
Abstract
Metabolic and mitochondrial dysfunction has been implicated in Parkinson’s disease, while exercise can induce essential pathways of mitochondrial biogenesis. Here, we tested whether long-term preventive treadmill training (16 weeks, 21 m/min, and 0° inclinations for 50 min/d, 5 d/week) effects the mitochondrial and neurodegeneration markers, in the striatum of rats in the 6-hydroxydopamine (6-OHDA) model of Parkinson’s disease. Following 16 weeks of exercise or no exercise period (n = 16 rats per group), the animals were divided into four experimental groups (n = 8 per group): (1) no exercise and saline (SED), (2) exercise and saline (EX), (3) no exercise and 6-OHDA (SED + 6-OHDA), and (4) exercise and 6-OHDA (EX + 6-OHDA). For the model, 8 μg of 6-OHDA (2 μg/μL prepared in a solution of 0.2% ascorbic acid and 0.9% saline) was injected into the right medial forebrain bundle. Exposure to 6-OHDA resulted in a significant reduction (P < 0.05) of mitochondrial factors AMP-activated protein kinase, peroxisome proliferator-activated receptor gamma coactivator-1 alpha, and tyrosine hydroxylase, and increased expression of silent information regulator T1, mitochondrial transcription factor A, and p53 in the SED + 6-OHDA group relative to SED group. By contrast, gene and protein expressions upon exercise were higher and p53 protein level was lower in the EX + 6-OHDA group compared with SED + 6-OHDA. Further, exercise reduced the extent of weight loss associated with the 6-OHDA injection. In conclusion, exercise might be used to reduce mitochondrial disorders in Parkinson’s disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Aging is the greatest risk factor for the development and progression of Parkinson’s disease (PD) (Lau et al. 2011). The mechanisms underlying the neurodegenerative processes in PD remain unknown, although neuronal oxidative stress induced by reactive oxygen species (ROS), mitochondrial dysfunction, and neuronal apoptosis caused by p53 accumulation, a tumor suppressor gene, have been demonstrated in experimental PD models (Patki and Lau 2011). The available evidence supports the essential role of mitochondrial dysfunction in PD pathogenesis. The dysfunction, in particular at the level of mitochondrial complex I, can lead to mitochondrial DNA alterations, mitochondrial ROS accumulation, and cell death, especially affecting the dopaminergic neurons (Phillipson 2014).
Peroxisome proliferator-activated receptor gamma coactivator-1alpha (Pgc1α) was initially identified as a transcriptional co-activator of mitochondrial biogenesis that plays a critical role in the oxidative metabolism in brown fat (Farshbaf et al. 2016). Subsequently, Pgc1α was shown to play a crucial role in the brain and to be associated with neuroprotection against oxidative stress and cell death (Wrann et al. 2013). Enzymes that act as energy sensors, such as the silent information regulator T1 (Sirt1) and AMP-activated protein kinase (Ampk), regulate Pgc1α activity (Oliveira et al. 2014). Ampk and Sirt1 were shown to directly affect Pgc1α activity by its phosphorylation and deacetylation, respectively (Kang et al. 2013). Ampk has αβγ subunits and is a Ser/Thr kinase that has an essential regulatory role in energy homeostasis, acting as a sensor of energy balance (Kim et al. 2013). It is observed that α-Ampk phosphorylation at the Thr172 amino acid leads to Ampk activation (Scharf et al. 2008). Ampk regulates fatty acid oxidation and has an important role in the whole-body energy balance. It also promotes mitochondrial biogenesis by increasing the levels of Pgc1αand other mitochondrial proteins (Oliveira et al. 2014). Sirt1 responds to cellular energy and redox status of the cell. They are dependent on the NAD+/NADH ratio, and act as metabolic sensors (Bayod et al. 2012). Interestingly, Sirt1 was shown to fine-tune cellular protection against oxidative stress in many diseases associated with neurodegeneration and metabolic disorders (Chong et al. 2012). Hence, since oxidative stress is a pathological process that can lead to PD, it is conceivable that Sirt1 plays a role in PD (Chong et al. 2012). Furthermore, mitochondrial dysfunctions may be linked to the upregulation of ROS, mainly in the brain (Bayod et al. 2012). Few studies investigated Ampk level in the brain; however, its activation appears to exert a similar effect in various tissues (Richter and Ruderman 2009). Activation of the Ampk–Sirt1–Pgc1α pathway by exercise is an accepted target of molecular changes that result in mitochondrial biogenesis (Bayod et al. 2012). Furthermore, since the mitochondrial transcription factor A (Tfam) and Pgc1α are essential for sustained mitochondrial biogenesis, deletion or downregulation of these genes render animals more susceptible to neurotoxicity and induction of PD-like symptoms (Patki and Lau 2011).
