
Abstract
Purpose of Review
Ductal carcinoma in situ (DCIS) is a non-obligate precursor of invasive breast cancer. Despite numerous studies investigating the progression from in situ to invasive disease, there is still controversy as to the mechanisms by which a DCIS comes to become an invasive cancer. Here, we reviewed the state-of-the-art of the pathologic and molecular characterization of DCIS.
Recent Findings
DCIS displays intra-lesion genetic heterogeneity. Single-cell sequencing studies have demonstrated that progression from DCIS to invasive breast cancer is a complex phenomenon, which can vary from lesion to lesion. While in some DCIS, multiple clones have the ability to invade, in others, clonal selection likely takes place.
Summary
The fact that DCIS displays intra-lesion genetic heterogeneity and that progression varies from patient to patient poses formidable challenges for the development of biomarkers to define the risk of progression to invasive breast cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ductal carcinoma in situ (DCIS) is a non-obligate precursor of invasive breast cancer that has been shown to originate from the terminal ductlobular units (TDLUs) [1]. The detection of DCIS has increased significantly since the 1970s owing to advances in stereotactic biopsy and adoption of mammography screening, to the point that, currently, DCIS accounts for approximately 25% of all diagnosed breast cancers.
The increase in incidence has spurred a greater understanding of the natural history and the clinical and pathologic features of DCIS. We are now cognizant of the fact that this disease is heterogenous and up to 40% of DCIS can progress to invasive disease if untreated [2]. Although there have been some advances in the development of tests to identify the patients at the greatest risk of progression to invasive disease, these assays have proven difficult to be used for the management of individual patients [2]. Understanding which patients are at greatest risk of progression is germane to the identification of those who are in need of further adjuvant treatment after surgical excision. It has been posited that the development of optimal biomarkers for the assessment of risk of progression of DCIS requires an understanding of the genomic alterations and/or epigenetic changes that are required sufficient for DCIS cells to breach the myoepithelial layer and basement membrane.
There are multiple hypotheses for progression from early lesions to invasive carcinoma that are biologically plausible in progression from DCIS to invasive ductal carcinoma (IDC). We [3•, 4, 5••] and others [6, 7••] have previously described three hypothetical models of DCIS progression: (1) a convergent phenotype model wherein multiple genomic and epigenomic events across independent subclones of disease converge onto similar pathways that drive progression of the disease as a whole, (2) an evolutionary bottleneck model wherein only one subclone develops the necessary combination of genomic alterations for progression, and (3) co-migration of multiple clones, whereby an invasive phenotype can result from multiple clones that escape from the duct and invade surrounding tissue [6, 7••]; in the latter context, it is plausible that the ability to invade was acquired rather early in the development of the DCIS or even that the molecular changes that resulted in the development of the DCIS were sufficient for the acquisition of an invasive phenotype.
In this review, we describe some of the extrinsic and intrinsic molecular factors associated with increased risk of DCIS to IDC and discuss which possible markers that can be useful in predicting DCIS behavior. We focus significantly on how the genomic data obtained through massively parallel sequencing and single-cell sequencing approaches have allowed for a better understanding of the genetic heterogeneity of DCIS and IDC, and the insights provided in understanding this complex path towards progression.
Clinical and Pathological Features of DCIS
Although DCIS was first described a century ago [8], it was the seminal work by Wellings and Jensen that postulated DCIS as a precursor lesion to IDC [9]. This was further supported by numerous clinical observations showing a location and temporal correlation of DCIS and IDC. DCIS is not only often found in close proximity to IDC at the time of diagnosis, but longitudinal studies looking at patient outcomes after surgical management of DCIS show a 20–50% risk of developing IDC in the same breast quadrant of the initial DCIS diagnosis [10, 11]. Collins et al. [12] also reviewed biopsies originally diagnosed as benign and identified 13 cases with previously undetected DCIS, of which 46% progressed to invasive carcinoma 5 to 18 years after the initial biopsy.
