mortality. Eur J Cancer Prev 4(4):329–332
Celinski K, Dworzanski T, Korolczuk A, Piasecki R, Slomka M, Madro A, Fornal R (2011) Effects of peroxisome proliferator-activated receptors-gamma ligands on dextran sodium sulphate-induced colitis in rats. J Physiol Pharmacol 62(3):347–356
Chantler PD, Lakatta EG (2012) Arterial-ventricular coupling with aging and disease. Front Physiol 3(90):1–12. https://doi.org/10.3389/fphys.2012.00090
Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P (2001) A PPARγ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 7(1):161–171. https://doi.org/10.1016/S1097-2765(01)00164-2
Chinetti G, Fruchart J-C, Staels B (2003) Peroxisome proliferator-activated receptors and inflammation: from basic science to clinical applications. Int J Obes Relat Metab Disord 3:S41–S45. https://doi.org/10.1038/sj.ijo.0802499
Chiu J-J, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91(1):327–387. https://doi.org/10.1152/physrev.00047.2009
Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M, Griffin PR, Spiegelman BM (2010) Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466(7305):451–456. https://doi.org/10.1038/nature09291
Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS (2000) Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem 275(42):32681–32687. https://doi.org/10.1074/jbc.M002577200\rM002577200 [pii]
Collino M, Aragno M, Castiglia S, Miglio G, Tomasinelli C, Boccuzzi G, Thiemermann C, Fantozzi R (2010) Pioglitazone improves lipid and insulin levels in overweight rats on a high cholesterol and fructose diet by decreasing hepatic inflammation. Br J Pharmacol 160(8):1892–1902. https://doi.org/10.1111/j.1476-5381.2010.00671.x
Collins AR, Meehan WP, Kintscher U, Jackson S, Wakino S, Noh G, Palinski W, Hsueh WA, Law RE (2001) Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 21(3):365–371. https://doi.org/10.1161/01.ATV.21.3.365
Corona JC, Duchen MR (2016) PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med 100:153–163. https://doi.org/10.1016/j.freeradbiomed.2016.06.023
Coste A, Lagane C, Filipe C, Authier H, Gales A, Bernad J, Douin-Echinard V, Lepert JC, Balard P, Linas MD, Arnal JF, Auwerx J, Pipy B (2008) IL-13 attenuates gastrointestinal candidiasis in normal and immunodeficient RAG-2(−/−) mice via peroxisome proliferator-activated receptor-gamma activation. J Immunol 180(7):4939–4947. https://doi.org/10.4049/jimmunol.180.7.4939
Croasdell A, Duffney PF, Kim N et al (2015) PPAR γ and the innate immune system mediate the resolution of inflammation. PPAR Res 2015:549691
Csiszar A (2009) Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Front Biosci 14:3128–3144. https://doi.org/10.2741/3440
Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Patel NSA, Paola RD, Genovese T, Chatterjee PK, Rosa MD, Caputi AP, Thiemermann C (2003) Rosiglitazone and 15-deoxy-Δ 12,14-prostaglandin J 2, ligands of the peroxisome proliferator-activated receptor-γ (PPAR-γ), reduce ischaemia/reperfusion injury of the gut. Br J Pharmacol 140(2):366–376. https://doi.org/10.1038/sj.bjp.0705419
D’Elia RV, Harrison K, Oyston PC et al (2013) Targeting the “cytokine storm” for therapeutic benefit. Clin Vaccine Immunol 20(3):319–327
De Silva TM, Kinzenbaw DA, Modrick ML et al (2016) Heterogeneous impact of ROCK2 on carotid and cerebrovascular function. Hypertension 68(3):809–817. https://doi.org/10.1161/HYPERTENSIONAHA.116.07430
De Silva TM, Li Y, Kinzenbaw DA et al (2018) Endothelial PPARγ (peroxisome proliferator–activated receptor-γ) is essential for preventing endothelial dysfunction with aging. Hypertension 72(1):227–234. https://doi.org/10.1161/HYPERTENSIONAHA.117.10799
Deeb SS, Fajas L, Nemoto M, Pihlajamäki J, Mykkänen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J (1998) A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet 20(3):284–287. https://doi.org/10.1038/3099
Delerive P, De Bosscher K, Besnard S et al (1999) Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem 274(45):32048–32054. https://doi.org/10.1074/jbc.274.45.32048
Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, Leibowitz MD, Colombel JF, Auwerx J (2001) Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J Exp Med 193(7):827–838. https://doi.org/10.1084/jem.193.7.827
Didion SP, Kinzenbaw DA, Schrader LI, Faraci FM (2006) Heterozygous CuZn superoxide dismutase deficiency produces a vascular phenotype with aging. Hypertension 48(6):1072–1079. https://doi.org/10.1161/01.HYP.0000247302.20559.3a
Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR (2008) Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 7(6):805–812. https://doi.org/10.1111/j.1474-9726.2008.00438.x
Duan SZ, Usher MG, Mortensen RM (2008) Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res 102(3):283–294. https://doi.org/10.1161/CIRCRESAHA.107.164384
Eisenach JH, Gullixson LR, Allen AR, Kost SL, Nicholson WT (2014) Cyclo-oxygenase-2 inhibition and endothelium-dependent vasodilation in younger vs. older healthy adults. Br J Clin Pharmacol 78(4):815–823. https://doi.org/10.1111/bcp.12397
El Assar M, Angulo J, Vallejo S et al (2012) Mechanisms involved in the aging-induced vascular dysfunction. Front Physiol 3(132):1–13. https://doi.org/10.3389/fphys.2012.00132
Emdin CA, Khera AV, Klarin D, Natarajan P, Zekavat SM, Nomura A, Haas M, Aragam K, Ardissino D, Wilson JG, Schunkert H, McPherson R, Watkins H, Elosua R, Bown MJ, Samani NJ, Baber U, Erdmann J, Gormley P, Palotie A, Stitziel NO, Gupta N, Danesh J, Saleheen D, Gabriel S, Kathiresan S (2018) Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 137(3):222–232. https://doi.org/10.1161/CIRCULATIONAHA.117.028021
Faraci FM (2011) Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol 300(5):H1566–H1582. https://doi.org/10.1152/ajpheart.01310.2010
Faveeuw C, Fougeray S, Angeli V, Fontaine J, Chinetti G, Gosset P, Delerive P, Maliszewski C, Capron M, Staels B, Moser M, Trottein F (2000) Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-12 production in murine dendritic cells. FEBS Lett 486(3):261–266
Ferrero-Miliani L, Nielsen OH, Andersen PS, Girardin SE (2007) Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1β generation. Clin Exp Immunol 147(2):227–235. https://doi.org/10.1111/j.1365-2249.2006.03261.x
Fleenor BS, Seals DR, Zigler ML, Sindler AL (2012) Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell 11(2):269–276. https://doi.org/10.1111/j.1474-9726.2011.00783.x
Gao M, Jiang Y, Xiao X, Peng Y, Xiao X, Yang M (2015) Protective effect of pioglitazone on sepsis-induced intestinal injury in a rodent model. J Surg Res 195(2):550–558. https://doi.org/10.1016/j.jss.2015.02.007
Genovese T, Esposito E, Mazzon E, di Paola R, Muià C, Meli R, Bramanti P, Cuzzocrea S (2008) Effect of cyclopentanone prostaglandin 15-deoxy-delta12,14PGJ2 on early functional recovery from experimental spinal cord injury. Shock 30(2):142–152. https://doi.org/10.1097/SHK.0b013e31815dd381
Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14(2):121–141
Goh K, Xiao S-D (2009) Inflammatory bowel disease: a survey of the epidemiology in Asia. J Dig Dis 10(1):1–6. https://doi.org/10.1111/j.1751-2980.2008.00355.x
Gosset P, Charbonnier AS, Delerive P et al (2001) Peroxisome proliferator-activated receptor gamma activators affect the maturation of human monocyte-derived dendritic cells. Eur J Immunol 31(10):2857–2865. https://doi.org/10.1002/1521-4141(2001010)31:10<2857::AID-IMMU2857>3.0.CO;2-X
Hevener AL, Olefsky JM, Reichart D, Nguyen MTA, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S, Gonzalez FJ, Glass CK, Ricote M (2007) Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest 117(6):1658–1669. https://doi.org/10.1172/JCI31561
Hodis HN, Mack WJ, Zheng L, Li Y, Torres M, Sevilla D, Stewart Y, Hollen B, Garcia K, Alaupovic P, Buchanan TA (2006) Effect of peroxisome proliferator-activated receptor gamma agonist treatment on subclinical atherosclerosis in patients with insulin-requiring type 2 diabetes. Diabetes Care 29(7):1545–1553. https://doi.org/10.2337/dc05-2462
Hontecillas R, Bassaganya-Riera J (2007) Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol 178(5):2940–2949. https://doi.org/10.4049/JIMMUNOL.178.5.2940