Mitochondrial dysfunction plays an important role in various neurodegenerative diseases, such as PD. Exercise can increase mitochondrial biogenesis in the brain and probably attenuates the extent of these dysfunctions (Steiner et al. 2011). Further, exercise is the most effective intervention against deleterious processes associated with aging, such as sarcopenia, metabolic alterations, and neurodegeneration (Bayod et al. 2012). However, the cellular and molecular mechanisms that mediate training-induced adaptations in PD are not yet clear (Tuon et al. 2015). Furthermore, most studies on the mitochondrial response to exercise involve tissues with high metabolic activity, and not the brain. Considering the above, in the current study, we investigated the responses of body weight, p53, and tyrosine hydroxylase (TH) protein levels, and the expression of some of the mitochondrial factor genes to preventive exercise in the striatum in a rat model of PD.
Materials and Methods
Animals
Wistar rats (male, n = 32, 200 ± 30 g, 2 months old at the start of the study; from the Pasteur Institute of Iran) that could maintain a forward position on a treadmill belt for 5 min at 5.0 m/min were included in the study. The animals were housed in groups of four animals per cage, in a controlled environment, under12/12-h light/dark cycle, room temperature of 22 ± 2 °C, and ad libitum access to food and water. All experiments were performed in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals,” and were approved by the Ethic Committee for Animal Experiments at Isfahan University (IR.UI.REC.1396008).
Exercise Protocols
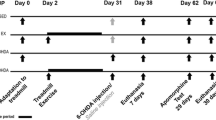
Rats were initially randomly assigned to two groups (n = 16 per group): (1) 16-week exercise (EX) group and (2) no exercise (SED) group. To familiarize the rats in EX group with the treadmill exercise, animals were placed on the treadmill (10 min, 5 m/min) for 1 week. The exercise regime on the treadmill was started following the familiarization period (five times per week, 16 weeks, at 0° inclination). The running protocol for each session was as follows: 5 min at 10 m/min for warm up, followed by 25 min at 15 m/min in the first week. The exercise intensity was increased to 50 min at 21 m/min by the second week, and was kept constant until the end of the exercise period. Then, 48 h after the completion of the exercise schedule, animals from each group were randomly assigned to two groups, and administered saline or 6-hydroxydopamine (6-OHDA) by surgery (see next section). There were four groups in total (n = 8 per group): (1) no exercise and saline (SED), (2) exercise and saline (EX), (3) no exercise and 6-OHDA (SED + 6-OHDA), and (4) exercise and 6-OHDA (EX + 6-OHDA). It should be noted that in this study, SED group is the control station, and other group’s data are corrected with it.
Surgical Procedures
Forty-eight hours after the last session of exercise, rats were anesthetized by an intraperitoneal injection of xylazine (10 mg/kg) and ketamine (80 mg/kg) (Costa et al. 2017). Animals from the 6-OHDA groups were then placed in a stereotaxic frame (Stoelting, USA) and administered 8 μg of 6-OHDA (Sigma-Aldrich; 2 μg/μL prepared in a solution of 0.2% ascorbic acid and 0.9% saline). A hole was drilled in the skull of each rat, and the drug was injected into the right medial forebrain bundle (Mabandla et al. 2004; Yoon et al. 2007; Carvalho et al. 2013), anteroposterior (AP): − 1.8 mm, lateral (LAT): 4.7 mm from the bregma, and vertical (DV): − 8.2 mm from the skull surface using a 5-μL Hamilton syringe attached to an infusion pump (BI Insight 2000), at a rate of 0.5 μL/min for 8 min. After the injection, the cannula was left in place for additional 5 min to prevent reflux of the drug. Then, the incision was sutured and the rats were kept isolated in cages until complete recovery. Rats from the saline groups (SED and EX), as the control animals, received an equal volume of a solution of 0.2% ascorbic acid and 0.9% saline (Tuon et al. 2015) in the same manner.
Weight Assessment
Because of the difference in the activity level in the SED and EX groups, and the possible effect of different weight at the start of the surgery on the experimental outcomes and cell death, animal weight was evaluated before and after the training period, and also 2 weeks after the surgery.
Euthanasia and Tissue Collection
One month after the surgery, the animals were anesthetized by an intraperitoneal injection of ketamine (5 mg/100 g of body weight) and xylazine (1 mg/100 g of body weight). Then, the animals were decapitated, and the right striatum was collected.
