
Abstract
α-Synuclein is a 140-amino acid protein produced predominantly by neurons in the brain which plays a role in the regulation of neurotransmitter release, synaptic function, and plasticity, thus making it the focus in understanding the etiology of a group of neurodegenerative diseases. We conducted genome-wide association studies (GWAS) of α-synuclein levels in cerebrospinal fluid (CSF) with 209 non-Hispanic white participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI-1) cohort using a linear regression model to identify novel variants associated with α-synuclein concentration. The minor allele (T) of rs7072338 in the long intergenic non-protein coding RNA 1515 (LINC01515) and the minor allele (T) of rs17794023 in clusterin-associated protein 1 (CLUAP1) were associated with higher CSF α-synuclein levels at genome-wide significance (P = 4.167 × 10–9 and 9.56 × 10–9, respectively). In addition, single nucleotide polymorphisms (SNPs) near amyloid beta precursor protein (APP) (rs1394839) (P = 2.31 × 10–7), Rap guanine nucleotide exchange factor 1 (RAPGEF1) (rs10901091) (P = 8.07 × 10–7), and two intergenic loci on chromosome 2 and 14 (rs11687064 P = 2.50 × 10–7and rs7147386 P = 4.05 × 10–7) were identified as suggestive loci associated with CSF α-synuclein levels. We have identified significantly associated SNPs for CSF α-synuclein. These associations have important implications for a better understanding of α-synuclein regulation and allow researchers to further explore the relationships between these SNPs and α-synuclein-related neurodegenerative disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
From the time of its discovery, α-synuclein, a 140-amino acid protein produced predominantly by neurons in the brain, has been the focus in understanding the etiology of a group of neurodegenerative diseases called α-synucleinopathies. These include Parkinson’s disease (PD), dementia with Lewy bodies (DLB) (Spillantini et al. 1997) and multiple system atrophy (MSA) (Gai et al. 1998). Moreover, α-synuclein regulates the fibrilization of both amyloid-β (Aβ) and tau, two key proteins in Alzheimer’s disease (AD) pathophysiology (Bachhuber et al. 2015; Giasson et al. 2003; Guo et al. 2013; Masliah et al. 2001; Yoshimoto et al. 1995), which suggests an important role for α-synuclein toxicity in neurodegeneration. The quantification of α-synuclein in CSF is in parallel with the measurement of proteins in CSF related to AD, namely total and phosphorylated tau protein and β-amyloid. Therefore, α-synuclein has gained much attention as a potential biomarker of α-synuclein-related neurodegenerative disorders in recent years. α-Synuclein was thought at first to be an exclusively intracellular protein and this notion was challenged when α-synuclein was detected in biological fluids, such as CSF (El-Agnaf et al. 2003; Mollenhauer et al. 2008). A number of studies have evaluated the potential of CSF α-synuclein as a diagnostic biomarker for α-synucleinopathies, but the results were inconsistent (Hong et al. 2010; Korff et al. 2013; Mollenhauer et al. 2008, 2010; Shi et al. 2011; Toledo et al. 2013; Wang et al. 2012). In general, patients with synucleinopathies, e.g., PD, DLB, and MSA often have reduced CSF α-synuclein compared to controls, while in AD patients, CSF α-synuclein levels were often higher as compared with cognitively healthy controls. Although the normal function of α-synuclein remains unclear, studies suggest that α-synuclein has a role in the regulation of neurotransmitter release, synaptic function, and plasticity (Lashuel et al. 2013). A pathological role for α-synuclein in these diseases is further supported by various genetic evidences. Multiplication of the gene encoding α-synuclein (SNCA) and six missense mutations ((A30P, E46K, H50Q, G51D, A53E, and A53T) in this gene are identified to be associated with dominant familial Parkinsonism (Appel-Cresswell et al. 2013; Kruger et al. 1998; Lesage et al. 2013; Pasanen et al. 2014; Polymeropoulos et al. 1997; Proukakis et al. 2013; Zarranz et al. 2004). In addition, multiple genome-wide association studies (GWAS) have identified SNPs in SNCA as major risk factors for sporadic PD (Simon-Sanchez et al. 2009). Nevertheless, the molecular mechanisms by which α-synuclein aggregation contributes to neurodegeneration remain unclear.