Hossain MS, Uddin MS, Asaduzzaman M et al (2016) Inquiry of analgesic and anti-inflammatory activities of Xanthosoma sagittifolium L.: an effective medicinal plant. J Coast Lif Med 5(1):22–26. https://doi.org/10.12980/jclm.5.2017J6-229
Hu E, Kim JB, Sarraf P, Spiegelman BM (1996) Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274(5295):2100–2103. https://doi.org/10.1126/science.274.5295.2100
Hu C, Lu K-T, Mukohda M, Davis DR, Faraci FM, Sigmund CD (2016) Interference with PPARγ in endothelium accelerates angiotensin II-induced endothelial dysfunction. Physiol Genomics 48(2):124–134. https://doi.org/10.1152/physiolgenomics.00087.2015
Huang FR, Zhan ZP, Luo J, Jiang SW, Peng J (2008) Duration of feeding linseed diet influences peroxisome proliferator-activated receptor γ and tumor necrosis factor gene expression, and muscle mass of growing-finishing barrows. Livest Sci 119(1–3):194–201. https://doi.org/10.1016/j.livsci.2008.04.003
Hyong A, Jadhav V, Lee S, Tong W, Rowe J, Zhang JH, Tang J (2008) Rosiglitazone, a PPAR gamma agonist, attenuates inflammation after surgical brain injury in rodents. Brain Res 1215:218–224. https://doi.org/10.1016/j.brainres.2008.04.025
Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, Nagy L, Yamamoto K, Schwabe JWR (2008) Structural basis for the activation of PPARγ by oxidized fatty acids. Nat Struct Mol Biol 15(9):924–931. https://doi.org/10.1038/nsmb.1474
Itzkowitz SH, Yio X (2004) Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 287(1):G7–17. doi: https://doi.org/10.1152/ajpgi.00079.2004
Jackson SM, Parhami F, Xi XP, Berliner JA, Hsueh WA, Law RE, Demer LL (1999) Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler Thromb. Vasc Biol 19(9):2094–2104
Jacobsen BA, Fallingborg J, Rasmussen HH et al (2006) Increase in incidence and prevalence of inflammatory bowel disease in northern Denmark: a population-based study, 1978-2002. Eur J Gastroenterol Hepatol 18(6):601–606. https://doi.org/10.1097/00042737-200606000-00005
Jaudszus A, Gruen M, Watzl B, Ness C, Roth A, Lochner A, Barz D, Gabriel H, Rothe M, Jahreis G (2013) Evaluation of suppressive and pro-resolving effects of EPA and DHA in human primary monocytes and T-helper cells. J Lipid Res 54(4):923–935. https://doi.org/10.1194/jlr.P031260
Jiang C, Ting AT, Seed B (1998) PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 391(6662):82–86. https://doi.org/10.1038/34184
Jin H, Gebska MA, Blokhin IO, Wilson KM, Ketsawatsomkron P, Chauhan AK, Keen HL, Sigmund CD, Lentz SR (2015) Endothelial PPAR- γ protects against vascular thrombosis by downregulating P-selectin expression. Arterioscler Thromb Vasc Biol 35(4):838–844. https://doi.org/10.1161/ATVBAHA.115.305378
Kajikawa M, Noma K, Maruhashi T, Mikami S, Iwamoto Y, Iwamoto A, Matsumoto T, Hidaka T, Kihara Y, Chayama K, Nakashima A, Goto C, Liao JK, Higashi Y (2014) Rho-associated kinase activity is a predictor of cardiovascular outcomes. Hypertension 63(4):856–864. https://doi.org/10.1161/HYPERTENSIONAHA.113.02296
Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N (2009) PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-beta. Lab Investig 89:47–58. https://doi.org/10.1038/labinvest.2008.156
Ketsawatsomkron P, Pelham CJ, Groh S, Keen HL, Faraci FM, Sigmund CD (2010) Does peroxisome proliferator-activated receptor-γ(PPARγ) protect from hypertension directly through effects in the vasculature? J Biol Chem 285(13):9311–9316
Kim T (2013) Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J Cardiol 5(6):164–174. https://doi.org/10.4330/wjc.v5.i6.164
Kim HJ, Jung KJ, Yu BP, Cho CG, Choi JS, Chung HY (2002) Modulation of redox-sensitive transcription factors by calorie restriction during aging. Mech Ageing Dev 123(12):1589–1595. https://doi.org/10.1016/S0047-6374(02)00094-5
Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, Alferink J, Novak N, Beyer M, Mayer G, Langhans B, Klockgether T, Waisman A, Eberl G, Schultze J, Famulok M, Kolanus W, Glass C, Kurts C, Knolle PA (2009a) The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell–intrinsic fashion and suppresses CNS autoimmunity. J Exp Med 206(10):2079–2089. https://doi.org/10.1084/jem.20082771