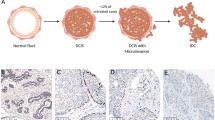
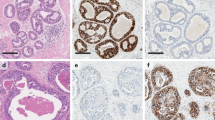
DCIS is not a single disease; rather, in a way akin to invasive breast cancer, it represents a heterogeneous group of cancers with likely distinct cells of origin, histopathologic features, molecular alterations, and clinical behavior [13]. From a histopathologic perspective, DCIS varies in their architecture, nuclear grade, presence of comedo necrosis, multifocality, and size. All these factors can help stratify the aggressiveness and consequently the risks for recurrence and progression of these lesions [13] (Fig. 1). Though historical classification systems attempted to stratify DCIS on the basis of growth patterns and architectural features, contemporary classification systems group lesions primarily on the basis of nuclear atypia, which appears to correlate to some extent with the subsequent risk of invasion [14, 15]. It should be noted, however, that women with DCIS, independent of the nuclear grade, have been shown to be at risk of having invasive cancer [15]. DCIS has been classified into the same intrinsic subtypes as invasive breast carcinoma, based on the expression of estrogen receptor (ER), progesterone receptor (PR), HER2, and using basal markers as EGFR and cytokeratin 5/6 [16,17,18,19]; however, the relative incidence and prognostic implications of these lesions are not as clear [18, 20].

Progression of ductal carcinoma in situ to invasive carcinoma from a histopathologic perspective. Representative micrographs and schematic representation of progressive stages of breast cancer including in situ carcinoma, microinvasive carcinoma and invasive carcinoma
Randomized clinical trials have not only clarified important aspects of the natural history and the pathologic factors involved in progression of DCIS, but also demonstrated that the risk of progression to IDC can be mitigated, at least in part, by radiation therapy and chemoprevention. The first clinical trial addressing DCIS therapy began in 1985, the NSABP B-17, that demonstrated a reduction of ipsilateral DCIS from 15.4% to 9% with surgery plus radiotherapy [21, 22]. Other clinical trials (EORTC 10853 and UK/ANZ) have also provided similar evidence that radiotherapy improved local control following excision [23, 24]. In addition to the clinical and therapeutic insights provided by these trials, they have also offered a wealth of samples to investigate the pathologic features associated with recurrence [21, 22]. DCIS with high nuclear grade, predominantly solid and with extensive comedo necrosis, was identified as a subgroup of poor prognosis [25]. The NSABP B-24 trial tested the addition of tamoxifen to radiotherapy in women submitted to breast conserving surgery for DCIS. The design did not require ER testing of DCIS. Tamoxifen was found to be effective in reducing the risk of ipsilateral and contralateral invasive cancer and DCIS [26]. Some years later, Allred et al. [27] analyzed the ER status of the NSABP B-24 cases and reported a significant decrease in breast cancer events after 10 years of follow-up, which was almost exclusive to the ER-positive lesions; thus, current practice is to recommend tamoxifen to patients with ER-positive DCIS. The frequency of HER2 overexpression in DCIS is around 35% and studies have had encouraging preliminary results of the NSABP B-43 trial regarding the use of trastuzumab for radiosensitization of DCIS lesions [24, 28, 29]. Though these studies help stratify risk, they fail to identify which patients can be spared from these treatments.
In an attempt to establish a method to predict which patients are at risk of relapse or of developing invasive cancer from DCIS, Oncotype DX, a multigene expression assay used in invasive breast cancer was pared down to 15 genes and tested in patients with DCIS from the ECOG E5194 trial [2, 30], a single-arm observational study to evaluate DCIS outcomes following excision with 3 mm margins and no radiation allowed. This assay was found to be effective to predict early but not late recurrences. Also, so far, there is no suggestion that this score predicts benefit from radiotherapy. In addition, it remains to be determined whether this multigene assay provides information above and beyond that offered by centralized and standardized pathologic and immunohistochemical assessment of DCIS [31]. The Prelude DCISionRT biological signature is another commercially available test that indicates the individual risk of recurrence and radiotherapy benefit after breast-conserving therapy [32]. The risk signature incorporates four clinicopathologic factors (age, size, margin status, and palpability) and seven immunohistochemically assessed biomarkers of hormone receptor and HER2 status, stress response, and proliferation (PR, COX2, FOXA1, HER2, Ki67, and p16). This test has been validated in the SweDCIS study population, and so far, it appears to provide predictive information regarding radiotherapy benefit [33]. The Prelude DCISionRT is on trial also in the USA (NCT03448926) with the estimated primary completion date for February 2023.