RNA Extraction and Real-Time Reverse-Transcription Quantitative PCR
One aliquot of each sample was homogenized in a buffer containing 1% Triton X-100, Tris 100 mM (pH 7.4), sodium pyrophosphate 100 mM, EDTA 100 mM, sodium vanadate 10 mM, PMSF 2 mM, and a protinine 0.1 mg/mL at 4 °C. The homogenate was centrifuged (11,000×g, 40 min, 4 °C) to remove insoluble material (Tuon et al. 2014, 2015). Total RNA was extracted from the striatum (n = 8 per group), using the RNeasy mini kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s instructions. RNA concentration and purity were determined using the NanoDrop1000 spectrophotometer (OD at 230 nm, 260 nm, and 280 nm). Then, 1 μg of total RNA was used for the synthesis of cDNA (cDNA synthesis kit, Fermentas, Lithuania) by utilizing oligodT primers. Real-time reverse-transcription quantitative PCR (RT-qPCR) was performed using SYBR Green PCR master mix (TaKaRa, Japan) and a Step One Plus thermocycler (ABI Applied Biosystems, USA). Sequences of the respective primer pairs are shown in Table 1. The data were analyzed based on the cycle threshold (CT). The CT value is defined as the fractional cycle number at which the fluorescence passes a fixed threshold. Ampk, Sirt1, Pgc1a, and Tfam mRNA levels were determined using the ΔΔCT method, and are presented as the percentage fold-expression change in the treated experimental groups relative to their corresponding control group after normalization to the Gapdh mRNA endogenous control (Patki and Lau 2011; Steiner et al. 2011).
Protein Extraction and Western Blotting
For protein extraction from the striatum samples (n = 5 per group), tissue pieces were processed in RIPA buffer (Cytomatin gene, Iran), supplemented with anti-protease cocktail (Sigma) according to the manufacturer’s protocol. Briefly, 1 mL of RIPA buffer was added to 100 mg of the brain tissue in 1.5-mL tube, and the samples homogenized at 4 °C. Insoluble material was removed by centrifugation (12,000×g for 20 min), and the protein concentration of the supernatant was determined using a protein assay kit (Bio-Rad, Hercules, CA).
Equal amounts of each protein samples (0.2 mg) were separated on 10% SDS-PAGE, and transferred to polyvinylidene difluoride membrane (Bio-Rad Laboratories, USA). The membranes were blocked overnight with 10% (w/v) skimmed milk (Merck, Germany) in phosphate-buffered saline, and then incubated overnight at 4 °C with primary antibodies. The antibodies used were anti-phospho (Thr172) Ampk (1:200, sc-33524), anti-Sirt1 (1:200, sc-15404), anti-Pgc1α (1:200, sc-55476), anti-Tfam (1:200, sc-23588), anti-TH (1:200, sc-25269), anti-p53 (1:200, sc-55476), and anti-β-actin (1:200, sc-47778) antibodies (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA). The membranes were then incubated with secondary antibodies conjugated with horseradish peroxidase (1:2500; Bio-Rad, 170–6516) for chemiluminescent detection. The signals were developed using the ECL kit (Bio-Rad Laboratories). Images were acquired using a gel imaging system (Bio-Rad Laboratories, USA) and the protein bands were quantified by densitometry using the ImageJ software (NIH, USA).
Data Analysis
All data are presented as the mean ± standard error of the mean (SEM). A two (time: before vs. after 16-week exercise period, i.e., “pre- vs. post-” comparisons) by two (group: EX vs. SED) mixed factorial analysis of variance (ANOVA) was used for comparison of the weight means. Data for treatment groups 1 month after the surgery were compared using one-way ANOVA, followed by the Tukey’s post hoc test (mRNA and protein levels); the same analyses were used for weight comparison 2 weeks after the surgery. All analyses were carried out using the SPSS (version 23.0) and GraphPad Prism 6 Software, La Jolla, CA, USA. The statistical significance threshold was set at P ≤ 0.05.
Results
Weight Analysis
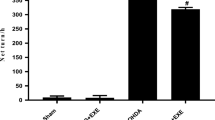
No significant interaction between the condition and time was observed, which suggested that the exercise did not differentially affect the weight gain [F(1,30) = 0.33, P = 0.5 (ηp2 = 0.005)]. However, there was a main effect for time. As expected, the rats gained significant weight during the study (P ≤ 0.001), both in the Ex group (pre-weight of 221.5 ± 3.2 g vs. post-weight of 350.6 ± 6.3 g) and SED group (pre-weight of 223.5 ± 5.05 g vs. post-weight of 354.0 ± 7.6 g). Further, 2 weeks after the surgery, the weight of animals in the SED + 6-OHDA group was significantly lower than that in the SED group, and a significant weight difference between the EX + 6-OHDA group and SED + 6-OHDA group was observed [F(3,28) = 20.7, P ≤ 0.001] (Fig. 1).

