
Abstract
Salidroside exhibits anti-inflammatory, anti-oxidative, and anti-apoptotic properties. To identify whether salidroside might be a candidate for treating ischemic stroke, we investigated the effects of salidroside or vehicle, given daily for 6 days, after middle cerebral artery occlusion (MCAO) for 2 h and reperfusion for either 1 or 48 h in rats. Salidroside reduced cerebral infarct volume and significantly improved neurological scores whether started after 1 or 48 h of reperfusion. Microarray analysis showed that 20 % (133/678) of the genes down-regulated by ischemia and 1 h of reperfusion were up-regulated by salidroside, whereas 13 % (105/829) of the genes induced by ischemia–reperfusion were inhibited by salidroside, suggesting that salidroside can reverse effects of ischemia–reperfusion on gene expression. The main enriched functional categories induced by salidroside were genes related to synaptic plasticity, whereas salidroside inhibited genes related to inflammation. Induction of Egr1, Egr2, Egr4, and Arc by salidroside was confirmed by qRT-PCR and western blotting in ischemic brains treated after either 1 or 48 h of reperfusion. The potential protective role of Egr4 in salidroside-mediated neuroprotection was subsequently investigated in CoCl2-treated PC12 cells. Egr4 was dose-dependently induced by salidroside in PC12 cells, and depleting Egr4 with target-specific siRNA increased caspase-3 activity and Bax, but decreased Bcl-xl, which were reversed by salidroside. Finally, we confirmed that salidroside inhibited the Bax/Bcl-xl-related apoptosis after MCAO with reperfusion. In conclusion, salidroside is highly neuroprotective with a wide therapeutic time window after ischemia–reperfusion injury in the rat, and this partially involves induction of Egrs, leading to inhibition of Bax/Bcl-xl-related apoptosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pathophysiological profile of ischemic stroke includes interruption of cerebral blood flow, leading to energy depletion, mitochondrial dysfunction, and neuronal cell death by necrosis or apoptosis (Muresanu et al. 2012). However, despite progress in our understanding, effective pharmacological treatments of ischemic stroke are still lacking. In the last two decades, more than 100 agents have been tested in clinical trials for their ability to reduce infarct size and, hence, improve clinical outcome (Goldstein 2007; Muresanu et al. 2012; Smith 2004). Nevertheless, tissue-type plasminogen activator remains the only FDA-approved drug for the treatment of ischemic stroke, although it is only effective in 3–5 % of patients due to its narrow therapeutic time and safety window (Goldstein 2007; Muresanu et al. 2012; Smith 2004). Thus, there is a critical need for the development of novel treatments of ischemic stroke.
Early growth response proteins (Egrs), a family of zinc finger transcription factors that include Egr1, Egr2, Egr3, and Egr4, are induced during neuronal activity and associated with synaptic plasticity (Bozon et al. 2003; DeSteno and Schmauss 2008; Williams et al. 1995). Egr transcripts are also increased in response to neuroprotective agents such as erythropoietin (EPO) and hypothermia after focal cerebral ischemia/reperfusion injury in the rat (Mengozzi et al. 2012; Ohta et al. 2007), and they are associated with neuronal survival in global cerebral ischemia in gerbils (Honkaniemi and Sharp 1996). In contrast, it has been reported that Egr1 induces apoptosis of rat cerebellar granule neurons via transactivation of Bim gene expression (Xie et al. 2011). Thus, the roles of Egrs in neuronal cell survival need further investigation.
Salidroside [2-(4-hydroxyphenyl)ethyl β-d-glucopyranoside, PubChem CID: 159278] is a bioactive component of Rhodiola rosea (Spasov et al. 2000). Salidroside is known to have anti-inflammatory, anti-oxidative, and anti-apoptotic effects in in vitro and in vivo. It exhibits anti-oxidative and anti-apoptotic effects on PC12 and SH-SY5Y cells following hypoglycemia and hydrogen peroxide insults (Yu et al. 2008; Zhang et al. 2007), and it exerts antioxidant effects on hematopoietic stem cells through a mechanism that involves activation of poly(ADP-ribose)polymerase-1 (Li et al. 2012). It attenuates myocardial ischemia–reperfusion injury in rabbits via activation of PI3K/Akt signaling (Xu et al. 2013), and it improves homocysteine-induced endothelial dysfunction in rats via a reduction in oxidative stress (Leung et al. 2013). Salidroside also reduces brain tissue loss, neuronal damage, and apoptosis following cerebral impact injury in mice (Chen et al. 2012), and rescues hippocampal neurones in rats exposed to the cytotoxic agent streptozotocin (Qu et al. 2012). In addition, animals pre-conditioned with salidroside exhibit smaller cerebral infarct volumes and lower neurological deficit scores after middle cerebral artery occlusion (MCAO) and reperfusion (Shi et al. 2012). However, the therapeutic time window and the underlying mechanisms of any effects of salidroside when administrated postoperatively to MCAO animals are unknown. Addressing these questions will help identify whether salidroside might be a candidate for the treatment for ischemic stroke. We therefore examined cerebral infarct volume, neurological deficit score, neuronal apoptosis, and gene expression profiles in the ischemic brains of rats given daily postoperative salidroside, or placebo, for 6 days after MCAO with reperfusion for either 1 or 48 h. The potential protective role of Egr4, one of the genes up-regulated by salidroside, was subsequently investigated in CoCl2-treated PC12 cells.
Materials and Methods
Animals
A total of 138 male Sprague–Dawley rats weighing 220 ± 20 g (7–8 weeks old) were purchased from the Slac Laboratory Animal Co. Ltd. (Shanghai, China). The animals were housed in groups of 5 rats/cage under controlled temperature (21–23 °C) and humidity (55–75 %) conditions, with a 12-h light/dark cycle, and acclimatized to for 7–10 days prior to surgery. Food and water were provided ad libitum throughout the study.
All animal experiments were conducted in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol for this study was approved by the Animal Care and Use Committee of Fujian University of Traditional Chinese Medicine.