Klotz L, Schmidt S, Heun R, Klockgether T, Kölsch H (2009b) Association of the PPARgamma gene polymorphism Pro12Ala with delayed onset of multiple sclerosis. Neurosci Lett 449(1):81–83. https://doi.org/10.1016/j.neulet.2008.10.066
Konkel L (2016) Inflammatory bowel disease in Asia: a second chance at uncovering environmental factors. Environ Health Perspect 124:A49–A54. https://doi.org/10.1289/ehp.124-A49
Kulkarni AA, Woeller CF, Thatcher TH et al (2012) Emerging PPARγ-independent role of PPARγ ligands in lung diseases. PPAR Res 2012:705352
Lesniewski LA, Durrant JR, Connell ML, Henson GD, Black AD, Donato AJ, Seals DR (2011) Aerobic exercise reverses arterial inflammation with aging in mice. AJP Hear Circ Physiol 301(3):H1025–H1032. https://doi.org/10.1152/ajpheart.01276.2010
Lewis JD, Lichtenstein GR, Deren JJ, Sands BE, Hanauer SB, Katz JA, Lashner B, Present DH, Chuai S, Ellenberg JH, Nessel L, Wu GD, Rosiglitazone for Ulcerative Colitis Study Group (2008) Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology 134(3):688–695. https://doi.org/10.1053/j.gastro.2007.12.012
Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK (2000a) Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest 106(4):523–531. https://doi.org/10.1172/JCI10370
Li M, Pascual G, Glass CK (2000b) Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol 20(13):4699–4707. https://doi.org/10.1128/MCB.20.13.4699-4707.2000.Updated
Liang HL, Ouyang Q (2008) A clinical trial of combined use of rosiglitazone and 5-aminosalicylate for ulcerative colitis. World J Gastroenterol 14(1):114–119. https://doi.org/10.3748/wjg.14.114
Marathe C, Bradley MN, Hong C, Chao L, Wilpitz D, Salazar J, Tontonoz P (2009) Preserved glucose tolerance in high-fat-fed C57BL/6 mice transplanted with PPARgamma−/−, PPARdelta−/−, PPARgammadelta−/−, or LXRalphabeta−/− bone marrow. J Lipid Res 50(2):214–224. https://doi.org/10.1194/jlr.M800189-JLR200
Marcu KB, Otero M, Olivotto E et al (2010) NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets 11(5):599–613. https://doi.org/10.1002/rcm.1690
Marion-Letellier R, Butler M, Déchelotte P, Playford RJ, Ghosh S (2008) Comparison of cytokine modulation by natural peroxisome proliferator–activated receptor γ ligands with synthetic ligands in intestinal-like Caco-2 cells and human dendritic cells—potential for dietary modulation of peroxisome proliferator–activated receptor. Am J Clin Nutr 87:939–948
Martens CR, Seals DR (2016) Practical alternatives to chronic caloric restriction for optimizing vascular function with ageing. J Physiol 594(24):7177–7195
Martin H (2010) Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat Res 690(1–2):57–63
Marx N, Bourcier T, Sukhova GK et al (1999) PPARgamma activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARgamma as a potential mediator in vascular disease. Arterioscler Thromb Vasc Biol 19(3):546–551. https://doi.org/10.1038/bjc.2011.434
Mayhan WG, Arrick DM, Sharpe GM, Sun H (2008) Age-related alterations in reactivity of cerebral arterioles: role of oxidative stress. Microcirculation 15(3):225–236. https://doi.org/10.1080/10739680701641421
Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D’Agostino RB, Perez A, Provost JC, Haffner SM (2006) Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. JAMA 296(21):2572–2581. https://doi.org/10.1001/jama.296.21.joc60158
Mistriotis P, Andreadis ST (2017) Vascular aging: molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res Rev 37:94–116
Modrick ML, Didion SP, Sigmund CD, Faraci FM (2009) Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol Heart Circ Physiol 296(6):H1914–H1919. https://doi.org/10.1152/ajpheart.00300.2009
Modrick ML, Kinzenbaw DA, Chu Y, Sigmund CD, Faraci FM (2012) Peroxisome proliferator-activated receptor-γ protects against vascular aging. Am J Phys Regul Integr Comp Phys 302(10):R1184–R1190. https://doi.org/10.1152/ajpregu.00557.2011
Mukohda M, Stump M, Ketsawatsomkron P, Hu C, Quelle FW, Sigmund CD (2016) Endothelial PPAR-γ provides vascular protection from IL-1β-induced oxidative stress. Am J Physiol Heart Circ Physiol 310(1):H39–H48. https://doi.org/10.1152/ajpheart.00490.2015
