
Abstract
The cytotoxic effects of methamphetamine (MA) are well established to be caused via induced oxidative stress which in turn compromises the core function of the blood–brain barrier (BBB) by reducing its ability to regulate the homeostatic environment of the brain. While most studies were conducted over a period of 24–48 h, this study investigated the mechanisms by which chronic exposure of MA adversely affect the endothelial cells of BBB over an extended period of 96 h. MA induced significant depression of cell numbers at 96 h. This result was supported by flow cytometric data on the cell cycle which showed that brain endothelial cells (bEnd5) at 96 h were significantly suppressed in the S-phase of the cell cycle. In contrast, at 24–72 h control cell numbers for G1, S and G2-M phases were similar to MA-exposed cells. MA (0–1,000 µM) did not, however, statistically affect the viability and cytotoxicity of the bEnd5 cells, and the profile of ATP production and DNA synthesis (BrdU) across 96 h did not provide a rationale for the suppression of cell division. Our study reports for the first time that chronic exposure to MA results in long-term disruption of the cell cycle phases which eventuates in the attenuation of brain capillary endothelial cell growth after 96 h, compounding and contributing to the already well-known adverse short-term permeability effects of MA exposure on the BBB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The blood–brain barrier (BBB) is a pivotal interface between the brain parenchyma and the circulating system. This barrier does not only play a vital role in protecting the CNS from toxic substances and pathogens from the systemic circulation, but is also responsible for the rigorous regulation of the homeostatic environment by restricting passive trans-capillary diffusion of ions and hydrophilic molecules (Abbott et al. 2010). Thus, the functional interruption of this barrier may cause the dysfunction of neurons and neural systems within the brain. Methamphetamine (MA) is a highly addictive psychostimulant and has demonstrated neurotoxic properties as well as the ability to compromise the BBB (Dietrich 2009). Most of the reports in the literature have shown that brain endothelial cells are compromised by MA with regards to their short-term permeability (within 24 h) by suppressing the expression of the paracellular tight junctions (TJ) proteins (Ramirez et al. 2009). Molecular studies indicated that the increased permeability across the paracellular TJs were as a result of decreases in the expression of the TJ proteins occludin (Ramirez et al. 2009; Park et al. 2012), claudin 5/3, and ZO1 (Mahajan et al. 2008).
In vitro studies on cultured brain endothelial cells have also shown that MA induces oxidative stress via a pathway producing NADPH oxidase (NOX2), which eventually leads to alteration in the expression of occludin, an integral TJ protein, ultimately resulting in an increase paracellular permeability of the BBB (Park et al. 2012). In addition, MA increases NO production (Martins et al. 2013) which leads to increase transcellular permeability and increased lymphocyte migration across confluent endothelial monolayers. The literature therefore points out that various MA-induced reactive oxidant pathways contribute to a variety of mechanisms which may compromise the crucial regulation of substances across the BBB.
Aside from the afore mentioned short-term (24–48 h) physiological effects of MA on the endothelial cells of the brain, little is known about the longer-term effects on the functional structure of these cells. Several reports in the literature indicated that MA exposure induces long-term structural changes in the brain (Ernst et al. 2000; Jedynak et al. 2007; Todd et al. 2013). These MA-induced changes in the plasticity of the brain may be as a result of cell cycle arrest of the neuronal components crucial for repair and regeneration (Yuan et al. 2011; Van der Wouwer et al. 2012). While there were several in vitro and in vivo studies which reports on the MA-induced cytotoxicity of brain endothelial cells caused by increase oxidative stress (Ramirez et al. 2009; Park et al. 2012; Martins et al. 2013; Sharma and Kiyatkin 2009), all these authors have indicated that MA affects the brain endothelial cells by compromising the “gate-keeper” function, mostly through the suppression of paracellular TJs formation. It was, therefore, relevant to investigate whether brain endothelial cells chronically exposed to MA may also exhibit longer-term metabolic and physiological effects (i.e., over a period of 96 h).
Currently, no studies have reported any mechanism by which MA affects the structure of the cellular components of BBB. Our study shows for the first time that MA has long-term disruptive effects on the division of brain endothelial cells. The study further shows that these changes are not immediate but is only discernible after 96 h, while it endorsed that MA is not directly toxic to the brain endothelial cells. We propose a hypothesis that MA affects the brain endothelial cells via a mechanism that involves the induced generation of ROS which leads to the interruption of cell division after 96 h.