Stroke Model, Neurological Deficit Scoring, and Drug Treatments
The animals were weighed before surgery and every day thereafter. Transient MCAO was conducted as described previously with some modifications (Kiyota et al. 1993). Briefly, the animals were anesthetized with 10 % chloral hydrate (3 ml kg−1). A silicone-coated nylon monofilament was inserted from the left common carotid artery to the origin of the middle cerebral artery. After 2 h of occlusion, reperfusion was induced by withdrawing the filament. Sham animals were operated on in the same manner except that the middle cerebral artery was not occluded. During the operation, animals’ body temperatures were monitored continuously and maintained at 37 ± 0.5 °C with a heat lamp and a heating pad.
Behavioral neurological deficit testing was performed 1 h after reperfusion and daily thereafter. Neurological deficit (Longa et al. 1989) was scored by blinded observers according to the following five-point scale: 0, no neurological deficit; 1, failure to extend the left forepaw fully; 2, circling to the left; 3, unable to bear weight on the left side; and 4, no spontaneous walking and/or complete immobility. In a pilot study, we monitored regional cerebral blood flow (rCBF) during MCAO using a laser Doppler flowmeter (AD Instruments, Australia). We found that our technique produced a >70 % decrease from baseline of rCBF after insertion of the occluding filament and a >60 % recovery after filament withdrawal, and that these changes were associated in the MCAO animals with a neurological deficit score of ≥2, 1 h after the start of reperfusion. We therefore subsequently selected a neurological deficit score of <2 at 1 h after reperfusion as exclusion criteria to exclude any animals in which MCAO might have been ineffective.
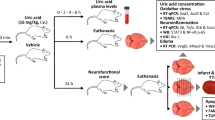
In our main study, 96 animals were subjected to MCAO and 36 rats were sham operated. MCAO rats with neurological deficit scores of 2–4 were randomly assigned to our experiments. Animals received vehicles (saline) or 50 mg kg−1 salidroside intraperitoneally once daily for 6 days, commencing 1 h (Groups 1, 2, 3) or 48 h (Groups 4, 5, 6) after reperfusion. Groups 1 and 4 were sham-operated rats that received vehicle (n = 12/group). Groups 2 and 5 were MCAO rats that received vehicle (n = 22 and 23, respectively). Groups 3 and 6 were MCAO rats that received salidroside (n = 22/group). After 6 days, 12, 16, 17, 12, 16, and 16 animals had survived in Group 1–6, respectively. A further sham-operated group (n = 12) was treated with 50 mg kg−1 salidroside intraperitoneally, starting 1 h after operation and continued for 6d. All surviving animals in each group were then sacrificed. For determination of infarct size by histology and TUNEL assays, brains from 6 animals of each group were perfusion-fixed with a mixture of 40 % formaldehyde, glacial acetic acid, and methanol (1:1:8 by volume) under a constant pressure of 110 mmHg, and then dissected and embedded in paraffin, as previously described (Nakayama et al. 1988; Belayev et al. 1996). Left cerebral hemispheres from the remaining animals were dissected, 3 of which from each group were frozen at −80 °C for protein extraction and the others were kept in RNAlater (Life Technologies, Grand Island, NY) for RNA extraction.
Assessment of Infarct Volume
Total cerebral volume and infarct volume were determined from coronal 10 μm sections cut from paraffin-embedded brains using a microtome (Thermo Scientific HM325), at 2 mm intervals throughout each brain except that sections were taken at 1 mm intervals from bregma +2 mm to bregma −3 mm, the region of the main infarct, as described by Fujimoto et al. (2008). Sections were stained with 0.1 % cresyl violet (Leagene, Beijing) for 10 min followed by quick differentiation in 1 % glacial acetic acid/70 % ethanol, dehydration, and mounting. The infarct area that was defined as the unstained region, the right hemisphere area, and the left hemisphere area in each section was measured using a digital medical image analysis system (Motic Med 6.0, Motic China Group, Xiamen, China). The volume was calculated as the integrated product of the cross-sectional area and the inter-sectional distance. The effect of edema on apparent infarct volume was corrected as described previously (Belayev et al. 1996), using the formula below:
Corrected Infarct volume (%) = Right hemisphere volume − (left hemisphere volume − measured infarct volume)/right hemisphere volume × 100 %.
TUNEL Staining
Apoptotic cells were detected in the section (5 μm) adjacent to the ones used for Nissl staining, using a DeadEnd™ Fluorometric TUNEL System kit in accordance with the manufacturer’s instructions (Promega, Wisconsin). The labeled cells were observed under a laser confocal microscope (ZEISS LSM710), with a 488-nm excitation light, and cells emitting green fluorescence in their nuclei were considered apoptotic. The average number of apoptotic cells across five random visual fields (200× magnification) per section in the peri-infarct zone was recorded by a blinded observer.
RNA Extraction, RNA Labeling, and Array Hybridization
Ischemic cerebral hemispheres were individually homogenized in TRIzol (Life Technologies, Grand Island, NY). Total RNA was extracted with an RNeasy Mini Kit, in accordance with the manufacturer’s instructions (Qiagen, Hilden, Germany). Three individual RNA samples from each group (sham-operated, untreated MCAO with 1 h of reperfusion, or MCAO with 1 h of reperfusion + salidroside) were submitted to microarray analysis on an Agilent Array platform (9 samples in total). Sample preparation and microarray hybridization were performed according to the manufacturer’s standard protocols (Agilent Technology, Santa Clara, CA).
Microarray Data Analysis
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v12.0 software package (Agilent Technologies). After quantile normalization of the raw data, genes that were flagged in all samples were chosen for further analysis. Genes whose expression levels differed significantly (1.5-fold change, p < 0.05, Student’s t test) between two groups (sham vs. MCAO or MCAO vs. MCAO + salidroside) were identified through volcano plot filtering (Li 2012). Functional annotation and biological term enrichment analysis was carried out with reference to the DAVID database, using a significance threshold of p < 0.01 (Huang et al. 2009). Our microarray results were deposited in the public database NCBI GEO: GSE52001 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52001.