Natarajan C, Bright JJ (2002) Peroxisome proliferator-activated receptor-gamma agonist inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun 3(2):59–70. https://doi.org/10.1038/sj.gene.6363832
Natarajan C, Muthian G, Barak Y et al (2003) Peroxisome proliferator-activated receptor-gamma-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J Immunol 171(11):5743–5750
Neri T, Cordazzo C, Carmazzi Y, Petrini S, Balìa C, Stefanelli F, Amoruso A, Brunelleschi S, Breschi MC, Pedrinelli R, Paggiaro P, Celi A (2012) Effects of peroxisome proliferator-activated receptor-γ agonists on the generation of microparticles by monocytes/macrophages. Cardiovasc Res 94(3):537–544. https://doi.org/10.1093/cvr/cvs125
Ng SC (2016) Emerging trends of inflammatory bowel disease in Asia. Gastroenterol Hepatol (N Y) 12:193–196
Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoé K (2001) Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol 116(1):40–48
Nissen SE, Wolski K (2007) Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356:2457–2471. https://doi.org/10.1056/NEJMoa072761
Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, Jure H, de Larochellière R, Staniloae CS, Mavromatis K, Saw J, Hu B, Lincoff AM, Tuzcu EM, PERISCOPE Investigators (2008) Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA 299(13):1561–1573. https://doi.org/10.1001/jama.299.13.1561
Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Eagle AR, Vats D, Brombacher F, Ferrante AW, Chawla A (2007) Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447(7148):1116–1120. https://doi.org/10.1038/nature05894
Oeseburg H, De Boer RA, Buikema H et al (2010) Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase a. Arterioscler Thromb Vasc Biol 30(7):1407–1414. https://doi.org/10.1161/ATVBAHA.110.206425
Oh S-H, Park S-M, Lee YH, Cha JY, Lee JY, Shin EK, Park JS, Park BL, Shin HD, Park CS (2009) Association of peroxisome proliferator-activated receptor-gamma gene polymorphisms with the development of asthma. Respir Med 103:1020–1024. https://doi.org/10.1016/j.rmed.2009.01.015
Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB (2010) Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol 30(11):2301–2309. https://doi.org/10.1161/ATVBAHA.110.209460
Padilla J, Leung E, Phipps RP (2002) Human B lymphocytes and B lymphomas express PPAR-γ and are killed by PPAR-γ agonists. Clin Immunol 103(1):22–33. https://doi.org/10.1006/clim.2001.5181
Park L, Anrather J, Girouard H, Zhou P, Iadecola C (2007) Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab 27(12):1908–1918. https://doi.org/10.1038/sj.jcbfm.9600491
Parnell GP, Booth DR (2017) The multiple sclerosis (MS) genetic risk factors indicate both acquired and innate immune cell subsets contribute to MS pathogenesis and identify novel therapeutic opportunities. Front Immunol 8(425):1–6
Pascual G, Sullivan AL, Ogawa S et al (2007) Anti-inflammatory and antidiabetic roles of PPARγ. Novartis Found Symp 286:183–196 discussion 196-203
Pelham CJ, Keen HL, Lentz SR, Sigmund CD (2013) Dominant negative PPARγ promotes atherosclerosis, vascular dysfunction, and hypertension through distinct effects in endothelium and vascular muscle. Am J Physiol Integr Comp Physiol 304(9):R690–R701. https://doi.org/10.1152/ajpregu.00607.2012
Pendse AA, Johnson LA, Kim HS, McNair M, Nipp CT, Wilhelm C, Maeda N (2012) Pro-and antiatherogenic effects of a dominant-negative P465L mutation of peroxisome proliferator-activated receptor- γ in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 32(6):1436–1444. https://doi.org/10.1161/ATVBAHA.112.248682
Penyige A, Poliska S, Csanky E, Scholtz B, Dezso B, Schmelczer I, Kilty I, Takacs L, Nagy L (2010) Analyses of association between PPAR gamma and EPHX1 polymorphisms and susceptibility to COPD in a Hungarian cohort, a case-control study. BMC Med Genet 11:152. https://doi.org/10.1186/1471-2350-11-152
Pereira PAT, Da Silva BC, Dos Santos DF et al (2015) Prostaglandin D2-loaded microspheres effectively activate macrophage effector functions. Eur J Pharm Sci 78:132–139. https://doi.org/10.1016/j.ejps.2015.07.001
Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, Carta AR (2014) Dynamic changes in pro-and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis 71:280–291. https://doi.org/10.1016/j.nbd.2014.08.011
Rao J, Ye Z, Tang H, Wang C, Peng H, Lai W, Li Y, Huang W, Lou T (2017) The RhoA/ROCK pathway ameliorates adhesion and inflammatory infiltration induced by AGEs in glomerular endothelial cells. Sci Rep 7:39727. https://doi.org/10.1038/srep39727
Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, Standiford TJ (2008) Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-{gamma}. Blood 112(10):4250–4258. https://doi.org/10.1182/blood-2007-12-128967
Reddy AT, Lakshmi SP, Dornadula S, Pinni S, Rampa DR, Reddy RC (2013) The nitrated fatty acid 10-nitro-oleate attenuates allergic airway disease. J Immunol 191(5):2053–2063. https://doi.org/10.4049/jimmunol.1300730
Regieli JJ, Jukema JW, Doevendans PA, Zwinderman AH, van der Graaf Y, Kastelein JJ, Grobbee DE (2009) PPARγ variant influences angiographic outcome and 10-year cardiovascular risk in male symptomatic coronary artery disease patients. Diabetes Care 32(5):839–844. https://doi.org/10.2337/dc08-1819
Ricote M, Glass CK (2007) PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta 1771(8):926–935
Ricote M, Li AC, Willson TM et al (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391(6662):79–82. https://doi.org/10.1038/34178
Rispens E, Bron A, Lee J, Fukumoto J (2012) The pathophysiology of inflammation in cell injury. Pathophysiol Cell Inj J 3:99–126
Roach REJ, Lijfering WM, Flinterman LE, Rosendaal FR, Cannegieter SC (2013) The increased risk of arterial cardiovascular disease after venous thrombosis is determined by common etiologic factors. Blood 121(24):4948–4954. https://doi.org/10.1182/blood-2013-01-479238
Rodríguez-Mañas L, El-Assar M, Vallejo S et al (2009) Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 8(3):226–238. https://doi.org/10.1111/j.1474-9726.2009.00466.x
Rothwell PM, Coull AJ, Silver LE, Fairhead JF, Giles MF, Lovelock CE, Redgrave JNE, Bull LM, Welch SJV, Cuthbertson FC, Binney LE, Gutnikov SA, Anslow P, Banning AP, Mant D, Mehta Z (2005) Population-based study of event-rate, incidence, case fatality, and mortality for all acute vascular events in all arterial territories (Oxford Vascular Study). Lancet 366(9499):1773–1783. https://doi.org/10.1016/S0140-6736(05)67702-1
Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, Wahli W, Desvergne B, Naccari GC, Chavatte P, Farce A, Bulois P, Cortot A, Colombel JF, Desreumaux P (2005) Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator–activated receptor-γ. J Exp Med 201(8):1205–1215. https://doi.org/10.1084/jem.20041948
Sánchez-Hidalgo M, Martín AR, Villegas I, Alarcón De La Lastra C (2005) Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces chronic colonic inflammation in rats. Biochem Pharmacol 69(12):1733–1744. https://doi.org/10.1016/j.bcp.2005.03.024
Sasaki M, Jordan P, Welbourne T, Minagar A, Joh T, Itoh M, Elrod JW, Alexander JS (2005) Troglitazone, a PPAR-gamma activator prevents endothelial cell adhesion molecule expression and lymphocyte adhesion mediated by TNF-alpha. BMC Physiol 5(1):3. https://doi.org/10.1186/1472-6793-5-3
Sawada N, Liao JK (2014) Rho/rho-associated coiled-coil forming kinase pathway as therapeutic targets for statins in atherosclerosis. Antioxid Redox Signal 20(8):1251–1267. https://doi.org/10.1089/ars.2013.5524
Seals DR, Jablonski KL, Donato AJ (2011) Aging and vascular endothelial function in humans. Clin Sci (Lond) 120:357–375. https://doi.org/10.1042/CS20100476
Seals DR, Kaplon RE, Gioscia-Ryan RA, LaRocca TJ (2014) You’re only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology 29:250–264. https://doi.org/10.1152/physiol.00059.2013
Shah YM, Morimura K, Gonzalez FJ (2007) Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am J Physiol Gastrointest Liver Physiol 292(2):G657–G666. https://doi.org/10.1152/ajpgi.00381.2006