The use of quantitative traits in GWAS has been shown to increase statistical power over case-control designs (Cruchaga et al. 2013; Kim et al. 2011). Here, on the basis of adequate evidence on the role of CSF α-synuclein in neurodegenerative disorders, we conducted a GWAS of CSF from ADNI database. Further examinations of the variants that we have identified in different datasets may lead to a deeper understanding of α-synuclein regulation and provide important insights into its effects on α-synuclein-related function and disorders.
Methods
ADNI Study Design
Data used in this study were obtained from the ADNI database (http://adni.loni.usc.edu). The most recent information from the ADNI is available online (http://www.adni-info.org). The ADNI is a large, multicenter, longitudinal neuroimaging study, launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies, and nonprofit organizations. The study gathered and analyzed thousands of brain scans, genetic profiles, and biomarkers in blood and cerebrospinal fluid. This study was approved by institutional review boards of all participating institutions and written informed consent was obtained from all participants or authorized representatives.
Participants and CSF Measurement
Our study population consisted of all CN, MCI, and AD dementia group participants from the ADNI-1. In this study, 686 (CN = 194, MCI = 330, AD = 160 at baseline) non-Hispanic Caucasian individuals from the ADNI cohort whose data met all quality control criteria were included, which would reduce the likelihood of population stratification effects in the GWAS.
CSF samples were collected from individuals in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Levels of CSF α-synuclein concentration were measured using LuminexMicroPlex (Luminex Corp, Austin, TX). The α-synuclein Luminex assay demonstrated low day-to-day as well as plate-to-plate signal variability. The accuracy for the assay, as determined by recovery of spiked α-synuclein, was ~ 93%(Toledo et al. 2013).
Genotyping and Quality Control in GWAS
The ADNI samples were genotyped with the Illumina 610 chip. Given the smaller size of the current sample as compared to previous analyses, several quality control measures were applied to the 620,901 SNPs to detect potential biases in genotyping using the PLINK software package. Only SNPs with a minor allele frequency (MAF) > 5%, call rates > 98%, and Hardy-Weinberg equilibrium P > 0.001 were retained for analysis. Finally, a maximum of 519,442 SNPs were retained after these procedures. On the basis of data for all of these SNPs, we excluded 151 individuals who had more than 5% missing genotypes within 757 samples. This more stringent threshold was chosen to reduce the likelihood of false-positive results in the context of modest sample size. In order to decrease CSF contamination by RBC, a human hemoglobin ELISA quantitation kit was used (https://ida.loni.usc.edu/pages/access/studyData.jsp), which has sensitivity well beyond the cut-off value of 1000 ng/ml (Hall et al. 2012). For this reason, 326 samples were removed. In addition, we excluded 71 samples that were non-Hispanic Caucasians. Finally, 209 individuals with CSF α-synuclein were retained at last.
Statistical Analyses
The distribution of α-synuclein levels were approximately considered as normal distribution after log transformation. One-way ANOVA models were used for quantitative normally distributed variables. Rank-based two-way methods were used for non-normally distributed quantitative variables (Toledo et al. 2013). Chi-square text was applied to categorical data (Wang et al. 2016). To examine the main effect of each SNP on the CSF α-synuclein biomarker, GWAS was performed with additive genetic model. We used a multiple linear regression model to estimate possible correlation between genotypes and CSF α-synuclein concentration (e.g., dose-dependent effect of the minor allele). Covariates such as age, gender, APOE ε4 status, educational level, and baseline disease status were considered and retained in the final models if P < 0.05. We focused on SNPs with uncorrected P < 5 × 10−8 (or Bonferroni correction correct P < 0.01) as genome-wide significant and secondarily examined SNPs with P values less than 1 × 10−5 to identify potential candidates (Risch and Merikangas 1996). All statistical analyses were performed by R 3.4.0 and PLINK (http://pngu.mgh.harvard.edu/wpurcell/plink/).
Results
Demographic Characteristic and CSF α-Synuclein Concentration
The detailed demographics of 209 (CN = 59, MCI = 101, AD = 49 at baseline) non-Hispanic Caucasian participants at baseline diagnosis were summarized in Table 1. No difference was found across the diagnostic groups for age, education, and sex (P > 0.05). Compared to CN and MCI subjects, AD individuals have higher CSF α-synuclein concentration, higher frequency of APOE ε4 allele, and worst cognitive function displayed by neuropsychological scales (MMSE and CDR-SB) (P < 0.05). In addition, associations were detected between baseline demographics (e.g., APOE ε4 status, disease status, and educational years) and CSF α-synuclein level (P < 0.05), which were considered as the evidence of covariates.