Quantitative Real-Time Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
First-strand cDNAs were synthesized from total RNA in the Superscript First-Strand Synthesis System in accordance with the manufacturer’s instructions (Life Technologies, Grand Island, NY). cDNAs were quantified by PCR using SYBR Green real-time PCR master mix (Life Technologies), as per the manufacturer’s instructions. GAPDH was used as an internal control. Results were expressed as fold change relative to sham control, after normalization to GAPDH. The sequences of PCR primers are available upon request.
Western Blot Analysis
Western blot analysis was performed as described previously, with some modifications (Hong et al. 2003). Briefly, protein samples from mitochondria isolated using a Tissue Mitochondria Isolation Kit or a Cell Mitochondria Isolation Kit (Beyotime, China), or from total cell lysates, were separated by SDS-PAGE and probed with the following antibodies purchased from Santa Cruz Biotechnology (Dallas, TX)—anti-Egr1 antibody (1:600 dilution), anti-Egr2 antibody (1:800 dilution), anti-Egr4 antibody (1:800 dilution), and anti-Arc antibody (1:600 dilution)—or with the antibodies from Cell Signaling (Danvers, MA)—anti-caspase-3 antibody (1:1000 dilution) or anti-cleaved-caspase-3 antibody (1:1000 dilution). The blots were stripped and re-probed with anti-β-actin antibody (1:1000 dilution, Cell Signaling) or with mitochondrial loading control anti-Cox IV antibody (1:1000, Abcam, UK).
Images of western blots were captured using a ChemiDoc XRS + imaging system (Bio-Rad, Hercules, CA), and the target protein bands were analyzed with Image J software. β-actin was used as an internal loading control. The results are expressed as fold changes relative to sham-operated controls after normalization of the data to β-actin.
Cell Culture and Treatments of Cells
Rat PC12 cells (American Type Culture Collection, ATCC) were maintained and cultured in accordance with the supplier’s instructions. Cells were seeded in 6-well plates (1 × 105 cells/plate), and chemically induced hypoxia was performed following the protocol described previously with modifications (Hu et al. 2010). Briefly, cells were incubated with vehicle or salidroside at the indicated concentrations in the presence of 200 µM CoCl2 for 48 h, and then harvested for western blot analysis or for caspase-3 activity assay using Caspase-3 Activity Assay Kit (Cell Signaling).
Transfection of Small Interfering RNA (siRNA) into PC12 Cells
Either one of two Egr4-targeted siRNA sequences (Qiagen, Cat# SIO1509025 & SIO1509032), AllStars Control siRNA (Qiagen) or vehicle alone, was transfected into CoCl2-treated cells, using RNAi Human/Mouse Starter Kit (Qiagen) according to the manufacturer’s instructions. Both Egr4-targeted siRNA sequences specifically depleted Egr4 expression (data not shown), and the Egr4-targeted siRNA sequence (Cat# SIO1509025) was used for subsequent experiments.
CoCl2-treated cells were treated in 4 groups. One group was transfected with 10 pM Egr4-targeted siRNA sequence, the second received only 10 µM salidroside, the third received a combination of 10 pM Egr4-targeted siRNA with 10 µM salidroside, and the fourth received only vehicles. A group of cells that received vehicles without CoCl2 treatment was also included. After 48 h of treatment, cells were harvested for caspase-3 activity assay, RT-PCR, and western blot analysis.
Statistical Analysis
The data are presented as means ± standard errors of the mean (SEMs) and were analyzed using one-way ANOVA followed by multiple comparisons with post hoc Bonferroni testing (SPSS 8.0). Neurological deficit scores were analyzed using the Mann–Whitney U test. A p value less than 0.05 was considered statistically significant.
Results
Infarction and Neurological Deficit Scores
Nissl staining showed that neuronal injury was most extensive in the cortices of ischemic brains. This is consistent with previous findings that longer periods of MCAO, such as the 120 min occlusion used in the present experiments, cause extensive infarction in the cortex as well as in subcortical structures (Belayev et al. 1996). Nissl staining also showed that the infarct volume was significantly reduced by salidroside treatment whether started after a period of 1 or 48 h of reperfusion (Fig. 1a, b). In addition, Nissl staining revealed no infarcts in the brains of the sham-operated animals, whether these were treated with placebo or with salidroside (Online Resource 1). Salidroside treatment gradually improved neurological deficit scores after MCAO and reperfusion. After 6 days, salidroside significantly improved neurological deficit scores whether commenced after 1 h or 48 h of reperfusion (Fig. 1c, d).

Salidroside reduces infarct volume and decreases neurological deficit scores. a, b Salidroside reduces infarct volume of MCAO rats received treatment started either 1 h (a) or 48 h (b) of reperfusion, and then continued daily for 6 days. Representative Nissl-stained coronal sections (upper panel), cerebral infarct area from bregma +2 mm to −3 mm (left lower panel), and cerebral infarct volumes (right lower panel). Filled circle sham; open circle MCAO; filled inverted triangle MCAO + salidroside (Sal). The results are presented as mean fold changes relative to sham ± SEM (n = 6 per group), *0.01 < p < 0.05, **p < 0.01. c, d Neurological deficit scores of MCAO rats are decreased by salidroside treatment which is started after either 1 h (c) or 48 h (d) of reperfusion, and then continued daily for 6 days (n = 12–17 per group). **p < 0.01, Mann–Whitney U test
Identification of Genes Regulated by Salidroside in Ischemic Brain Injury
Next, we examined the effects of salidroside treatment on cerebral gene expression profiles. Without salidroside, MCAO and 1 h of reperfusion significantly up-regulated 829 genes and significantly down-regulated 678 genes compared to sham-operated animals (Online Resources 2&3). Salidroside treatment of MCAO rats after 1 h of reperfusion significantly enhanced 209 transcripts (Online Resource 4), of which 133 were from genes down-regulated by ischemia–reperfusion injury (Online Resource 5). These significantly up-regulated genes included Egr1, Egr2, Egr4, and Arc. Salidroside also significantly inhibited 159 transcripts (Online Resource 6), of which 105 derived from genes up-regulated by ischemia–reperfusion injury (Online Resource 7). These significantly down-regulated genes included CD44, CD14, C1s, Ccr5, and A2m, which are involved in inflammatory responses. Only 1 of the 678 transcripts down-regulated by MCAO with 1 h of reperfusion was further reduced by salidroside treatment. None of the 829 genes up-regulated by cerebral ischemia–reperfusion injury were further induced by salidroside. Thus, 20 % (133/678) of the genes down-regulated after ischemia and 1 h of reperfusion were up-regulated again by salidroside, whereas 13 % (105/829) of the genes induced by ischemia–reperfusion were inhibited by salidroside, suggesting that salidroside can reverse effects of ischemia–reperfusion on gene expression.