DCIS Progression: Intrinsic Factors
Genetic Alterations and Gene Expression Profiles
Genomic analyses of paired and almost invariably synchronous DCIS and invasive breast cancer samples have been performed to investigate whether recurrent somatic genetic alterations would be restricted to or overrepresented in the invasive disease. These studies have demonstrated that somatic genetic alterations of DCIS are remarkably similar to those of IDCs [34, 35••]. At the gene level, similar to invasive cancer, recurrent mutations in PIK3CA, TP53, and GATA3 have been identified in DCIS, demonstrating that, at diagnosis, DCIS is already a genetically advanced disease and that the driver genetic alterations identified in early stage invasive breast cancers are already present at the DCIS stage [36••]. These studies have failed to identify somatic mutations that would constitute a common denominator as a driver of progression from in situ to invasive disease. This is not surprising, as invasion may constitute a convergent phenotype. The analyses of specific pathways rather than specific genes may provide insight into the progression. For example, the Myc [37,38,39] or PI3K/AKT pathways have been shown to be important in the progression of a subset of ER-positive/HER2-negative DCIS, though this finding only explains a small subset of cases [40•].
Given that breast cancer is considered to be a C-class tumor (i.e., a tumor type where copy number alterations (CNAs) play a pivotal role in the biology of the disease) [41], CNAs have been postulated as potential drivers of DCIS progression. Quantitative multigene fluorescence in situ hybridization (QM-FISH) analysis of 66 synchronous DCIS and IDC detecting CNAs in 30 genes revealed frequent amplification of MDMx, CCNE2, ERBB2, IGF1R, CKS1BP7, and MYC and frequent deletion of TP53, CHEK1, RB1, CDH1, CHEK2, and NEK9 in breast cancers [39]. This study demonstrated that the levels of genomic alterations observed in DCIS are similar to those of invasive carcinoma. This also suggests that, in most cases, CNAs occur before the acquisition of an invasive phenotype and may not contribute to the development of invasion [39]. Burkhardt et al. used FISH to assess both pure DCIS and DCIS with associated IDC and found no significant differences in CNAs affecting HER2, ESR1, CCND1, or MYC [42]. A meta-analysis including 26 studies did not identify significant differences in CNAs between DCIS and IDC, further suggesting that if CNAs are a significant part of DCIS progression, they constitute an early event in the process [43]. In addition, no particular CNA was necessary or sufficient for the progression to IDC. This is not surprising, given that CNAs appear to develop in bursts of evolution that take place rather early in the development of a breast cancer [44].
Our group sought to investigate the patterns of progression from DCIS to invasive breast cancer on the basis of single-cell sequencing analysis [5••]. Our study revealed that both DCIS and invasive synchronous carcinoma harbored truncal and clonal genetic alterations. Subclonal events, however, were also found in both the DCIS and invasive components. Examples of both clonal selection and multiclonal invasion, however, were observed. This again implies that invasion and progression do not necessarily need to be related to CNAs, as some previous studies suggested [42, 45, 46], or that the ability to invade was an early event followed by genomic instability and the generation of DCIS resistant subclones [5••].
An analysis that used immunohistochemistry-based intrinsic subtyping and array-based comparative genomic hybridization of 22 DCIS and 30 IDC [47] resulted in the identification of 9 breast cancer-related genes, including TP53 and GATA3, that highly contributed to the discrimination of DCIS into two progression risk clusters. The cluster A (rapidly progressive) showed a greater number of gene and chromosome CNAs, a larger IDC/DCIS ratio, a higher frequency of non-luminal subtype, and a higher nuclear grade when compared with the other group (cluster B). These observations may contribute to triage cases by progression risk to more appropriate treatment [47]. All of these observations suggested that progression from DCIS to invasive breast carcinoma is not driven by highly recurrent genetic alterations in DCIS cells [4] and that some degree of stochasticity likely plays a role in the acquisition of molecular alterations prior to the invasion process.