The effect of treadmill running on the weight (in g) of animals injected with 6-OHDA 2 weeks after the exercise period. The values are expressed as the mean ± SEM (n = 8). *P ≤ 0.001 compared with the SED group; #P ≤ 0.001 compared with the SED + 6-OHDA group, according to one-way ANOVA followed by Tukey’s post hoc test. SED no exercise, EX exercise
The Effect of Exercise on Gene Expression in the Brain in the Rat PD Model
The effect of the preventive endurance training on the gene expression of mitochondrial factors in the rat 6-OHDA model was evaluated by RT-qPCR. Analyses revealed a significant difference in the Ampk mRNA levels [F(3,28) = 32.5, P ≤ 0.001]. As shown in Fig. 2a, 6-OHDA injection resulted in a significant reduction in Ampk levels in the SED + 6-OHDA group compared with the SED group (90%) and EX + 6-OHDA group (94%), as there was no difference between the last group and EX group. Unexpectedly, the striatal expression of Sirt1 (Fig. 2b) and Tfam (Fig. 2d) was significantly increased in the SED + 6-OHDA group compared with the SED group (20% and 23%, respectively). The expression of these genes in 6-OHDA sedentary group were nevertheless substantially lower than those in the EX + 6-OHDA group (Sirt1, 14% and Tfam, 20%). A significant difference in Sirt1and Tfam levels was also apparent when was analyzed by one-way ANOVA [F(3,28) = 144.06 and 84.9, respectively, P ≤ 0.0001]. A significant difference in Pgc1a levels associated with exercise was also confirmed [F(3,28) = 30.8, P ≤ 0.0001]. Accordingly, the Pgc1a levels in the SED + 6-OHDA group were significantly lower than those in the control animals in SED group (60%), while preventive exercise resulted in significantly higher Pgc1a levels in the EX groups than in the SED group animals, as Pgc1a mRNA expression in EX + 6-OHDA was 78% higher than SED + 6-OHDA group (Fig. 2c). Furthermore, TH mRNA level was significantly different between groups [F(3,28) = 309.8, P ≤ 0.0001]. As shown in Fig. 2e, in the SED + 6-OHDA group, TH mRNA significantly reduced compared with the SED group (46%) and EX + 6-OHDA group (68%). However, its level was significantly higher in the EX group than in the EX + 6-OHDA group (16%). A significant difference in p53 mRNA levels due to exercise was also shown when the interaction between all of experimental groups was analyzed [F(3,28) = 96.3, P ≤ 0.0001]. These results suggest that 6-OHDA injection significantly increased p53 level compared with other groups. Although exercise in EX + 6-OHDA group is associated with significant reduction in p53 mRNA compared with 6-OHDA sedentary group (35%), its mRNA is still significantly higher than normal level in SED group (38%) (Fig. 2f).

The effect of treadmill exercise on the expression of selected genes in the rat brain. Changes in the expression of Ampk (a), Sirt1 (b), Pgc1a (c), Tfam (d), TH (e), and P53 (f) genes in the striatum of animals from different groups were analyzed by RT-qPCR. Gene expression was normalized to Gapdh expression. All data are shown as the mean ± SEM (n = 8 per group). *P ≤ 0.001, **P ≤ 0.01, and ***P ≤ 0.05, compared with the SED group; #P ≤ 0.001 and ##P ≤ 0.01 compared with the SED + 6-OHDA group, according to one-way ANOVA followed by Tukey’s post hoc test. SED no exercise, EX exercise
The Effect of Exercise on Protein Levels in the Brain in the Rat PD Model
Our results, in the immunoblots, the SED + 6-OHDA group showed lower the p-Ampkα (Thr 172) and Pgc1α expression when compared to the SED group (~86%), the EX group (~90%), and the EX + 6-OHDA group (81% and 59%, respectively). Although these levels in the last group were still significantly lower than the SED group (18% and 68%, respectively) and the EX group (41% and 78%) (Fig. 3a, c). A significant difference in the p-Ampkα (Thr 172) and Pgc1α expression due to exercise were also demonstrated [F(3,16) = 123.7 and 461.02, respectively, P ≤ 0.0001].

The effect of treadmill exercise on Ampk, Sirt1, Pgc1α, Tfam, TH, and p53 levels in the rat brain. Protein levels of Ampk (a), Sirt1 (b), Pgc1α (c), Tfam (d), TH (e), and p53 (f) in the striatum of animals from different groups were assayed by western blotting. All data are shown as the mean ± SEM (n = 5 per group). *P ≤ 0.001 compared with the SED group; #P ≤ 0.001 and ##P ≤ 0.01 compared with the SED + 6-OHDA group, according to one-way ANOVA followed by Tukey’s post hoc test. SED no exercise, EX exercise
However, the results were contrary to our initial expectation in the Sirt1 and the Tfam expression. The immunoblotting results showed an increased the Sirt1 (Fig. 3b) and the Tfam (Fig. 3d) protein expression in the SED + 6-OHDA group when compared to the SED group (~23%). Those levels in the EX + 6-OHDA group were equivalent to the EX group and significantly higher than the SED + 6-OHDA group (~30%). A significant difference in the Sirt1 and the Tfam protein expression due to exercise was also demonstrated [F(3,16) = 79.3 and 72.9, respectively, P ≤ 0.0001].