Materials and Methods
Chemicals and Reagents
MA (CAS: 51-57-0) was purchased from Sigma-Aldrich. Suspended bEnd5 cells were used for cell viability analyses using trypan blue (Cat No. T-8154, Sigma-Aldrich). Promega CytoTox 96® Non-Radioactive Cytotoxicity Assay (Cat No. G1780) and CellTiter-Glo® Luminescent Cell viability assay (Cat No. G7571) (Promega, SA) was used for cytotoxicity and ATP production determination, respectively. DNA proliferation was assessed using the BrdU cell proliferation ELISA chemiluminescence assays (Cat No. 11669915001) (Roche, Germany). Cell cycle analysis incorporated propidium iodide (PI) (CAS: 255355-16-4) and ConcertTM RNase A (Cat. No. 1209 1 -039) purchased from Sigma-Aldrich.
Cell Lines and Culturing Conditions
Cell lines established from brain endothelial cells of BALB/c mice, bEnd5 cells purchased from ECACC, Sigma-Aldrich (Cat No. 96091930) were maintained in Dulbecco’s modified Eagle medium (DMEM)/Nut medium (Mix F–12 with glutamax) (Invitrogen, Germany), supplemented with 10 % fetal bovine serum (FBS) 1 % l-pyruvate, 1 % non-essential amino acids (Invitrogen, Germany), and 1 % penicillin, streptomycin (pen-strep) (Sigma-Aldrich), at 37 °C, 5 % CO2 in a humidified atmosphere. Before conducting experiments, cells were washed in 1 X phosphate buffer solution (PBS) (Invitrogen, Germany). Adherent cells were brought into suspension by the addition of 0.25 % Trypsin/EDTA (Invitrogen, Germany). Cell passages used for all analysis ranged between P18 and P40.
Analysis of Percent Cell Viability and Viable Cell Counts
For the trypan blue exclusion viability and cell count analysis, bEnd5 cells were seeded at 2 × 104 cells per 35 mm tissue culture-treated petri-dish. After 24 h, media was removed and cells were incubated with concentrations of 0.1 µM, 1 µM, 0.01 mM, 0.1 mM, and 1 mM MA. Control cells were exposed to supplemented culture media only. Cells were trypsinated at the selected time intervals of 24, 48, 72, and 96 h. Media aspirated prior to the addition of trypsin was also collected and constituted as part of the sample analyzed. Cell viability and percent cell growth was determined using bEnd5 cell suspension, trypan blue and culture media (1:3:6 ratio, respectively). Viable and non-viable cells were quantified on a haemocytometer using phase contrast microscopy. Percent viability was determined by live MA-exposed cells were expressed against those of controls.
Cytotoxic Effects of MA on bEnd5 Cells
The Promega CytoTox 96® Non-Radioactive Cytotoxicity Assay quantitatively measured lactate dehydrogenase (LDH) release. Percentage Toxicity is a reflection of the ratio of the amount of LDH spontaneously released into the supernatant, bathing the cultured cells to the total amount of LDH found in the cytoplasm as well as the cultured medium. Using a 30-min coupled enzymatic assay, the measurement of the released LDH in culture supernatants is based on the reduction of a tetrazolium salt, in a NADH-coupled enzymatic reaction, to a reduced form (a red formazan product), where the amount of color formed is directly proportional to the number of lysed cells measured. 2 × 103 bEnd5 cells/100 μl culture media were seeded in clear-walled 96-well microtiter plates and incubated for 24 h. Media was removed and incubated with selected concentrations of MA for 24, 48, 72, and 96 h. Wells were prepared for (1) spontaneous LDH release from bEnd5 cells formed the control group. For this group, cells were added to the wells at a final combined volume of 100 μl/well, (2) experimental groups contained MA-exposed cells made up to a final volume equal to those of control wells, (3) Maximum LDH release from cells were determined by lysing cells with 10 μl 0.8 % Triton® X-100 per 100 μl of culture medium. This yielded complete lysis of cells when incubated for 45 min prior to harvesting the supernatants, (4) volume correction control was prepared in order to correct for volume changes caused by the addition of the lysis solution. 10 μl of lysis solution was added to a set of wells containing 100 μl of culture medium (without cells), and (5) culture medium background (blanks) corrects for any LDH activity contributed by serum in culture medium and for the presence of phenol red in the culture medium. 100 μl of culture medium only was added to a set of wells. Following the incubation periods stipulated in this study, 50 μl supernatant was transferred to enzymatic assay plates, 50 μl reconstituted substrate mix was added to each well and incubated for 30 min at RT (in a dark room). 50 μl stop solution was then added to each well and the absorbance read at 490 nm.
Cell Titer Glo® Luminescent ATP Cell Viability Analysis
2 × 103 bEnd5 cells were seeded in 100 μl culture media per well in opaque-walled 96-well microtiter plates and incubated for 24 h. Media was removed and incubated with selected concentrations of MA for 24, 48, 72, and 96 h. Wells containing medium without cells were prepared in order to obtain a value for background luminescence, while control wells contained cells supplemented with media only. After selected incubation time intervals, cultured microtiter plates were allowed to equilibrate to RT for approximately 30 min, and a volume of reconstituted CellTiter-Glo Reagent (prepared as per manufacturer’s instructions) equal to the volume of cell culture medium was added to each well. The well contents were gently agitated for 2 min on an orbital shaker to induce cell lysis. The microtiter plates were incubated for an additional 10 min at RT in order to stabilize luminescent signal (with minimal light exposure), and luminescence was recorded using the Glomax multidetection system (Serial No. 51187002, Whitehead Scientific).