Sharma AM, Staels B (2007) Review: peroxisome proliferator-activated receptor gamma and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab 92(2):386–395. https://doi.org/10.1210/jc.2006-1268
Sharma R, Kaundal RK, Sharma SS (2009) Amelioration of pulmonary dysfunction and neutrophilic inflammation by PPARγ agonist in LPS-exposed guinea pigs. Pulm Pharmacol Ther 22(3):183–189. https://doi.org/10.1016/j.pupt.2008.11.011
Shrestha UK, Karimi O, Crusius JBA et al (2010) Distribution of peroxisome proliferator-activated receptor-gamma polymorphisms in Chinese and Dutch patients with inflammatory bowel disease. Inflamm Bowel Dis 16(2):312–319. https://doi.org/10.1002/ibd.21059
Sigmund CD (2010) Endothelial and vascular muscle PPARgamma in arterial pressure regulation: lessons from genetic interference and deficiency. Hypertens (Dallas, Tex 1979) 55:437–444. https://doi.org/10.1161/HYPERTENSIONAHA.109.144170
Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD (1999) A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. J Clin Invest 104(4):383–389. https://doi.org/10.1172/JCI7145
Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, Hong SH, Castro GL, Yin YQ, Nelson MC, Hsiao G, Greaves DR, Downes M, Yu RT, Olefsky JM, Evans RM (2009) PPAR activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci 106(52):22504–22509. https://doi.org/10.1073/pnas.0912487106
Sung B, Park S, Yu BP, Chung HY (2004) Modulation of PPAR in aging, inflammation, and calorie restriction. J Gerontol A Biol Sci Med Sci 59(10):997–1006. https://doi.org/10.1093/gerona/59.10.B997
Syrovets T, Schüle A, Jendrach M, Büchele B, Simmet T (2002) Ciglitazone inhibits plasmin-induced proinflammatory monocyte activation via modulation of p38 MAP kinase activity. Thromb Haemost 88(2):274–281
Szatmari I, Pap A, Rühl R, Ma JX, Illarionov PA, Besra GS, Rajnavolgyi E, Dezso B, Nagy L (2006) PPARgamma controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Exp Med 203(10):2351–2362. https://doi.org/10.1084/jem.20060141
Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2014) Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and downregulation of NADPH oxidase. AJP Hear Circ Physiol 306(3):H299–H308. https://doi.org/10.1152/ajpheart.00744.2013
Touyz RM, Briones AM (2011) Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res 34(1):5–14. https://doi.org/10.1038/hr.2010.201
Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR, Kim JK, Maeda N (2004) Hypertension and abnormal fat distribution but not insulin resistance in mice with P465L PPARγ. J Clin Invest 114(2):240–249. https://doi.org/10.1172/JCI200420964
Tyagi S, Gupta P, Saini AS et al (2011) The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res 2(4):236–240. https://doi.org/10.4103/2231-4040.90879
Ungvari Z, Kaley G, De Cabo R et al (2010) Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 65(10):1028–1041. https://doi.org/10.1093/gerona/glq113
Varga T, Czimmerer Z, Nagy L (2011) PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta 1812(8):1007–1022
Villegas I, Martín AR, Toma W, Alarcón De La Lastra C (2004) Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, protects against gastric ischemia-reperfusion damage in rats: role of oxygen free radicals generation. Eur J Pharmacol 505(1–3):195–203. https://doi.org/10.1016/j.ejphar.2004.10.020
Waku T, Shiraki T, Oyama T, Fujimoto Y, Maebara K, Kamiya N, Jingami H, Morikawa K (2009) Structural insight into PPARgamma activation through covalent modification with endogenous fatty acids. J Mol Biol 385(1):188–199. https://doi.org/10.1016/j.jmb.2008.10.039
Wan Y, Saghatelian A, Chong LW, Zhang CL, Cravatt BF, Evans RM (2007) Maternal PPARγ protects nursing neonates by suppressing the production of inflammatory milk. Genes Dev 21(15):1895–1908. https://doi.org/10.1101/gad.1567207
Wang N, Verna L, Chen NG, Chen J, Li H, Forman BM, Stemerman MB (2002) Constitutive activation of peroxisome proliferator-activated receptor-gamma suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J Biol Chem 277(37):34176–34181. https://doi.org/10.1074/jbc.M203436200