The striatal level of TH is shown in Fig. 3e. TH level in the SED + 6-OHDA group was significantly lower than its level in the SED group (60%). Moreover, preventive exercise in the EX + 6-OHDA group resulted the TH level that was significantly higher than the SED + 6-OHDA group (30%); however, the TH level was not as high as that in the control group. A significant difference in TH level associated with exercise was also evidenced [F(3,16) = 316.4, P ≤ 0.0001]. Further, the striatal p53 protein level was considerably elevated in the SED + 6-OHDA group. However, p53 protein level in exercised rats exposed to 6-OHDA was significantly lower than its expression in the 6-OHDA sedentary group (32%), although the former was significantly higher than the SED group (42%) (Fig. 3f). A significant difference in the p53 protein level associated with exercise was revealed [F(3,16) = 112.6, P ≤ 0.0001].
Discussion
PD leads to weight disorder, mitochondrial dysfunction, and oxidative stress. Consequently, therapeutic approaches for targeting mitochondrial biogenesis in PD, such as exercise, hold great promise (Aguiar Jr et al. 2016). The current study was designed to investigate the effects of preventive physical training, prior to 6-OHDA treatment and onset of PD-like state, on the mitochondrial biogenesis markers in the striatum in the experimental rat model of PD.
In the rat model used in the current study, the weight of all animals increased during the preventive training period, regardless of the exercise. A logical assumption is that the increase in physical activity resulted in a concurrent increase in food intake (Landers et al. 2013). However, weight gain in the EX group did not differ from that in the SED group before the surgery. This observation is supported by some studies (Choe et al. 2012; Landers et al. 2013). For example, Landers et al. (2013) reported that running animals started out lighter and finished lighter than non-runners. Hence, differential weight gain did not affect the results of the current study. Furthermore, as shown in Fig. 1, 6-ODHA injection in the SED group resulted in weight loss in comparison with the control animals (SED + 6-OHDA group vs. SED group). However, preventive exercise prior to the PD induction in the EX + 6-OHDA group resulted in weight recovery and increase relative to the SED + 6-OHDA group. These findings may be partially explained by the interplay among the genes under investigation. Ampk plays an important physiological role in responses to the energy-related signals to the body, and it has been suggested that Ampk activity can be managed pharmacologically to affect the food intake and body weight (Ronnett et al. 2009). Accordingly, reduction in Ampk activity results in weight loss (Andersson et al. 2005). One prominent hypothesis is that impaired energy metabolism may underlie slow and progressive neurodegeneration (Kim et al. 2013). Further, Ampk activation leads to increased Pgc1a expression. The current findings (Figs. 2a, c and 3a, c) indicate that alteration in Ampk expression is associated with similar changes in Pgc1a level, supporting the link between these genes. However, why, where, and how that happens are still elusive (Cantó and Auwerx 2009). Pgc1a plays an important role in preventing or delaying the onset of muscle atrophy by suppressing the expression of atrophy-related genes (Kim et al. 2017). On the other hand, the anti-apoptotic effect of training is consistent with the protective effect of Pgc1α, i.e., suppression of the apoptotic process in the muscle upon 6-OHDA induction (Kang et al. 2013). Indeed, these observations were supported by the changes in mRNA and protein levels observed in the current study: Ampk, Pgc1a, and TH levels were reduced and P53 was increased by the 6-OHDA treatment, and the effect was alleviated by exercise. According to studies, dopamine is one of the neurotransmitters involved in feeding behavior that modulates the rewarding properties of food and the motivation for food intake (Kistner et al. 2014). In the present study, the findings indicate that the exercise protects of dopamine against 6-OHDA and can be useful in the regulation of body weight (Fig. 3e, f).
Pathological apoptosis because of dysregulated p53 expression could potentially lead to a reduced number of DA neurons (Qi et al. 2016). Our data support the notion that p53 is involved in the process of dopaminergic neuronal cell death in this PD animal model. This study, similar to previous research (Real et al. 2017), described that dopaminergic neurons showed a marked decrease in sedentary compression to exercised animals after 6-OHDA induction, suggesting that the nervous system may be protected by decrease oxidative stress and neuroinflammatory markers by exercise (Fig. 3e). Corroborating with this data, an increase in other mitochondrial factors due to exercise in animal models of PD (Figs. 2 and 3) suggests that the exercise protocol was able to modulate mechanisms that may participate in neurodegenerative processes, and thus prevent the development of PD.