DNA Proliferation Detection Using 5-Bromo-2′-deoxyuridine (BrdU) Chemiluminescent Assay
Chemiluminescent Assay2x103 bEnd5 cells were seeded in 100 μl culture medium per well in black-walled 96-well microtiter plates and incubated for 24 h. Media was removed and incubated with selected concentrations of MA for 24, 48, 72, and 96 h. Wells containing media without cells were prepared in order to obtain a value for background luminescence, while control wells contained cells supplemented with media only. After selected incubation time intervals, 10 µl of BrdU labeling solution was added to all wells and incubated for 2 h at 37 °C, 5 % CO2 in a humidified atmosphere. Subsequently the labeling medium was removed, and 200 µl FixDenat was added and incubated at RT for 30 min (with minimal light exposure). After fixation, FixDenat was removed and 100 µl anti-BrdU-POD (antibody) added to each well and incubated for a further 90 min at RT. The anti-BrdU-POD was removed and the microtiter plate washed three times with washing buffer (200 µl/well). Following the final wash, 100 µl of reconstituted substrate was added to each well and the contents were gently shaken for 10 min on an orbital shaker and luminescence was quantified.
Flow Cytometry Analysis of Cell Cycles
bEnd5 cells exposed to selected concentrations of MA at various time intervals were trypsinated and suspended in ice-cold 70 % EtOH to a final volume of 10 ml and were placed at −20 °C for a minimum of 2 h. Cells were centrifuged at 1,000 rpm for 5 min at RT, and EtOH was carefully removed and the pellet resuspended in 1 ml EtOH. The resuspended pellet was transferred to an eppendorf tube and centrifuged with a bench top microfuge for 1 min to ensure pellet formation. The remaining EtOH was removed and the pellet was resuspended in 1 ml of PBS. 20 mg/ml RNase was diluted in PBS at a ratio 1:199 ,respectively, and the required volume of 50 µl per 50 × 104 cells was added to the pellet and incubated for 30 min or RT. PI staining solution was prepared (0.1 % Triton-X100, 2 mM MgCl2, 0.1 M NaCl2, 0.01 M PIPES buffer, pH 6.8, 0.01 mg/ml PI) and made up to the final volume with distilled water. Approximately 20 min preceding flow cytometry analysis, the required volume of PI staining solution was added to the cells (9X the volume of RNase/PBS solution).
Cell Cycle Analysis
Individual samples were subjected to a Becton–Dickinson FACS Calibur flow cytometer with a 488-nm Coherent laser. Nile Red fluorescent particles were used for instrument standardization, stability, and reproducibility. Each analysis was based on at least 10,000 events and each sample was analyzed in triplicate. The software, used for acquisition of data, was the Cellquest Pro version 5.2.1. The cell population was identified and gated (R1) on a forward scatter (FSC) versus side scatter (SSC) dot plot in acquisition mode. Fluorescent Channel 2(FL2) at 575 nm was used for PI detection. A dot plot of FL2A (area) versus FL2 W (width) was used to identify single cells (R2) and thus eliminated doublets. A histogram plot of FL2A was used to enumerate G1/G0, S-phase, and G2/M populations. The combined parameters of FSC, SSC, FL2A, and FL2 W were used to display the results. A threshold on the FSC channel was set to remove sample debris. Analysis and results obtained of acquired data were performed using Modfit version 2.0, software.
Statistical Analysis
The statistical package Medcalc (version 11.5.1) was used to calculate the statistics in this study. The t test was employed for normally distributed samples, while the Wilcoxin Rank Sum Test was used for statistical analysis of samples which were not normally distributed.
Results
Analysis on bEnd5 Cell Growth
Cell number is often a crucial indicator that an experimental vector has implicated the normal physiology of a cell. Over the period of 24–72 h, both control and MA-exposed cells follow a normal and predictable growth profile (see Fig. 1). During this period, statistical differences between controls and MA-exposed cells are limited one or two concentrations and were relatively small. At 24 h, lower concentrations of 0.1, 1, and 10 µM MA displayed higher counts of viable cells when compared to controls (P ≤ 0.014), while higher MA concentrations (100 and 1,000 µM) were not different (P ≥ 0.24). At 48 and 72 h, cell numbers of control cells were statistically similar to MA-exposed cells (P ≥ 0.086) with the exception of cells treated with 10 µM MA which displayed a statistical increase at 48 h (P = 0.019) and at 72 h (P = 0.021).