Loci Associated with CSF α-Synuclein Levels
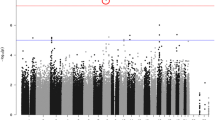
Relationships between 519,442 SNPs and CSF α-synuclein levels were shown in a Manhattan plot, with APOE ε4 status, disease status, and educational years included as covariates (Fig. 1). The obtained genomic inflation factors of CSF biomarker associations (λ = 1.00) indicated a low risk of confounding due to population stratification. Six SNPs in the regions of long intergenic non-protein coding RNA 1515 (LINC01515) and clusterin-associated protein 1 (CLUAP1) reached genome-wide significance (unadjusted P < 10−7, adjusted P < 0.01). In addition, SNPs near APP (rs1394839) (P = 2.31 × 10−7), RAPGEF1 (rs10901091) (P = 8.07 × 10−7), and two intergenic loci on chromosome 2 and 14 (rs11687064 P = 2.50 × 10−7 and rs7147386 P = 4.05 × 10−7) were identified as suggestive loci associated with CSF α-synuclein levels (Table 2). All the annotation information of SNPs that did not reach genome-wide significance or uncorrected P values less than 10−5 were listed in Supplementary Table 1.

Manhattan plot for the GWAS of CSF α-synuclein biomark. Observed − log10 P values (y-axis) are displayed for all tested SNPs on each autosomal chromosome (x-axis). The red horizontal line at 10−7 indicates genome-wide significance
Among all the SNPs, rs7072338, which is located in the intron region of LINC01515 on chromosome 10, showed the strongest association with CSF α-synuclein (uncorrected P = 4.167 × 10−9, Bonferroni corrected P = 2.164 × 10−3). Another four SNPs located near rs7072338 also reached a GWAS significant P value (uncorrected P = 1.909 × 10−8, Bonferroni corrected P = 9.917 × 10−3). Besides that, three SNPs around rs7072338 showed a P value lower than 10–5(Supplementary Table 1). We confirmed the most significant SNP in this locus and other seven SNPs in linkage disequilibrium (LD, r2 > 0.8) (Fig. 2a). However, after controlling for rs7072338 genotype, no SNPs in this region showed an association with CSF α-synuclein levels indicating that all the association in this locus was driven by rs7072338 (Fig. 2b). In addition, the linkage disequilibrium (LD) pattern between rs7072338 and nearby SNPs was almost identical in the ADNI cohort compared with 1000 Genomes European subjects (Supplementary Fig. 1), suggesting that the SNP genotypes from this study were accurate. The minor allele (T) of rs7072338 was associated with higher CSF α-synuclein levels in a dose-dependent effect within both combined groups and each diagnostic group (normal group, p = 5.14 × 10–5; pMCI group, p = 3.77 × 10–3; sMCI group, P = 0.034 and AD group, P = 8.56 × 10–4) (Supplementary Fig. 2).

a Regional association results for the LINC01515 region of chromosome 10 Fig. b Association results for 10q21.3 controlling for rs7072338
Moreover, rs17794023, located in CLUAP1, also showed a genome-wide significant association with CSF α-synuclein levels (P = 9.56 × 10−9). This locus survived even after Bonferroni corrections for multiple testing (Bonferroni corrected P = 4.964 × 10–3). The minor allele (T) of rs17794023 was associated with higher CSF α-synuclein levels in a dose-dependent effect within both combined groups and each diagnostic group (normal group, P = 2.81 × 10–3; pMCI group, P = 3.32 × 10–3; sMCI group, p = 0.56 and AD group, p = 3.14 × 10–5) (Supplementary Fig. 3).