Interestingly, Sirt1 and Tfam levels in the 6-OHDA sedentary group were higher than that of controls (Figs. 2 and 3). According to Patki and Lau (2011), this increase might indicate an attempt to compensate for the loss of mitochondrial function and to improve mitochondrial biogenesis to repair cellular damage caused by 6-OHDA injection. However, as observed in the current study, the compensatory increase of Sirt1and Tfam expression was not able to rescue the increase of p53 levels and the reduction of TH levels, neurodegeneration, and mitochondrial abnormalities (Fig. 3). However, 1 month after the surgery, when 6-OHDA still caused cell death (SED + 6-OHDA group), in the 6-OHDA-treated exercised rats, all mRNAs and protein levels were higher and the dopaminergic neuroprotection (via reduced p53 levels and enhanced TH levels) in the striatum was more pronounced than those in the 6-OHDA-treated sedentary group (Figs. 2 and 3). Hence, the employed exercise protocol partially alleviated the destructive effect of 6-OHDA, suggesting lesser DNA damage. These findings were supported by previous studies (Patki and Lau 2011; Tuon et al. 2015; Garcia et al. 2017; de Jager et al. 2018). However, because of the difference in the dose of administered 6-OHDA and local of the toxin injection, no changes in TH levels in the striatum in the exercised animal model of PD compared with the control group were reported in some studies (Tajiri et al. 2010; Garcia et al. 2017). In the current study, the preventive exercise was longer more intense than previous study. On the other hand, because of the amount of injected 6-OHDA used to induce the PD-like state, the exercised animals may have taken longer than 1 month to recover the dopaminergic system and mitochondrial factors to baseline levels. Garcia et al. (2017) reported that 3 and 4 months after the 6-OHDA injection, results were better than evaluations at shorter intervals.
The outcomes of the current study do not appear to support the notion that a therapeutic approach to treating PD targets Tfam or Sirt1compensatory gene regulation. Nevertheless, exercise can be useful strategy for metabolic and mitochondrial disorders. It is not clear why exercise provides these effects; probably, it enhances the release of neurotrophins in order to neuroprotective effects, and may also increase oxygen supply and blood flow for the removal of the neurotoxin from the striatum (Mabandla et al. 2004). Many studies confirmed that exercise is a powerful intervention that upregulates important factors of mitochondrial biogenesis (Patki and Lau 2011; Kang et al. 2013; Oliveira et al. 2014; LaHue et al. 2016). Repetitive muscle contraction changes the metabolic status of cell, e.g., increases the AMP/ATP and NAD+/NADH ratios, that activate Ampk and Sirt1, respectively, leading to transcriptional activation and modification of Pgc1α (Kang et al. 2013). However, Bayod et al. (2012) proposed that exercise does not reduce the effect of aging on Ampk expression. Similarly, Landers et al. (2013) reported that exercise stress reduces TH levels in animals injected with 6-OHDA compared with a sedentary 6-OHDA-treated group. Generally, it appears that the reduction of p53 protein levels and apoptosis, increase of TH, and mitochondrial factor levels after a preventive endurance exercise in the striatum of rats in the PD model can play a neuroprotective role, alleviate weight loss, and attenuate atrophy.
Generally, expressions of the proteins of the investigated factors, in the EX + 6-OHDA group, were recovered compared with the SED + 6-OHDA group, but those were significantly lower than the control group. Probably, recovery to normal status needs to longer time than 1 month that has to be analyzed in the future. It is important to attention that all of biochemical assessments are analyzed in intact hemisphere where may be indicate compensatory effects following the lesion. However, Ronnett et al. (2009) proposed that pAmpk was high in both the healthy and the disordered hemisphere, suggesting that the metabolic derangements and compensatory responses in one hemisphere resulted in global Ampk activation.
Generally, it is important which hemisphere is analyzed. In this study, we investigated the lesioned side. However, comparison of both lesion and non-lesioned hemispheres, in next studies, could be interesting data.
Conclusion
In the current study, we demonstrated the loss of weight and striatal dopaminergic neurons in association with changes in the expression of specific mitochondrial factors upon 6-OHDA injection. We also showed that exercise-dependent neuroprotection in the 6-OHDA animal model of PD involves the balancing of expression of biochemical factors that affect mitochondrial function. The presented results not only confirm the increase of p53 level in the experimental model of PD but also indicate the protective role of exercise in counteracting neurodegeneration, leading to a reduction of weight loss, and neuronal and mitochondrial deficits observed in PD. In addition, the compensatory increase of Sirt1 and Tfam levels following the 6-OHDA injection appeared to be inadequate for reduction of the disorders, since weight loss, reduction of TH levels, and induction of p53 levels in the striatum were not reduced in the 6-OHDA sedentary group after the compensatory responses. Nevertheless, the findings indicate that preventive endurance running triggers many signals to reduce the mitochondrial and metabolic disturbances and through neuroprotection can be effective in reduce the level of symptom and increase the quality of life of Parkinson’s patients.