The effects of MA on endothelial cell numbers. Control bEnd5 cell numbers were compared to MA-exposed bEnd5 cells over 96 h. Only at 96 h did MA-exposed cells display significant suppression of cell growth, where values reflect mean ± SEM. Significant differences (t test; P < 0.05) compared to controls are indicated with an asterisk
At 96 h, all MA-exposure cells produced markedly lower (P ≤ 0.033) viable cell numbers when compared to controls (Fig. 1). 0.1, 1, 10, 100, and 1,000 µM MA resulted in a 35.25, 49.76, 32.33, 21.07, and 47.05 % decrease in viable cell numbers, respectively.
Trypan Blue (TB) Cell Exclusion Viability Analysis
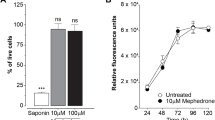
In view of the significant decrease of MA-exposed cell numbers at 96 h, we investigated whether MA affected the viability of cells. The exclusion of TB from cells indicated that they were functionally alive, while those cells which had allowed entry of TB through their compromised membranes were considered as not viable. The percent viability was expressed as a ratio between viable MA-exposed cells and viable cells under controlled conditions. Although the range of MA extended from low physiological concentrations (0.1–10 µM) to supra-physiological concentrations of 100–1,000 µM, no statistically significant difference between control viability and MA-exposed cells were observed (Fig. 2). The lowest viability, also not significantly different (P > 0.05) from controls, was 95.39 ± 0.92 at 48 h and at 1,000 µM for MA-exposed cells. The data indicated that MA, even at supra-physiological concentrations did not affect the viability of the bEnd5 brain endothelial cells.

The effects of MA on brain endothelial cell viability (%). The trypan blue exclusion assay displays that the effects of the selected MA concentrations (µM) had no statistical significant effect on bEnd5 cell viability across the time range of 24–96 h, where values reflect mean ± SEM (Wilcoxin; P < 0.05)
Analysis of MA Toxicity in bEnd5 Cells
As TB exclusion assay is an extremely basic assay, an additional test for cellular toxicity was utilized. Using the LDH assay, toxicity was expressed as a percentage which was reflection of the amount of the cytoplasmic enzyme, LDH, release into the supernatant by critically compromised cells. bEnd5 cells exposed to selected concentrations of MA did not display toxicity levels higher than 6.536 ± 0.460 %, and across the entire range were not statistically different from control values. Thus, despite supra-physiological concentrations to MA (100 and 1,000 µM), no significant increase in the release of LDH to the media was observed, indicating that the bEnd5 endothelial cells across the MA concentration range was not compromised. See Fig. 3 below.

The non-cytotoxic effect of MA on bEnd5 cells is illustrated and values reflect mean ± SEM of percent LDH release into the media bathing by the cells relative to the total LDH present within the cells and that within the media bathing the cells. All means were not significantly different (t test; P < 0.05) from controls, which indicated that MA at the selected concentration did not elicit toxicity
Analysis of DNA Synthesis Using BrdU Incorporation
The possibility that MA, a highly permeable, lipophilic molecule, had entered the nucleus and subsequently obstructed DNA replication was investigated using the BrdU assay (Fig. 4). However, after 24 h, no statistical difference in levels of DNA synthesis was observed. At 48 h, 10 and 100 µM MA-exposed cells had lower DNA synthesis levels when compared to controls (P ≤ 0.043), while in the other concentrations (0.1, 1 and 1,000 µM), percent DNA synthesis (P ≥ 0.25) was similar to control values. At 72 h, only cells exposed to 1 µM MA had statistically lower DNA synthesis levels compared to controls (P = 0.034). At 96 h, all concentrations of MA resulted in similar (P ≥ 0.15) DNA synthesis levels when compared to controls, with the exception of 0.1 µM MA which resulted in slightly lower percent DNA synthesis (P = 0.034).

bEnd5 cells were exposed to selected concentrations (µM) of MA on DNA synthesis, where DNA which had incorporated BrdU during the process of replication (cell division) were expressed in relative light units (RLU) ± SEM (n = 3). Significant differences (Wilcoxin; P < 0.05) to controls are indicated with an asterisk
Overall, there was no discernable pattern in which MA-exposed cells displayed any aligning evidence to clearly implicate DNA replication in a manner which would have decreased cellular proliferation at 96 h.
Effect of MA on ATP Production
The availability of ATP for various energetic demands of the dividing cell is crucial. It is plausible that MA is implicated in the uncoupling of mitochondrial ATP synthesis. We therefore investigated whether MA is linked to the non-availability of ATP. ATP production in bEnd5 cells exposed to MA for 24 h was statistically similar (P ≥ 0.38) to those of controls with the exception of 0.1 µM MA-exposed cells which displayed lower (P = 0.044) levels (Fig. 5).