Analysis employing DAVID functional annotation of the genes that were up-regulated by salidroside treatment of rats subjected to MCAO and 1 h of reperfusion showed that the top-ranking enriched categories were genes concerned with “regulation of neuronal synaptic plasticity,” “regulation of synaptic transmission,” and “regulation of neurological system process,” including the genes such as Egr1, Egr2, Arc, and Vgf (all p < 0.01; Table 1). The main genes down-regulated by salidroside after MCAO and 1 h of reperfusion were genes involved in “copper ion binding” including the genes Sparc, Mt1a, Heph,Cp, and Afp; “inflammatory responses” including the genes CD14, CD44, C1s, Ccr5, Scn9a, and A2m; and “responses to wounding” including the genes CD14, CD44, C1s, Ccr5, Scn9a, A2m, Arg1, Lamb2, and Sparc (all p < 0.01; Table 1).
Validation of Expressions of Egr1, Egr2, Egr4, and Arc in Ischemic Brain
Expressions of Egr1, Egr2, Egr4, and Arc were selected for further validation, as these genes are known to be significantly associated with synaptic plasticity and are induced in ischemic brain by EPO and hypothermia (Mengozzi et al. 2012; Ohta et al. 2007). qRT-PCR results demonstrated that Egr1, Egr2, Egr4, and Arc were down-regulated in ischemic brains compared to sham, and this ischemic effect was reversed in MCAO rats treated with salidroside commencing 1 h after reperfusion (Fig. 2a–d). Western blot analysis showed that the protein levels of Egr1, Egr2, Egr4, and Arc were reduced in the untreated MCAO group compared to the levels observed in the sham-operated group, but normalized in the salidroside-treated MCAO group (Fig. 2a–d), in parallel to the mRNA levels from these genes. In contrast, salidroside did not alter the expressions of Egr1, Egr2, Egr4, and Arc in sham-operated rats (Online Resource 8).

Egr1, Egr2, Egr4, and Arc are induced by salidroside in ischemic brain treated after 1 h of reperfusion. a left hand panel Egr1 mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Egr1 antibody and anti-β-actin antibody and, below, the Egr1 protein level normalized to β-actin level. b left hand panel Egr2 mRNA level normalized to GAPDH mRNA; right hand panel representative western blot with anti-Egr2 antibody and anti-β-actin antibody and, below, the Egr2 protein level normalized to β-actin level. c left hand panel Egr4 mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Egr4 antibody and anti-β-actin antibody and, below, the Egr4 protein level normalized to β-actin level. d left hand panel Arc mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Arc antibody and anti-β-actin antibody and, below, the Arc protein level normalized to β-actin level. All values are expressed as mean fold changes relative to the sham group ± SEM (n = 3 per group). *0.01 < p < 0.05, **p < 0.01
Expressions of Egr1, Egr2, Egr4, and Arc were also investigated in ischemic brain from the animals received salidroside for the first time after 48 h of reperfusion. The mRNA and protein levels of these genes were also down-regulated in the untreated MCAO and 48 h reperfusion group compared to that in the sham-operated controls, but were still normalized if salidroside treatment was started even at the end of the 48 h reperfusion period (Fig. 3a–d).

Egr1, Egr2, Egr4, and Arc are induced by salidroside in ischemic brain treated after 48 h of reperfusion. a Left hand panel Egr1 mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Egr1 antibody and anti-β-actin antibody and, below, the Egr1 protein level normalized to β-actin level. b Left hand panel Egr2 mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Egr2 antibody and anti-β-actin antibody and, below, the Egr2 protein level normalized to β-actin level. c Left hand panel Egr4 mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Egr4 antibody and anti-β-actin antibody and, below, the Egr4 protein level normalized to β-actin level. d Left hand panel Arc mRNA level normalized to GAPDH mRNA; right hand panel Representative western blot with anti-Arc antibody and anti-β-actin antibody and, below, the Arc protein level normalized to β-actin level. All values are expressed as mean fold changes relative to the sham group ± SEM (n = 3 per group). *0.01 < p < 0.05, **p < 0.01
Salidroside-Induced Egr4 is Associated with its Anti-apoptotic Effects in CoCl2-Treated PC12 Cells
As the effects of Egrs on apoptosis of neuronal cells are either controversial (e.g., Egr1 & Egr2) (Honkaniemi and Sharp 1996; Mengozzi et al. 2012; Ohta et al. 2007; Xie et al. 2011) or have not been studied (e.g., Egr4), we investigated the action of Egr4 in CoCl2-treated PC12 cells. CoCl2 treatment significantly increased caspase-3 activity, and decreased the protein levels of both Egr4 and Egr1 in PC12 cells, and all of these effects were dose-dependently reversed by salidroside (Fig. 4a–c).