References
Aguiar AS Jr, Duzzioni M, Remor AP, Tristão FSM, Matheus FC, Raisman-Vozari R, Latini A, Prediger RD (2016) Moderate-intensity physical exercise protects against experimental 6-hydroxydopamine-induced hemiparkinsonism through Nrf2-antioxidant response element pathway. Neurochem Res 41(1–2):1–9. https://doi.org/10.1007/s11064-015-1709-8
Andersson U, Treebak JT, Nielsen JN, Smith KL, Abbott CR, Small CJ, Carling D, Richter EA (2005) Exercise in rats does not alter hypothalamic AMP-activated protein kinase activity. Biochem Biophys Res Commun 329(2):719–725. https://doi.org/10.1016/j.bbrc.2005.02.027
Bayod S, Del Valle J, Lalanza J, Sanchez-Roige S, de Luxan-Delgado B, Coto-Montes A, Canudas A, Camins A, Escorihuela RM, Pallas M (2012) Long-term physical exercise induces changes in sirtuin 1 pathway and oxidative parameters in adult rat tissues. Exp Gerontol 47(12):925–935. https://doi.org/10.1016/j.exger.2012.08.004
Cantó C, Auwerx J (2009) PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20(2):98–105. https://doi.org/10.1097/MOL.0b013e328328d0a4
Carvalho MM, Campos FL, Coimbra B, Pêgo JM, Rodrigues C, Lima R, Rodrigues AJ, Sousa N, Salgado AJ (2013) Behavioral characterization of the 6-hydroxidopamine model of Parkinson’s disease and pharmacological rescuing of non-motor deficits. Mol Neurodegener 8:14. https://doi.org/10.1186/1750-1326-8-14
Choe M, Koo B-S, An GJ, Jeon S (2012) Effects of treadmill exercise on the recovery of dopaminergic neuron loss and muscle atrophy in the 6-ohda lesioned parkinson's disease rat model. Korean J Physiol Pharmacol 16(5):305–312. https://doi.org/10.4196/kjpp.2012.16.5.305
Chong ZZ, Shang YC, Wang S, Maiese K (2012) SIRT1: new avenues of discovery for disorders of oxidative stress. Expert Opin Ther Targets 16(2):167–178. https://doi.org/10.1517/14728222.2012.648926
Costa RO, Gadelha-Filho CVJ, Costa AEM, Feitosa ML, Jo DP, Lucena JD, Aquino PEA, Lima FAV, Neves KRT, Barros Viana GS (2017) The treadmill exercise protects against dopaminergic neuron loss and brain oxidative stress in parkinsonian rats. Oxidative Med Cell Longev 2017:1–10. https://doi.org/10.1155/2017/2138169
de Jager L, Amorim EDT, Lucchetti BFC, Lopes FNC, Crestani CC, Pinge-Filho P, Martins-Pinge MC (2018) Nitric oxide alterations in cardiovascular system of rats with Parkinsonism induced by 6-OHDA and submitted to previous exercise. Life Sci 204:78–86. https://doi.org/10.1016/j.lfs.2018.05.017
Farshbaf MJ, Ghaedi K, Megraw TL, Curtiss J, Faradonbeh MS, Vaziri P, Nasr-Esfahani MH (2016) Does PGC1α/FNDC5/BDNF elicit the beneficial effects of exercise on neurodegenerative disorders? NeuroMolecular Med 18(1):1–15. https://doi.org/10.1007/s12017-015-8370-x
Garcia PC, Real CC, Britto LR (2017) The impact of short and long-term exercise on the expression of arc and AMPARs during evolution of the 6-Hydroxy-dopamine animal model of Parkinson’s disease. J Mol Neurosci 61(4):542–552. https://doi.org/10.1007/s12031-017-0896-y
Kang C, Chung E, Diffee G, Ji LL (2013) Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: role of PGC-1α. Exp Gerontol 48:1343–1350
Kim TW, Cho HM, Choi SY, Suguira Y, Hayasaka T, Setou M, Koh HC, Hwang EM, Park JY, Kang SJ, Kim HS, Kim H, Sun W (2013) (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson's disease. Cell Death Dis 4(11):e919. https://doi.org/10.1038/cddis.2013.447
Kim Y, Triolo M, Hood DA (2017) Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxidative Med Cell Longev. https://doi.org/10.1155/2017/3165396
Kistner A, Lhommée E, Krack P (2014) Mechanisms of body weight fluctuations in Parkinson’s disease. Front Neurol 2(5):84. https://doi.org/10.3389/fneur.2014.00084.