The effects of MA (µM) on ATP concentrations (expressed in relative light units (RLU) ± SEM). Note the physiological dose response to MA at 48 h, followed by the self-regulated suppression of ATP production at 72 h. ATP concentrations at 96 h are elevated indicating sufficient energy available for the completion of cellular division. Experiments were done in triplicate. Significant differences (t test; P < 0.05) compared to controls are indicated with an asterisk
At 48 h, all cells exposed to MA resulted in significantly higher ATP levels (P ≤ 0.009) when compared to controls, except cells exposed to the lowest concentration of MA (0.1 µM) which was similar to control (P = 0.46). MA appeared to exert a dose response effect on the production of ATP (Fig. 5). It was interesting that at 72 h, those categories of cells which had amplified concentrations of ATP at 48 h, now were significantly suppressed (P ≤ 0.034), while ATP production in 0.1 µM MA-exposed cells remained similar (P = 0.18) to that of controls. The mitochondria seemed to be able to recognize when there was an oversupply of ATP, and subsequently had the ability to suppress production within a 24-h period.
At 96 h, ATP production in MA-exposed cells was markedly higher (P ≤ 0.03) when compared to controls with only the lowest concentration of MA displaying similar values to controls (Fig. 5). This clearly indicated that the cells were not in an energy deficient state and therefore not a plausible rationale for the MA-induced decrease in cell numbers seen at 96 h.
The ATP profile across the 96 h indicates that the mitochondrial machinery to produce ATP was not compromised by exposure to MA, and could respond to the physiological energy requirements of the cell. In all groups that had displayed the decrease in cellular proliferation at 96 h (see Fig. 1), ATP production had been substantially increased, while the cells exposed to the lowest concentration produced similar levels of ATP to controls.
Effects of MA on Cell Cycles Using Propidium Iodide
Flow cytometry was used to determine if MA was implicated in a particular stage in the mitotic cycle of the cell. Each of the graphs in Fig. 6(a–f) represents a snap-shot of a minimum of 10,000 cells (0 (control)-1,000 µM MA) at a distinct point in time: i.e., after being exposed to MA at 96 h. These cells are then categorized into histograms to represent the various stages of the mitotic cell cycle, viz., phases G1, G2-M, and S (Fig. 6g–j). These results indicated that there were no significant differences between controls and MA-exposed cells between 24 and 72 h, as indicated by Fig. 6g–i. However, at 96 h (Fig. 6a–f, j), a distinct divergence was observed in that cells tended to be detained at the G1-phase, and to some extent also at the G2 phase. Data on the S-phase endorsed this finding in that all MA-exposed cells had 24.17–36.14 % less cells than in the control group (Fig. 6j). Relative cell numbers at 96 h showed that the G1 population of MA-exposed cells had increased (52.37–93.4 %), while the S-phase population was lower (24.17–36.14 %) when compared to percent relative numbers between 24 and 72 h (P < 0.05).

A graphical representation of control groups a of bEnd5 cells as well as those exposed to 0.1 µM (b), 1 µM (c), 10 µM (d), 100 µM (e), and 1,000 µM (f) MA for 96 h which were stained with PI and their cell cycles phases analyzed using the FACS Calibur flow cytometer. The graphs g–j presents the combined parameters of FSC, SSC, FL2A, and FL2 W used to display the PI and FL2A used to enumerate G1, S-phase, and G2-M cell populations over the period 24–96 h. Significant differences to controls were not observed between 24 and 72 h (g, h, i). At 96 h (j), significant amounts of MA-exposed cells are detained in the phases G1/G2-M, while statistically less cells were found in the S-phase (P < 0.05). The relative cell numbers detected at the different time intervals were expressed as a percent ±SEM. The histograms (g–j) represent experiments done in triplicate (n = 3)
Discussion
The endothelial cells of the BBB are crucial for the regulation of the internal milieu of the brain and therefore any substance which compromises its gate-keeping function will by default place the brain at risk to the continuous changes in the chemical makeup of the systemic blood vascular system as well as to exposure of blood borne pathogens (Culot et al. 2008). Numerous studies have indicated that MA is indeed such a substance which has the ability to affect the short-term neural functioning of the brain (psychosis) (Bowyer and Ali 2006) and also cause longer-term structural damage to brain tissue (Yuan et al. 2011). Its chemical structure also imbues it with lipophilic characteristics which allow it to passively permeate across the BBB bringing about its primary neurological affects as well as detrimental secondary neuronal morphological and structural effects (Martins et al. 2011). It is not clear from the literature; however, as to the precise mechanism by which MA impacts the functionality of the BBB and if it impairs the functional structure of the endothelial cells. The central hypothesis of this study is that MA impairs the functionality of the BBB by affecting the long-term physiology of the brain endothelial cells. We tested this hypothesis by subjecting MA-exposed bEnd5 cells to a series of assays over a 96-h period.