Discussion
To our knowledge, we firstly performed a GWAS of CSF α-synuclein levels in the ADNI cohort. Six SNPs in the regions of LINC01515 and CLUAP1 were identified to be genome-wide significant loci associated with CSF α-synuclein levels. Among them, the significant association of SNPs in LINC01515 was driven by rs7072338. Ultimately, we detected two SNPs (LINC01515 (rs7072338) and CLUAP1 (rs17794023)) are associated with CSF α-synuclein levels. Moreover, SNPs near APP (rs1394839), RAPGEF1 (rs10901091), and two intergenic loci on chromosome 2 and 14 (rs11687064 and rs7147386) were identified as suggestive loci associated with CSF α-synuclein levels. Function of LINC01515 has never been explored yet, while CLUAP1 appears to be involved in AD-linked cognitive deterioration as a consequence of their interactions with Aβs (Armato et al. 2013). Previous studies indicated that CLUAP1 is involved in ciliogenesis and impacts cognitive deterioration in AD as a consequence of the neurogenesis process occurring in the hippocampus (Armato et al. 2013; Botilde et al. 2013). Besides causing cognition impairment, missense mutation in the CLUAP1 gene was also found to modify the age of onset in PSEN1 E280A AD (Velez et al. 2016). Beyond that, CLUAP1 plays an important role in carcinogenesis of multiple types of tumors such as osteosarcomas, ovarian, colon, and lung cancers and may be useful as a tumor-associated antigen or a novel therapeutic intervention for treatment in multiple malignancies (Ishikura et al. 2007; Takahashi et al. 2004). Interestingly, our analysis identified one SNP near APP gene as a suggestive locus. Mutations in APP that increase production of APP-derived Aβ cause autosomal dominant forms of familial AD (FAD) (Selkoe 2001). Aβ plaques and α-synuclein-rich Lewy bodies are the major neuropathological hallmarks of Alzheimer’s disease (AD) and Parkinson’s disease. Evidence from animal models shows that Aβ may contribute to the development of Lewy body diseases by promoting the aggregation of α-synuclein and exacerbating α-synuclein-dependent neuronal pathologies (Masliah et al. 2001). In addition, α-synuclein may lead to inhibition of Aβ deposition and reduced plaque formation (Bachhuber et al. 2015). The relationships between Aβ and α-synuclein still need further research.
Rs7072338 and rs17794023 are intronic SNPs which can affect protein structure by regulation of alternative splicing, positive regulation of gene expression, and regulation of nonsense-mediated decay (Jo and Choi 2015), and have even been experimentally shown to affect transcription (Greenwood and Kelsoe 2003). Therefore, they may play an important role in α-synuclein levels. In fact, most of the SNPs detected by traditional case-control GWASs have been mapped to intron regions rather than exonic or nonsynonymous sites (Li et al. 2012; Welter et al. 2014). Investigation of the functional implication of these intronic SNPs will thus be an important research subject in the future. However, our data are not whole exome or genome and full sequencing data within the region may reveal other candidate causal variants. Further exploration in larger populations will be necessary to assess whether and how these SNPs contribute to α-synuclein-related functions and disorders. In addition, participants included in our study were AD oriented; according to the results of subgroup analysis, our findings could be generalized to cognitively normal population. However, whether these findings could be generalized to other populations (e.g., Parkinson disease) has never been assessed and still needs further exploration.
Conclusion
We have identified an association between two genetic significant variants and four suggestive loci with CSF α-synuclein levels. Our results have important implications for a better understanding of α-synuclein regulation and allow researchers to further explore the relationships between these SNPs and α-synuclein-related neurodegenerative disorders.