LaHue SC, Comella CL, Tanner CM (2016) The best medicine? The influence of physical activity and inactivity on Parkinson's disease. Mov Disord 31(10):1444–1454. https://doi.org/10.1002/mds.26728
Landers MR, Kinney JW, Allen DN, van Breukelen F (2013) A comparison of voluntary and forced exercise in protecting against behavioral asymmetry in a juvenile hemiparkinsonian rat model. Behav Brain Research 248:121–128. https://doi.org/10.1016/j.bbr.2013.04.002
Lau Y-S, Patki G, Das-Panja K, Le W-D, Ahmad SO (2011) Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson’s disease with moderate neurodegeneration. Eur J Neurosci 33(7):1264–1274. https://doi.org/10.1111/j.1460-9568.2011.07626.x
Mabandla M, Kellaway L, Gibson ASC, Russell VA (2004) Voluntary running provides neuroprotection in rats after 6-hydroxydopamine injection into the medial forebrain bundle. Metab Brain Dis 19(1–2):43–50. https://doi.org/10.1023/B:MEBR.0000027416.13070.c3
Oliveira NR, Marques SO, Luciano TF, Pauli JR, Moura LP, Caperuto E, Pieri BL, Engelmann J, Scaini G, Streck EL (2014) Treadmill training increases SIRT-1 and PGC-1α protein levels and AMPK phosphorylation in quadriceps of middle-aged rats in an intensity-dependent manner. Mediat Inflamm. https://doi.org/10.1155/2014/987017
Patki G, Lau Y-S (2011) Impact of exercise on mitochondrial transcription factor expression and damage in the striatum of a chronic mouse model of Parkinson's disease. Neurosci Lett 505(3):268–272. https://doi.org/10.1016/j.neulet.2011.10.036
Phillipson OT (2014) Management of the aging risk factor for Parkinson's disease. Neurobiol Aging 35(4):847–857. https://doi.org/10.1016/j.neurobiolaging.2013.10.073
Qi X, Davis B, Chiang Y-H, Filichia E, Barnett A, Greig NH, Hoffer B, Luo Y (2016) Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson's disease model. J Neurochem 138(5):746–757. https://doi.org/10.1111/jnc.13706
Real CC, Garcia PC, Britto LRG (2017) Treadmill exercise prevents increase of Neuroinflammation markers involved in the dopaminergic damage of the 6-OHDA Parkinson’s disease model. J Mol Neurosci 63(1):36–49. https://doi.org/10.1007/s12031-017-0955-4
Richter EA, Ruderman NB (2009) AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J 418(2):261–275. https://doi.org/10.1042/BJ20082055
Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S (2009) AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem 1:17–23. https://doi.org/10.1111/j.1471-4159.2009.05916.x.
Scharf MT, Mackiewicz M, Naidoo N, O'Callaghan JP, Pack AI (2008) AMP-activated protein kinase phosphorylation in brain is dependent on method of killing and tissue preparation. J Neurochem 105(3):833–841. https://doi.org/10.1111/j.1471-4159.2007.05182.x
Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM (2011) Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol (1985) 111(4):1066–1071. https://doi.org/10.1152/japplphysiol.00343.2011.
Tajiri N, Yasuhara T, Shingo T, Kondo A, Yuan W, Kadota T, Wang F, Baba T, Tayra JT, Morimoto T, Jing M, Kikuchi Y, Kuramoto S, Agari T, Miyoshi Y, Fujino H, Obata F, Takeda I, Furuta T, Date I (2010) Exercise exerts neuroprotective effects on Parkinson's disease model of rats. Brain Res 1310:200–207. https://doi.org/10.1016/j.brainres.2009.10.075
Tuon T, Valvassori SS, Dal Pont GC, Paganini CS, Pozzi BG, Luciano TF, Souza PS, Quevedo J, Souza CT, Pinho RA (2014) Physical training prevents depressive symptoms and a decrease in brain-derived neurotrophic factor in Parkinson's disease. Brain Res Bull 108:106–112. https://doi.org/10.1016/j.brainresbull.2014.09.006
Tuon T, Souza PS, Santos MF, Pereira FT, Pedroso GS, Luciano TF, De Souza CT, Dutra RC, Silveira PC, Pinho RA (2015) Physical training regulates mitochondrial parameters and Neuroinflammatory mechanisms in an experimental model of Parkinson’s disease. Med Cell Longev. https://doi.org/10.1155/2015/261809
Wrann Christiane D, White James P, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, Lin Jiandie D, Greenberg Michael E, Spiegelman Bruce M (2013) Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab 18(5):649–659. https://doi.org/10.1016/j.cmet.2013.09.008
Yoon M-C, Shin M-S, Kim T-S, Kim B-K, Ko I-G, Sung Y-H, Kim S-E, Lee H-H, Kim Y-P, Kim C-J (2007) Treadmill exercise suppresses nigrostriatal dopaminergic neuronal loss in 6-hydroxydopamine-induced Parkinson's rats. Neurosci Lett 423:12–17. https://doi.org/10.1016/j.neulet.2007.06.031
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rezaee, Z., Marandi, S.M., Alaei, H. et al. Effects of Preventive Treadmill Exercise on the Recovery of Metabolic and Mitochondrial Factors in the 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Neurotox Res 35, 908–917 (2019). https://doi.org/10.1007/s12640-019-0004-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-0004-x