This is the first study to present data which demonstrate that a MA exposure affects the proliferation of brain endothelial cells dramatically after 96 h. The effects of MA are not prominent between 24 and 72 h, and although small, statistically significant biphasic results are exhibited. At 24 h MA-exposed cells showed a small, but statistical, increase at lower concentrations of MA, this trend only continued at only the 10 µM MA concentration at 48 and 72 h. It was interesting to note that over the whole concentration range, between 24 and 72 h, a biphasic effect was observed (see Fig. 1). It is possible that at the lower concentrations, MA has the ability to potentiate the proliferation of cells by subtlely affecting certain cell cycle proteins, while at higher concentrations the same proteins may undergo further conformational changes, attenuating the cell-cycle process, and thus presenting a biphasic response. However, these effects were not as striking as the effects at 96 h.
At 96 h, the rate of cell division was significantly suppressed relative to controls across the whole concentration range of MA (see Fig. 1). This is the first time this effect is described, in that, between 24–72 h (short term) effects appear to be minimal, but 4 days after MA exposure (long term; 96 h), there is clearly a statistically significant suppressive effect on cell division. This effect is not seen in the short term and is indicative of the long-term influence of MA, which induces a constraint on proliferation of the bEnd5 cells. Subsequently, we used a trypan blue assay to indicate whether MA induced a decrease in the viability while also monitoring the toxicity of MA on the bEnd5 cells, both to obtain a possible rationale for the decrease in the cell numbers at 96 h.
Some studies in the literature indicate that MA does not affect the viability of endothelial cells (Dietrich 2009; Ramirez et al. 2009). To verify these reports, we used a range of MA concentrations, which fell into both the physiological (0.01–10 µM) and also supra-physiological concentrations (100–1,000 µM), to analyse its effects on the viability and toxicity on the bEnd5 endothelial cells (Ramirez et al. 2009; Martins et al. 2011). It was surprising that MA, across both concentration ranges, did not statistically affect either the viability (Fig. 2) or toxicity (Fig. 3) of the bEnd5 cells. These results were corroborated by (Martins et al. 2013) who also showed that treatment with MA (≤100 µM) did not affect the viability of primary rat brain microvascular endothelial cells. Our study both endorses this finding, and in addition, showed that supra-physiological concentrations of MA (1,000 µM) also had no effect on viability, confirming the extremely robust nature of the endothelial cells of the BBB.
These results appear contrary to other studies which have reported that MA indeed is toxic to brain endothelial cells, and does affect the viability of endothelial cells. In contrast, in most of these in vitro studies. the dose of MA was far higher than what is found in the plasma of addicts (plasma concentrations: 2-11 μM), (Melega et al. 2007). For instance, Zhang et al. (2009) used a supra-physiological range of MA of 100 μM to 10 mM, and showed a 20 % toxic effect on HBMVEC cells starting at the 2,500 μM concentration. While in in vivo studies, MA concentrations of 30–100 mg/kg are routinely used (Li et al. 2008; Martins et al. 2011) making it difficult to ascribe the effects on the endothelial cells of the BBB as primary but rather as secondary to the MA-induced extreme hyperthermia and seizures, commonly observed at these concentrations (Dietrich 2009). These MA doses would translate to extremely high concentrations in addicts as a 30-mg/kg dose would translate to a 2.1-g dose in a 70-kg person. It is clear from the literature that these high doses are the exceptional rather than the norm in addicts and recreational users, where using more than 1 g could be fatal (Melega et al. 2007).
BrdU analysis also did not indicate that MA compromised the replication of DNA, although the data showed a depression of DNA synthesis at certain MA concentrations which varied between 24 and 72 h. At 96 h, however, no significant difference was found between controls and the varying MA concentrations, thus indicating that suppression of DNA synthesis is not a plausible rationale for the changes in the proliferation of MA-exposed bEnd5 cells at 96 h (see Fig. 4).
Recently, studies have pointed to MA having an effect on the progression of cells through the cell cycle. Yuan et al. (2011) demonstrated that the long-term MA exposure (4–13 days) resulted in a decrease number of hippocampus immature neurons, which resulted from a decrease proliferation in neural progenitor cells. Furthermore, these authors further reported a reduction in the transition of cells from the G1-phase to the S-phase. Our study reports for the first time that bEnd5 endothelial cells echoed these findings, in that during 24–72 h MA-treated cells did not differ statistically from controls (Fig. 6g–i), but at 96 h, the MA-exposed bEnd5 cells exhibited increased cell numbers in the G1 phase relative to controls and significantly lower cell numbers in the S-phase (see Fig. 6j: 96 h). This demonstrated that dividing cells were not progressing normally through the mitotic phase of the cell cycle, but appeared to be delayed from entering the S-phase. This result provides a rationale for the significant decrease observed in the proliferation rate of MA-exposed cells at 96 h (see Fig. 1). In support of this hypothesis, (Yuan et al. 2011) reported that MA inhibited hippocampal neuronal cell proliferation by decreasing the proliferating pool of progenitor cells that are in the synthesis (S)-phase of the cell cycle, and that reduced number of S-phase cells were not due to alterations in the length of the S-phase. Thus, it appears that the long-term mechanism (i.e., after 96 h) whereby MA affects the inhibition of cells in the G1-phase may be generic and infers that different cell types may be implicated via a similar mechanism.