References
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ (2013) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 28:811–813
Armato U, Chakravarthy B, Pacchiana R, Whitfield JF (2013) Alzheimer’s disease: an update of the roles of receptors, astrocytes and primary cilia (review). Int J Mol Med 31:3–10
Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F, Abou-Ajram C, Nuscher B, Serrano-Pozo A, Muller A, Prinz M, Steiner H, Hyman BT, Haass C, Meyer-Luehmann M (2015) Inhibition of amyloid-beta plaque formation by alpha-synuclein. Nat Med 21:802–807
Botilde Y, Yoshiba S, Shinohara K, Hasegawa T, Nishimura H, Shiratori H, Hamada H (2013) Cluap1 localizes preferentially to the base and tip of cilia and is required for ciliogenesis in the mouse embryo. Dev Biol 381:203–212
Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC, Consortium G, Alzheimer's Disease Neuroimaging I, Alzheimer Disease Genetic C, Goate AM (2013) GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78:256–268
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 17:1945–1947
Gai WP, Power JH, Blumbergs PC, Blessing WW (1998) Multiple-system atrophy: a new alpha-synuclein disease? Lancet 352:547–548
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640
Greenwood TA, Kelsoe JR (2003) Promoter and intronic variants affect the transcriptional regulation of the human dopamine transporter gene. Genomics 82:511–520
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM (2013) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117
Hall S, Ohrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F, Nilsson C, Hakan W, Decraemer H, Nagga K, Minthon L, Londos E, Vanmechelen E, Holmberg B, Zetterberg H, Blennow K, Hansson O (2012) Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol 69:1445–1452
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Leverenz JB, Baird G, Montine TJ, Hancock AM, Hwang H, Pan C, Bradner J, Kang UJ, Jensen PH, Zhang J (2010) DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133:713–726
Ishikura H, Ikeda H, Abe H, Ohkuri T, Hiraga H, Isu K, Tsukahara T, Sato N, Kitamura H, Iwasaki N, Takeda N, Minami A, Nishimura T (2007) Identification of CLUAP1 as a human osteosarcoma tumor-associated antigen recognized by the humoral immune system. Int J Oncol 30:461–467
Jo BS, Choi SS (2015) Introns: the functional benefits of introns in genomes. Genomics Inform 13:112–118
Kim S, Swaminathan S, Shen L, Risacher SL, Nho K, Foroud T, Shaw LM, Trojanowski JQ, Potkin SG, Huentelman MJ, Craig DW, DeChairo BM, Aisen PS, Petersen RC, Weiner MW, Saykin AJ, Alzheimer's Disease Neuroimaging I (2011) Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology 76:69–79
Korff A, Liu C, Ginghina C, Shi M, Zhang J, Alzheimer's Disease Neuroimaging I (2013) alpha-Synuclein in cerebrospinal fluid of Alzheimer’s disease and mild cognitive impairment. J Alzheimer's Dis 36:679–688
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48
Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, Verny C, Brice A, French Parkinson’s Disease Genetics Study G (2013) G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73:459–471
Li MJ, Wang P, Liu X, Lim EL, Wang Z, Yeager M, Wong MP, Sham PC, Chanock SJ, Wang J (2012) GWASdb: a database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res 40:D1047–D1054
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L (2001) beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98:12245–12250
Mollenhauer B, Cullen V, Kahn I, Krastins B, Outeiro TF, Pepivani I, Ng J, Schulz-Schaeffer W, Kretzschmar HA, McLean PJ, Trenkwalder C, Sarracino DA, Vonsattel JP, Locascio JJ, El-Agnaf OM, Schlossmacher MG (2008) Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol 213:315–325
Mollenhauer B, El-Agnaf OM, Marcus K, Trenkwalder C, Schlossmacher MG (2010) Quantification of alpha-synuclein in cerebrospinal fluid as a biomarker candidate: review of the literature and considerations for future studies. Biomark Med 4:683–699
Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, Paetau A (2014) Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 35(2180):e2181–e2185
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH (2013) A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80:1062–1064
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Shi M, Bradner J, Hancock AM, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Kang UJ, Cain KC, Wang Y, Aasly J, Goldstein D, Zhang J (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 69:570–580
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Takahashi M, Lin YM, Nakamura Y, Furukawa Y (2004) Isolation and characterization of a novel gene CLUAP1 whose expression is frequently upregulated in colon cancer. Oncogene 23:9289–9294
Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J (2013) CSF alpha-synuclein improves diagnostic and prognostic performance of CSF tau and Abeta in Alzheimer’s disease. Acta Neuropathol 126:683–697
Velez JI, Rivera D, Mastronardi CA, Patel HR, Tobon C, Villegas A, Cai Y, Easteal S, Lopera F, Arcos-Burgos M (2016) A mutation in DAOA modifies the age of onset in PSEN1 E280A Alzheimer’s disease. Neural Plast 2016:9760314
Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, Srulijes K, Trojanowski JQ, Lee VM, Siderowf AD, Hurtig H, Litvan I, Schiess MC, Peskind ER, Masuda M, Hasegawa M, Lin X, Pan C, Galasko D, Goldstein DS, Jensen PH, Yang H, Cain KC, Zhang J (2012) Phosphorylated alpha-synuclein in Parkinson’s disease. Sci Transl Med 4:121ra120
Wang HF, Wan Y, Hao XK, Cao L, Zhu XC, Jiang T, Tan MS, Tan L, Zhang DQ, Tan L, Yu JT, As DNI (2016) Bridging integrator 1 (BIN1) genotypes mediate Alzheimer’s disease risk by altering neuronal degeneration. J Alzheimer's Dis 52:179–190
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42:D1001–D1006
Yoshimoto M, Iwai A, Kang D, Otero DA, Xia Y, Saitoh T (1995) NACP, the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc Natl Acad Sci U S A 92:9141–9145
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173
Funding
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. This work was supported by grants from the Taishan Scholars Program of Shandong Province (ts201511109 and tsqn20161079), Qingdao Key Health Discipline Development Fund, Qingdao Outstanding Health Professional Development Fund, and Qingdao Innovation and Entrepreneurship Leading Talent Program.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
The study procedures were approved by the institutional review boards of all participating centers (https://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf). Ethics approval was obtained from the institutional review boards of each institution involved: Oregon Health and Science University; University of Southern California; University of California—San Diego; University of Michigan; Mayo Clinic, Rochester; Baylor College of Medicine; Columbia University Medical Center; Washington University, St. Louis; University of Alabama at Birmingham; Mount Sinai School of Medicine; Rush University Medical Center; Wien Center; Johns Hopkins University; New York University; Duke University Medical Center; University of Pennsylvania; University of Kentucky; University of Pittsburgh; University of Rochester Medical Center; University of California, Irvine; University of Texas Southwestern Medical School; Emory University; University of Kansas, Medical Center; University of California, Los Angeles; Mayo Clinic, Jacksonville; Indiana University; Yale University School of Medicine; McGill University, Montreal-Jewish General Hospital; Sunnybrook Health Sciences, Ontario; U.B.C.Clinic for AD & Related Disorders; Cognitive Neurology—St. Joseph’s, Ontario; Cleveland Clinic Lou Ruvo Center for Brain Health; Northwestern University; Premiere Research Inst (Palm Beach Neurology); Georgetown University Medical Center; Brigham and Women’s Hospital; Stanford University; Banner Sun Health Research Institute; Boston University; Howard University; Case Western Reserve University; University of California, Davis—Sacramento; Neurological Care of CNY; Parkwood Hospital; University of Wisconsin; University of California, Irvine—BIC; Banner Alzheimer’s Institute; Dent Neurologic Institute; Ohio State University; Albany Medical College; Hartford Hospital, Olin Neuropsychiatry Research Center; Dartmouth-Hitchcock Medical Center; Wake Forest University Health Sciences; Rhode Island Hospital; Butler Hospital; UC San Francisco; Medical University South Carolina; St. Joseph’s Health Care Nathan Kline Institute; University of Iowa College of Medicine; Cornell University; and University of South Florida: USF Health Byrd Alzheimer’s Institute.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Informed Consent
Informed consent was obtained from all participants or their authorized representatives.
Additional information
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Electronic supplementary material
Supplementary Table 1
Title: Association results for CSF α-synuclein in ADNI; Description: Abbreviations: CHR = Chromosome; SNP=Single nucleotide polymorphism; BP = base pair location in release 19, build 135 of the human genome in the dbSNP database; A1 = The minor allele; MAF = Minor allele frequency in ADNI; SNP.Type = Type of SNP; BETA = Change CSF α-synuclein per copy of the minor allele, in which positive numbers indicate more rapid decline and negative numbers indicate slower decline; P = relationship between SNPs and CSF α-synuclein using multiple linear regression model adjust for educational years, APOE ε4 status and baseline disease status. (XLSX 12 kb)
Supplementary Fig.1
Title: Linkage disequilibrium surrounding rs7072338 in both the ADNI cohort and the 1000 Genomes cohort; Description: Abbreviations: BP = base pair location in release 19, build 135 of the human genome in the dbSNP database. (pdf 588 kb). (PDF 588 kb)
Supplementary Fig.2
Multiple linear regression models were used to estimate possible correlation between genotypes of rs7072338 and CSF α-synuclein concentration (PDF 111 kb)
Supplementary Fig.3
Multiple linear regression models were used to estimate possible correlation between genotypes of rs17794023 and CSF α-synuclein concentration (PDF 112 kb)
Rights and permissions
About this article

Cite this article
Zhong, Xl., Li, JQ., Sun, L. et al. A Genome-Wide Association Study of α-Synuclein Levels in Cerebrospinal Fluid. Neurotox Res 35, 41–48 (2019). https://doi.org/10.1007/s12640-018-9922-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-018-9922-2