Salidroside suppresses caspase-3 activity and induces Egr4 and Egr1 in CoCl2-induced PC12 cells. a Caspase-3 activity. b Representative western blots probed with anti-Egr4 antibody and anti-β-actin antibody and, below, Egr4 protein levels normalized to β-actin levels. c Representative western blots probed with anti-Egr1 antibody and anti-β-actin antibody and, below, Egr 1 protein levels normalized to β-actin levels. Cells were treated with vehicle (N), 200 µM CoCl2 (C), or salidroside (Sal) with indicated concentrations in the presence of 200 µM CoCl2 for 48 h. All values are mean ± SEM and expressed as fold changes relative to their respective controls from three independent experiments. *0.01 < p < 0.05, **p < 0.01
When CoCl2-treated PC12 cells were transfected with an Egr4-targeted siRNA, the mRNA and protein levels of Egr4 were decreased (Fig. 5a, b) and the cells developed significantly higher caspase-3 activity (Fig. 5c) compared to cells transfected with a control siRNA. This pro-apoptotic effect of Egr4-targeted siRNA was associated with increases of Bax and decreases of Bcl-xl protein in mitochondria (Fig. 5d). All of these pro-apoptotic effects were reversed by salidroside (Fig. 5c, d). These results suggested that the induction of Egr4 by salidroside may play a critical role in inhibition of the Bax/Bcl-xl-related pathway of apoptosis by salidroside in CoCl2-treated PC12.

Effects of Egr4 siRNA and salidroside on Egr4 levels, caspase-3 activity, mitochondrial Bax, and Bcl-xl levels in CoCl2-treated PC12 cells. a Egr4 mRNA levels normalized to GAPDH mRNA. b Representative western blots probed with anti-Egr4 antibody and anti-β-actin antibody and, below, Egr4 protein levels normalized to β-actin levels. c caspase-3 activity. d Representative western blots probed with anti-Bax antibody and anti-Cox IV antibody and, below, Bax protein levels normalized to Cox IV levels. e Representative western blots probed with anti-Bcl-x1 antibody and anti-Cox IV antibody and, below, Bcl-x1 protein levels normalized to Cox IV levels. Cells were treated with vehicle (N), 200 µM CoCl2 (C), or 10 pM negative control siRNA (NC), or 10 pM Egr4-targeted siRNA, or 10 µM salidroside (Sal), or a combination of 10 pM Egr4-targeted siRNA + Sal in the presence of 200 µM CoCl2 for 48 h. The mRNA transcripts were normalized to GAPDH mRNA. The levels of Egr4 and the mitochondrial Bax and Bcl-x1 were normalized to β-actin and Cox IV, respectively. All values are mean ± SEM and expressed as fold changes relative to their respective controls from three independent experiments. *0.01 < p < 0.05, **p < 0.01
Salidroside Inhibits the Bax/Bcl-xl Mitochondrial Apoptosis in Ischemic Brain
We then investigated whether salidroside inhibits the Bax/Bcl-xl-related mitochondrial pathway of apoptosis in ischemic brains. Whether the sham-operated animals were treated with placebo or with salidroside, no TUNEL-positive cells were detected in the cerebral region that corresponds to the infarcted region in MCAO rats (Online Resource 9). In contrast, salidroside significantly reduced the number of TUNEL-positive cells in the peri-infarct zone of MCAO rats with 1 h of reperfusion (Fig. 6). At the same time, salidroside did not affect total caspase-3 protein expression but did markedly decrease the level of the cleaved, activated form of caspase-3 (Fig. 7a). Moreover, mitochondrial Bax was increased in MCAO rats after 1 h of reperfusion, and this increase was abolished by salidroside (Fig. 7b). In contrast, mitochondrial Bcl-xl was decreased in the MCAO rats, and this decrease was abolished by salidroside (Fig. 7b). Similar inhibition of the Bax/Bcl-xl-related pathway of apoptosis by salidroside was also observed in ischemic brain of MCAO rats with 48 h of reperfusion (data not shown).

Salidroside reduces apoptotic cells in ischemic brain treated after 1 h of reperfusion. Representative images showing TUNEL-positive cells in the peri-infarct zone of MCAO and MCAO + salidroside groups, and the corresponding area for the sham-operated group. The images were captured using laser confocal microscope. Merged images are an overlay of TUNEL (green) and DAPI (blue) staining (magnification, ×400; bar = 50 μm); and, below, the average number of TUNEL-positive cells as a percentage of total cells in 5 random fields from the peri-infarct zone (or, for sham-operated group, the corresponding area) of each section. Data are mean ± SEM (n = 6 per group)

Salidroside inhibits apoptosis involving the Bax/Bcl-xl-related mitochondrial pathway in ischemic brain. a Upper panel Representative western blots probed with anti-caspase-3 antibody, anti-cleaved caspase-3 antibody, and anti-β-actin antibody; middle panel Caspase-3 levels normalized to β-actin levels; lower panel Cleaved caspase-3 levels normalized to β-actin levels. b Upper panel Representative western blots probed with anti-Bax antibody, anti-Bcl-xl antibody, and anti-Cox IV antibody; middle panel Bax levels normalized to Cox IV levels; lower panel Bcl-xl levels normalized to Cox IV levels. The results in the middle and lower panels are presented as mean fold changes relative to the sham group ± SEM (n = 3 per group). *0.01 < p < 0.05, **p < 0.01
Discussion
The present study demonstrates that treatment with salidroside markedly reduced cerebral infarct volume and significantly improved neurological function in rats injured by 2 h of MCAO followed by reperfusion. Oxygen-free radicals and other mechanisms triggered by reperfusion after cerebral ischemia produce cumulative neuronal damage after a period of cerebral ischemia (Traystman et al. 1991), so it is important that salidroside was able to exert these protective effects when treatment was started after reperfusion for 1 h or even after reperfusion that had continued for 48 h. Thus, our results, together with the previous report that salidroside pre-conditioning reduces brain injury in MCAO rats (Shi et al. 2012), indicate that salidroside is effective for the treatment of cerebral ischemia with reperfusion injury over a wide therapeutic time window extending from pre-conditioning before the injury to for periods of up to at least 48 h of reperfusion. A wide therapeutic time window is an important prerequisite for agents that might be used clinically because of the uncertainty over reperfusion times after cerebral ischemia in patients. In this regard, therefore, salidroside, or other agents that share its mechanisms of action, fulfills an important criterion of potential therapeutics for clinical ischemic stroke.