MA is well known for affecting metabolic pathways and thus the status of cellular metabolism is crucial for the cell’s ability to divide. It was, therefore, logical to investigate whether the interruption at the G1 phase of the cell cycle, which resulted in the reduction in cell proliferation at 96 h, was caused by metabolic exhaustion or failure. We profiled ATP production over 96 h and observed a dose related increase between 24 and 48 h, and at 72 h, a reverse in this trend were observed, while at 96 h, ATP concentration was significantly higher than control values (see Fig. 5). Although it is clear that MA affects ATP production, the results indicate that this does not appear to be the mechanism by which MA induces its inhibitory effects on the cell cycle. Indeed at 96 h, it appears as if the production/stores of ATP had increased in preparation of the metabolic requirements for the cells entering into the S-phase of cell division. Thus, the decrease in cells entering the S-phase is not likely to be as a result of compromised metabolic machinery of the endothelial cells. These findings endorse earlier reports by (Abdul Muneer et al. 2011) who found that MA exposure (up to 100 µM) did not affect the Kerbs cycle of brain endothelial cells.
Furthermore, many studies have reported (Martins et al. 2013; Ramirez et al. 2009; Busu et al. 2013) that MA induces ROS production in brain endothelia cells, and triggers certain molecular processes which ultimately results in the blockages in the cell cycle, and thus cell proliferation is negatively impacted. Busu et al. (2013) showed that MA induces the production of nitric oxide (NO) in BBB endothelial cells. Van der Wouwer et al. (2012) reported that in human neuroblastoma NB69 cells NO induced hypophosphorylation of pRb and down regulated cyclin D1 which contributed to the cell cycle arrest at the G1/S-phase transition. It is, therefore, very plausible that MA may use this generic mechanism to bring about long-term morphological effects in brain tissue, including the endothelial cells of the BBB.
The data presented in this paper clearly indicate that chronic use of MA has long-term adverse effects on the most important cellular component of the BBB, the endothelial cell. In the face of compromised cell division of the endothelial cell, the BBB, as a functional tissue, will not only be adversely implicated in the face of injury, pathogenic infection and the interruption of the normal cycle of cell replacement within the endothelial layer of the capillary, but most importantly in its core function to regulate the homeostatic environment of the neurons of the brain.
Conclusion
In summary, we report for the first time that both low and high concentrations of MA induced suppression of cell division at 96 h, which was collaborated by our cell cycle data. Surprisingly, this affect is only detected after 96 h and not before. Furthermore, we demonstrated that MA does not kill the endothelial cells as established by the toxicology study and supported by the viability, and that metabolically the endothelial cells were not negatively affected by MA. Our data, therefore, support the hypothesis that the mechanism whereby MA exerts its long-term effects may involve the cumulative effect of ROS species on the cell cycle, in particular the blockage of cells in the G1 phase and consequently inhibiting them from entering into the S-phase. These data suggest a mechanism for MA-induced altered BBB morphology in addition to the described altered brain morphology prominent in addicts (Todd et al. 2013; Jedynak et al. 2007; Robinson and Kolb 2004). In view of the presented data, we propose the hypothesis whereby MA compromises the endothelial cell division of the BBB via a ROS induced mechanism, after 96 h.
Furthermore, in view that our study demonstrates that MA produces long-term effects (at 96 h) on the BBB, we caution the proposed use of MA as a therapeutic agent to transiently increase the BBB permeability to temporally allow the entry of drugs, which under normal physiological conditions do not cross (Rau et al. 2011). The data also alerts recreational MA users as to the long-term detrimental effects on the BBB.