Wang L, Waltenberger B, Pferschy-Wenzig EM, Blunder M, Liu X, Malainer C, Blazevic T, Schwaiger S, Rollinger JM, Heiss EH, Schuster D, Kopp B, Bauer R, Stuppner H, Dirsch VM, Atanasov AG (2014a) Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): a review. Biochem Pharmacol 92(1):73–89. https://doi.org/10.1016/j.bcp.2014.07.018
Wang M, Jiang L, Monticone RE, Lakatta EG (2014b) Proinflammation: the key to arterial aging. Trends Endocrinol Metab 25(2):72–79
Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK (2003) PPAR{gamma} and PPAR{delta} negatively regulate specific subsets of lipopolysaccharide and IFN-{gamma} target genes in macrophages. Proc Natl Acad Sci 100(11):6712–6717. https://doi.org/10.1073/pnas.1031789100
Williams C, Panaccione R, Ghosh S, Rioux K (2011) Optimizing clinical use of mesalazine (5-aminosalicylic acid) in inflammatory bowel disease. Ther Adv Gastroenterol 4(4):237–248. https://doi.org/10.1177/1756283X11405250
Williamson K, Stringer SE, Alexander MY (2012) Endothelial progenitor cells enter the aging arena. Front Physiol 3(30):1–7
Wojciechowska W, Li Y, Stolarz-Skrzypek K et al (2012) Cross-sectional and longitudinal assessment of arterial stiffening with age in European and Chinese populations. Front Physiol 3(209):1–5
Xu X, Wang B, Ren C et al (2017) Recent progress in vascular aging: mechanisms and its role in age-related diseases. Aging Dis 8(4):486–505. https://doi.org/10.14336/AD.2017.0507
Yamamoto K, Takeshita K, Kojima T et al (2005) Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res 66(2):276–285
Yazdani SK, Tillman BW, Berry JL, Soker S, Geary RL (2010) The fate of an endothelium layer after preconditioning. J Vasc Surg 51(1):174–183. https://doi.org/10.1016/j.jvs.2009.08.074
Yin Y, Hou G, Li E, Wang Q, Kang J (2014) PPAR gamma agonists regulate tobacco smoke-induced toll like receptor 4 expression in alveolar macrophages. Respir Res 15(28):1–14. https://doi.org/10.1186/1465-9921-15-28
Youssef J, Badr M (2004) Role of peroxisome proliferator-activated receptors in inflammation control. J Biomed Biotechnol 2004(3):156–166. https://doi.org/10.1155/S1110724304308065
Yu JH, Kim KH, Kim H (2008) SOCS 3 and PPAR-γ ligands inhibit the expression of IL-6 and TGF-β1 by regulating JAK2/STAT3 signaling in pancreas. Int J Biochem Cell Biol 40(4):677–688. https://doi.org/10.1016/j.biocel.2007.10.007
Zachary I, Mathur A, Yla-Herttuala S, Martin J (2000) Vascular protection: a novel nonangiogenic cardiovascular role for vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 20(6):1512–1520. https://doi.org/10.1161/01.RES.0000218275.54089.12
Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, Szalkowski D, Moller DE (1996) Insulin-and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor γ. J Biol Chem 271(50):31771–31774
Zhang Q, Hu W, Meng B, Tang T (2010) PPAR γ agonist rosiglitazone is neuroprotective after traumatic spinal cord injury via anti-inflammatory in adult rats. Neurol Res 32(8):852–859. https://doi.org/10.1179/016164110X12556180206112
Zingarelli B, Sheehan M, Hake PW, O'Connor M, Denenberg A, Cook JA (2003) Peroxisome proliferator activator receptor-γ ligands, 15-deoxy-delta (12,14) -prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol 171(12):6827–6837
Acknowledgments
The authors are grateful to the Department of Pharmacy, Southeast University, Dhaka, Bangladesh.
Author information
Authors and Affiliations
Contributions
This work was carried out in collaboration between all authors. MSU and GMA designed the study, wrote the protocol, and managed the analyses of the study. MSU, MTK, MJ, GEB, and AAM prepared the draft of the manuscript. MSU prepared the figures of the manuscript. KN, MSA, and GEB reviewed the scientific contents of the manuscript. All authors read and approved the final submitted version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Uddin, M.S., Kabir, M.T., Jakaria, M. et al. Endothelial PPARγ Is Crucial for Averting Age-Related Vascular Dysfunction by Stalling Oxidative Stress and ROCK. Neurotox Res 36, 583–601 (2019). https://doi.org/10.1007/s12640-019-00047-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-00047-5