Microarray analysis drew attention to a variety of genes responding to salidroside and therefore, potentially, to its mechanisms of action. Generally, salidroside prevented or reversed many of the effects of ischemia–reperfusion on gene expression. Thus, 20 % (133/678) of the genes down-regulated by ischemia–reperfusion were up-regulated again by salidroside. Conversely, 13 % (105/829) of genes induced by ischemia–reperfusion were inhibited by salidroside treatment. Of course, the expression levels of mRNA expression do not necessarily correlate to protein levels or to enzymic activity, and this may explain why no core component of the apoptosis machinery was identified as responding to salidroside by microarray analysis, although we found that salidroside inhibited apoptosis in ischemic brain. Nevertheless, we found that salidroside clearly modulates transcription of several relevant classes of genes, including those involved in synaptic plasticity and inflammation. The increases in the expressions of Egr1, Egr2, Egr4, and Arc we observed caused by salidroside are particularly interesting given the well-documented roles of these genes in neuronal synaptic plasticity (Bozon et al. 2002, 2003; DeSteno and Schmauss 2008; Williams et al. 1995). The immediate early gene Arc is a master regulator of synaptic plasticity, involved in maintaining LTP and consolidation of long-term memory (Guzowski et al. 2000; Shepherd and Bear 2011), and is highly responsive to synaptic activity and can also be induced by Egr1 or Egr3 (Li et al. 2005). Hence, our data support the notion that salidroside could enhance synaptic plasticity after ischemic brain injury through involvement of Egrs and Arc.
We further investigated a potential protective role for Egrs using CoCl2-treated PC12 cells. Both hypoxia and CoCl2 stimulate the levels of erythropoietin (EPO) through a common pathway, probably involving the same conformational change in an upstream O2-sensing heme protein in Hep3B cells (Goldberg et al. 1988). In addition, CoCl2, like hypoxia, induces hypoxia-inducible factor-1 (HIF-1) expression, and this has been used to model hypoxic responses in HepG2 and Hep3B (Piret et al. 2002; Chandel et al. 1998; Jiang et al. 1997). Similarly, exposure to CoCl2 is known to trigger a spectrum of hypoxia-related responses in PC12 cells, including induction of HIF-1α protein levels, induction of mitochondrial DNA damage, and increased generation of reactive oxygen species (ROS) and apoptosis (Hartwig et al. 2014; Hu et al. 2010; Kotake-Nara et al. 2005; Wang 2000; Wang et al. 2001, 2009; Zou et al. 2001). We also confirmed in the present study that CoCl2 mimicked important cellular responses that otherwise characterize the response of PC12 cells to hypoxia. Thus, treatment of PC12 cells with CoCl2 in vitro induced caspase-3 activity and Bax expression, inhibited expressions of Bcl-xl and Egrs, and caused apoptosis, and that all of these effects were paralleled in vivo by ischemia/reperfusion injury. It is also consistent with the idea that treatment with CoCl2 can model responses to hypoxia, in which we found that salidroside reversed all of these hypoxia-related pro-apoptotic effects in both CoCl2-treated PC12 cells in vitro and in vivo after ischemia/reperfusion injury. Moreover, we showed, using specific RNAi, that the up-regulation of Egr4 by salidroside can reduce apoptosis in PC12 cells exposed to CoCl2 and that, therefore, the up-regulation of Egr4 by salidroside may at least partly explain the neuroprotective effects of salidroside. Up-regulation of Egrs has already been associated with the efficacy of other neuroprotective agents, such as EPO and hypothermia, in studies of cerebral ischemia/reperfusion in rats (Mengozzi et al. 2012; Ohta et al. 2007), and these results, taken together with our in vitro results directly implicating Egr4 in salidroside-mediated neuroprotection, strongly suggest that the in vivo up-regulation of Egr4 caused by salidroside is at least partly responsible for the neuroprotective effects of salidroside in cerebral ischemia/reperfusion injury.
Salidroside inhibition of apoptosis is associated with decrease of mitochondrial Bax and increase of mitochondrial Bcl-xl in CoCl2-treated PC12 and in ischemia/reperfusion brain, which are in good agreement with a previous report that salidroside inhibits the Bax/Bcl-2-related mitochondrial pathway of apoptosis in cerebral impact injury (Chen et al. 2012). Salidroside-mediated inhibition of the Bax/Bcl-xl-related mitochondrial pathway of apoptosis may depend at least partially on the induction of Egrs. Further investigations are required to understand the precise role of Egr4 and other Egrs in salidroside-mediated neuroprotection in vivo. Nevertheless, our results report for the first time that induction of Egr4 could contribute significantly to the mechanisms of the robust neuroprotective effects of salidroside in various in vitro and in vivo experimental models.
The observations that the transcript levels of Egrs and Arc were significantly reduced in ischemic brains 6 days after reperfusion, compared to the sham group, contrast with previous studies showing the rapid induction of these genes following brain ischemia (Honkaniemi and Sharp 1996; Mengozzi et al. 2012). This discrepancy could be due to the different time points tested. Indeed, we have observed that Egr1, Egr2, Egr4, and Arc mRNA levels were increased markedly 1 h after reperfusion, and then progressively decreased at 4, 8, 24 h, and 6 days after reperfusion (data unpublished).
Microarray analysis also revealed that salidroside treatment inhibited a group of genes associated with inflammation such as CD14, CD44, C1s, Ccr5, and A2m. The expression of these genes was confirmed by qRT-PCR (data not shown). Inflammation plays a critical role in the pathophysiology of stroke (Jin et al. 2010). CD44, the mRNA of which was down-regulated by salidroside, is a cell-surface glycoprotein involved in lymphocytic activation and cytokine production. CD44 is induced in microglia, macrophages, and microvessels by cerebral ischemia in rats and could play a role in cerebral inflammation and remodeling (Wang et al. 2001). Moreover, CD44 deficiency protects mice from cerebral ischemia (Wang et al. 2001, 2002). CD14 is another gene that was down-regulated by salidroside. CD14 is a co-receptor with toll-like receptor 4, detecting bacterial lipopolysaccharide, reactive oxygen species, and other triggers to enhance inflammation (Zhou et al. 2013). CD14 is predominately expressed in microglia in the CNS and contributes to neuroinflammatory responses in brain diseases, including cerebral ischemia (Beschorner et al. 2002). Therefore, the reduced expression of these inflammatory genes by salidroside could contribute to its neuroprotective properties. We are currently investigating whether the reduced expression of these inflammatory genes is associated with Egrs induced by salidroside.