References
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37(1):13–25
Abdul Muneer PM, Alikunju S, Szlachetka AM, Murrin LC, Haorah J (2011) Impairment of brain endothelial glucose transporter by methamphetamine causes blood-brain barrier dysfunction. Mol Neurodegener. 6:23
Bowyer JF, Ali S (2006) High doses of methamphetamine that cause disruption of the blood-brain barrier in limbic regions produce extensive neuronal degeneration in mouse hippocampus. Synapse 60:521–532
Busu C, Li W, Caldito G, Aw TK (2013) Inhibition of glutathione synthesis in brain endothelial cells lengthens S-phase transit time in the cell cycle: implications for proliferation in recovery from oxidative stress and endothelial cell damage. Redox Biol 1:131–139
Culot M, Lundquist S, Vanuxeem D, Nion S, Landry C, Delplace Y, Dehouck MP, Berezowski V, Fenart L, Cecchelli R (2008) An in vitro blood-brain barrier model for high throughput (HTS) toxicological screening. Toxicology In Vitro 22:799–811
Dietrich JB (2009) Alteration of blood-brain barrier function by methamphetamine and cocaine. Cell Tissue Res 336:385–392
Ernst T, Chang L, Leonido-Yee M, Speck O (2000) Evidence for long-term neurotoxicity associated with methamphetamine abuse: a 1H MRS study. Neurology 54:1344–1349
Jedynak JP, Uslaner JM, Esteban JA, Robinson TE (2007) Methamphetamine-induced structural plasticity in the dorsal striatum. Eur J Neurosci 25(3):847–853
Li X, Wang H, Qiu P, Luo H (2008) Proteomic profiling of proteins associated with methamphetamine-induced neurotoxicity in different regions of rat brain. Neurochem Int 52(1–2):256–264
Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, Qi M, Toh J, Xu G, Prasad PN, Schwartz SA (2008) Methamphetamine alters blood brain barrier permeability via the modulation of tight junction expression: implication for HIV-1 neuropathogenesis in the context of drug abuse. Brain Res 1203:133–148
Martins T, Baptista S, Gonçalves J, Leal E, Milhazes N, Borges F, Ribeiro CF, Quintela O, Lendoiro E, López-Rivadulla M, Ambrósio AF, Silva AP (2011) Methamphetamine transiently increases the blood-brain barrier permeability in the hippocampus: role of tight junction proteins and matrix metalloproteinase-9. Brain Res 64(1441):28–40
Martins T, Burgoyne T, Kenny BA, Hudson N, Futter CE, Ambrósio AF, Silva AP, Greenwood J, Turowski P (2013) Methamphetamine-induced nitric oxide promotes vesicular transport in blood-brain barrier endothelial cells. Neuropharmacology 65:74–82
Melega WP, Cho AK, Harvey D, Lacan G (2007) Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse 61:216–220
Park M, Hennig B, Toborek M (2012) Methamphetamine alters occludin expression via NADPH oxidase-induced oxidative insult and intact caveolae. J Cell Mol Med 16(2):362–375
Ramirez SH, Potula R, Fan S, Eidem T, Papugani A, Reichenbach N, Dykstra H, Weksler BB, Romero IA, Couraud PO, Persidsky Y (2009) Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J Cereb Blood Flow Metab 29(12):1933–1945
Rau TF, Kothiwal AS, Rova AR, Brooks DM, Rhoderick JF, Poulsen AJ, Hutchinson J, Poulsen DJ (2011) Administration of low dose methamphetamine 12 h after a severe traumatic brain injury prevents neurological dysfunction and cognitive impairment in rats. Exp Neurol 253C:31–40
Robinson TE, Kolb B (2004) Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 1:33–46
Sharma HS, Kiyatkin EA (2009) Rapid morphological brain abnormalities during acute methamphetamine intoxication in the rat: an experimental study using light and electron microscopy. J Chem Neuroanat 37(1):18–32
Todd G, Noyes C, Flavel SC, DellaVedova CB, Spyropoulos P, Chatterton B, Berg D, White JM (2013) Illicit stimulant use is associated with abnormal substantia nigra morphology in humans. PLoS One 8(2):e56438
Van der Wouwer MV, Couzinié C, Serrano-Palero M, González-Fernández O, Galmés-Varela C, Menéndez-Antolí P, Grau L, Villalobo A (2012) Activation of the BRCA1/Chk1/p53/p21(Cip1/Waf1) pathway by nitric oxide and cell cycle arrest in human neuroblastoma NB69 cells. Nitric Oxide 26(3):182–191
Yuan CJ, Quiocho JMD, Kim A, Wee S, Mandyam CD (2011) Extended access methamphetamine decreases immature neurons in the hippocampus which results from loss and altered development of neuronal progenitors without altered dynamics of the S-phase of the cell cycle. Pharmocol Biochem Behav 100:98–108
Zhang X, Banerjee A, Banks WA, Ercal N (2009) N-Acetylcysteine amide protects against methamphetamine-induced oxidative stress and neurotoxicity in immortalized human brain endothelial cells. Brain Res 1275:87–95
Acknowledgments
The present study was supported by a Grant from the Senate Research Committee of University of the Western Cape, Cape Town South Africa. This study was ethically approved by UWC Senate Research Ethics Committee prior to execution of the research proposal.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fisher, D., Gamieldien, K. & Mafunda, P.S. Methamphetamine is not Toxic but Disrupts the Cell Cycle of Blood–Brain Barrier Endothelial Cells. Neurotox Res 28, 8–17 (2015). https://doi.org/10.1007/s12640-015-9520-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-015-9520-5