In summary, our study provides evidence that salidroside is highly neuroprotective across a wide therapeutic time window of reperfusion after MCAO in the rat. Salidroside has the ability to reverse many effects of ischemia–reperfusion on gene expression, including the ability to re-induce a group of genes, Egr1, Egr2, Egr4, and Arc, associated with synaptic plasticity. In addition, we establish that Egr4 can prevent apoptosis in an in vitro model of oxidative neuronal damage and that its increased expression could contribute to salidroside-mediated neuroprotection in ischemic brain. These findings suggest that salidroside may be an attractive candidate to treat ischemic stroke.
References
Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD (1996) Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke 27:1616–1623
Beschorner R, Schluesener HJ, Gozalan F, Meyermann R, Schwab JM (2002) Infiltrating CD14+ monocytes and expression of CD14 by activated parenchymal microglia/macrophages contribute to the pool of CD14+ cells in ischemic brain lesions. J Neuroimmunol 126:107–115
Bozon B, Davis S, Laroche S (2002) Regulated transcription of the immediate-early gene Zif268: mechanisms and gene dosage-dependent function in synaptic plasticity and memory formation. Hippocampus 12:570–577
Bozon B, Davis S, Laroche S (2003) A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron 40:695–701
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95:11715–11720
Chen SF, Tsai HJ, Hung TH, Chen CC, Lee CY, Wu CH, Wang PY, Liao NC (2012) Salidroside improves behavioral and histological outcomes and reduces apoptosis via PI3K/Akt signaling after experimental traumatic brain injury. PLoS One 7:e45763
DeSteno DA, Schmauss C (2008) Induction of early growth response gene 2 expression in the forebrain of mice performing an attention-set-shifting task. Neuroscience 152:417–428
Fujimoto M, Takagi Y, Aoki T, Hayase M, Marumo T, Gomi M, Nishimura M, Kataoka H, Hashimoto N, Nozaki K (2008) Tissue inhibitor of metalloproteinases protect blood–brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab 28:1674–1685
Goldberg MA, Dunning SP, Bunn HF (1988) Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science 242:1412–1415
Goldstein LB (2007) Acute ischemic stroke treatment in 2007. Circulation 116:1504–1514
Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA (2000) Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci 20:3993–4001
Hartwig K, Fackler V, Jaksch-Bogensperger H, Winter S, Furtner T, Couillard-Despres S, Meier D, Moessler H, Aigner L (2014) Cerebrolysin protects PC12 cells from CoCl2-induced hypoxia employing GSK3beta signaling. Int J Dev Neurosci 38:52–58
Hong G, Lockhart A, Davis B, Rahmoune H, Baker S, Ye L, Thompson P, Shou Y, O’Shaughnessy K, Ronco P, Brown J (2003) PPARgamma activation enhances cell surface ENaCalpha via up-regulation of SGK1 in human collecting duct cells. FASEB J 17:1966–1968
Honkaniemi J, Sharp FR (1996) Global ischemia induces immediate-early genes encoding zinc finger transcription factors. J Cereb Blood Flow Metab 16:557–565
Hu J, Zhao TZ, Chu WH, Luo CX, Tang WH, Yi L, Feng H (2010) Protective effects of 20-hydroxyecdysone on CoCl2-induced cell injury in PC12 cells. J Cell Biochem 111:1512–1521
Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL (1997) Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem 272:19253–19260
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779–789
Kiyota Y, Pahlmark K, Memezawa H, Smith ML, Siesjo BK (1993) Free radicals and brain damage due to transient middle cerebral artery occlusion: the effect of dimethylthiourea. Exp Brain Res 95:388–396
Kotake-Nara E, Takizawa S, Quan J, Wang H, Saida K (2005) Cobalt chloride induces neurite outgrowth in rat pheochromocytoma PC-12 cells through regulation of endothelin-2/vasoactive intestinal contractor. J Neurosci Res 81:563–571
Leung SB, Zhang H, Lau CW, Huang Y, Lin Z (2013) Salidroside improves homocysteine-induced endothelial dysfunction by reducing oxidative stress. Evid Based Complement Alternat Med 2013:679635
Li W (2012) Volcano plots in analyzing differential expressions with mRNA microarrays. J Bioinform Comput Biol 10(6):1231003
Li L, Carter J, Gao X, Whitehead J, Tourtellotte WG (2005) The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol Cell Biol 25:10286–10300
Li X, Sipple J, Pang Q, Du W (2012) Salidroside stimulates DNA repair enzyme Parp-1 activity in mouse HSC maintenance. Blood 119:4162–4173
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Mengozzi M, Cervellini I, Villa P, Erbayraktar Z, Gokmen N, Yilmaz O, Erbayraktar S, Manohasandra M, Van Hummelen P, Vandenabeele P, Chernajovsky Y, Annenkov A, Ghezzi P (2012) Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Acad Sci USA 109:9617–9622
Muresanu DF, Buzoianu A, Florian SI, von Wild T (2012) Towards a roadmap in brain protection and recovery. J Cell Mol Med 16:2861–2871
Nakayama H, Ginsberg MD, Dietrich WD (1988) (S)-emopamil a novel calcium channel blocker and serotonin S2 antagonist, markedly reduces infarct size following middle cerebral artery occlusion in the rat. Neurology 38:1667–1673
Ohta H, Terao Y, Shintani Y, Kiyota Y (2007) Therapeutic time window of post-ischemic mild hypothermia and the gene expression associated with the neuroprotection in rat focal cerebral ischemia. Neurosci Res 57:424–433
Piret JP, Mottet D, Raes M, Michiels C (2002) CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann NY Acad Sci 973:443–447
Qu ZQ, Zhou Y, Zeng YS, Lin YK, Li Y, Zhong ZQ, Chan WY (2012) Protective effects of a Rhodiola crenulata extract and salidroside on hippocampal neurogenesis against streptozotocin-induced neural injury in the rat. PLoS One 7:e29641
Shepherd JD, Bear MF (2011) New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci 14:279–284
Shi TY, Feng SF, Xing JH, Wu YM, Li XQ, Zhang N, Tian Z, Liu SB, Zhao MG (2012) Neuroprotective effects of salidroside and its analogue tyrosol galactoside against focal cerebral ischemia in vivo and H2O2-induced neurotoxicity in vitro. Neurotox Res 21:358–367
Smith WS (2004) Pathophysiology of focal cerebral ischemia: a therapeutic perspective. J Vasc Interv Radiol 15:S3–S12
Spasov AA, Wikman GK, Mandrikov VB, Mironova IA, Neumoin VV (2000) A double-blind, placebo-controlled pilot study of the stimulating and adaptogenic effect of Rhodiola rosea SHR-5 extract on the fatigue of students caused by stress during an examination period with a repeated low-dose regimen. Phytomedicine 7:85–89
Traystman RJ, Kirsch JR, Koehler RC (1991) Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J Appl Physiol 71:1185–1195
Wang G (2000) Mitochondrial DNA damage and a hypoxic response are induced by CoCl2 in rat neuronal PC12 cells. Nucleic Acids Res 28:2135–2140
Wang H, Zhan Y, Xu L, Feuerstein GZ, Wang X (2001) Use of suppression subtractive hybridization for differential gene expression in stroke: discovery of CD44 gene expression and localization in permanent focal stroke in rats. Stroke 32:1020–1027
Wang X, Xu L, Wang H, Zhan Y, Pure E, Feuerstein GZ (2002) CD44 deficiency in mice protects brain from cerebral ischemia injury. J Neurochem 83:1172–1179
Wang S, Hu C, Jiang D, Peng J, Zhou Z, Yuan Q, Nie S, Jiang J, Li Y, Huang K (2009) All-trans retinoic acid inhibits cobalt chloride-induced apoptosis in PC12 cells: role of the dimethylarginine dimethylaminohydrolase/asymmetric dimethylarginine pathway. J Neurosci Res 87:1938–1946
Williams J, Dragunow M, Lawlor P, Mason S, Abraham WC, Leah J, Bravo R, Demmer J, Tate W (1995) Krox20 may play a key role in the stabilization of long-term potentiation. Brain Res Mol Brain Res 28:87–93
Xie B, Wang C, Zheng Z, Song B, Ma C, Thiel G, Li M (2011) Egr-1 transactivates Bim gene expression to promote neuronal apoptosis. J Neurosci 31:5032–5044
Xu MC, Shi HM, Gao XF, Wang H (2013) Salidroside attenuates myocardial ischemia-reperfusion injury via PI3K/Akt signaling pathway. J Asian Nat Prod Res 15:244–252
Yu S, Liu M, Gu X, Ding F (2008) Neuroprotective effects of salidroside in the PC12 cell model exposed to hypoglycemia and serum limitation. Cell Mol Neurobiol 28:1067–1078
Zhang L, Yu H, Sun Y, Lin X, Chen B, Tan C, Cao G, Wang Z (2007) Protective effects of salidroside on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Eur J Pharmacol 564:18–25
Zhou M, Wang CM, Yang WL, Wang P (2013) Microglial CD14 activated by iNOS contributes to neuroinflammation in cerebral ischemia. Brain Res 1506:105–114
Zou W, Yan M, Xu W, Huo H, Sun L, Zheng Z, Liu X (2001) Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation. J Neurosci Res 64:646–653
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81473382), the Department of Science & Technology of Fujian Province (No. 2014Y4004), the Collaborative Innovation Center for Rehabilitation Technology of Fujian University of TCM and the TCM Rehabilitation Research Center of SATCM. The authors would like to thank Mr. Bin Chen for his support in the animal surgery.
Conflict of interest
No competing financial interests are associated with this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Wenfang Lai and Zhenwei Zheng have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12640_2015_9529_MOESM1_ESM.tif
Online Resource 1: Representative Nissl-staining coronal sections show no infarcts in the brains of the sham-operated animals treated with placebo or with salidroside, starting 1 h after operation and continued for 6 d (n = 3 per group). Supplementary material 1 (TIFF 2178 kb)
12640_2015_9529_MOESM2_ESM.xls
Microarray analysis of gene expression in ischemic brain of sham-operated, MCAO received vehicle after 1 h of reperfusion (MCAO) and MCAO received salidroside after 1 h of reperfusion (MCAO + Sal). Differential gene expression was identified based on 1.5-fold change, p < 0.05 difference threshold, Student’s t test. Supplementary material 2 (XLS 310 kb)
12640_2015_9529_MOESM3_ESM.tif
No effect of salidroside on expression of Egr1, 2, 4 and Arc in sham-operated brains. Sham-operated animals were treated with placebo or with salidroside, starting 1 h after operation and continued for 6 d. mRNA levels were measured by qRT-PCR, and expressed as fold change relative to sham control ± SEM (n = 3 per group), after normalization to GAPDH. Supplementary material 3 (TIFF 4426 kb)
12640_2015_9529_MOESM4_ESM.tif
Representative images show no TUNEL-positive cells in the region of sham-operated animals that corresponds to the infarcted region in MCAO rats. Sham-operated animals were treated with placebo or with salidroside, starting 1 h after operation and continued for 6d. The images were captured using laser confocal microscope. Merged images are an overlay of TUNEL (green) and DAPI (blue) staining (magnification, 400 × ; bar = 50 μm). Supplementary material 4 (TIFF 4137 kb)
Rights and permissions
About this article

Cite this article
Lai, W., Zheng, Z., Zhang, X. et al. Salidroside-Mediated Neuroprotection is Associated with Induction of Early Growth Response Genes (Egrs) Across a Wide Therapeutic Window. Neurotox Res 28, 108–121 (2015). https://doi.org/10.1007/s12640-015-9529-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-015-9529-9