Doebar et al. described distinct gene expression profiles in cases with pure DCIS compared to cases with DCIS with synchronous IDC [35••]. In addition, there was a high genomic concordance between synchronous DCIS and IDC (52 out of 92 mutations were present in both components). The remaining 40 mutated genes, however, were restricted to the invasive component. The proportion of tumor cells with these mutations was higher in the invasive component compared to the DCIS component in a subset of patients. The same authors had previously described genes that were highly expressed in DCIS with IDC, but not in the pure DCIS [48]. Validation in a large cohort would be required to confirm if the differently expressed genes (PLAU, COL1A1, SCGB1D2, S100A7, KRT81, KRT81, NOTCH3, CXCL14, EGFR) could be used to predict progression.
Clonality and Genomic Intratumor Heterogeneity
Subclonal genetic heterogeneity has been independently identified in both the DCIS and invasive components, and the prevalence of individual subclones has been shown to be different between the synchronous DCIS and invasive lesions [4, 5••, 7••]. Our group previously observed after pairwise analysis of mutations in DCIS and synchronous IDC, that 3/13 cases harbored genomic differences, which could potentially propagate an invasive phenotype, which is supportive of the convergent phenotype hypothesis [3•]. Phylogenetic studies have suggested that ancestral relationships between IDC and DCIS are complex and varied. When Newburger et al. assessed multiple lesions with six cases of IDC, they found that the genome of some ancestor cells had a strong predisposition to generate cancerous progeny, as seen by their ability to produce independent subclones with the invasive phenotype. When comparing numbers of single-nucleotide variation and degree of aneuploidy among progeny, it appeared that these events occurred gradually (not as a part of some acute genomic assault that results in invasion) suggesting other factors at play [49, 50].
Observational studies have shown that there are genetic alterations that are not highly shared in synchronous DCIS and IDC, such as TP53 mutations, which are more prevalent in the invasive component than in the DCIS [51], whereas the opposite was found for GATA3 [36]. These data suggest that TP53 mutations may be implicated in the progression of DCIS whereas GATA3 mutations are selected against progression.
Single-cell sequencing approaches have contributed substantially to our understanding of the progression from in situ to invasive breast cancer [5••]. Our study by Martelotto et al. described and validated a method for whole-genome sequencing-based copy number analysis of single cells derived from formalin-fixed paraffin-embedded (FFPE) tissue. Two cases of synchronously diagnosed DCIS and invasive breast cancer were separately microdissected and nuclei prepared. In one case, hierarchical clustering of all nuclei sequenced displayed, among other clonal events, gains of chromosome 1q and losses of chromosomes 11q and 22. Focal amplifications on chromosome 17q encompassing the ERBB2 (17q12-q21.2), PPM1D and BCAS3 (17q22-q23.2) loci were also observed [5••]. Subclonal events such as losses of chromosomes 1p and 8p and alterations on chromosomes 5 and 8 were found in both components, in situ and invasive. Six distinct but highly related subpopulations were identified also in both components. These findings are consistent with the notion that the ability to invade was acquired early in the in situ disease and followed by genomic instability and the development of multiple genetically heterogeneous DCIS subclones that in parallel progressed to invasive disease. In a second case, clonal alterations were identified, such as loss of chromosomes 13q (RB1) and 17p (TP53) as well as focal amplifications on chromosomes 8p11.2-p12 (FGFR1) and 11q13.3-q13.4 (CCND1). Subclonal events included chromosomes 20p–20q gain, loss of chromosome 9, and segmental losses at 3p21.31–p12.3 and 3q21.2–q24. These and other subclonal alterations defined several distinct clusters, which were restricted to either the DCIS or the invasive component. In this case, based on the identified subclones, the reconstructed phylogeny suggested a scenario where intra-tumor genetic heterogeneity occurred early in disease development and that progression from DCIS to invasive carcinoma might have occurred through the selection of a minor subclone of the DCIS. Interestingly, this putative evolutionary bottleneck may have resulted in the selection of a minor subclone of the DCIS harboring a homozygous deletion of PTEN [5••]. Subsequent validation based on FISH analyses confirmed the single-cell sequencing observations. These findings suggest that the DCIS progression models may be different from patient to patient.
To further address these hypotheses [7••], paired DCIS and invasive carcinoma samples were assessed using a technique for spatial mapping of single-cell copy number analysis called topographic single-cell sequencing (TSCS) combined with high-depth exome DNA sequencing analysis, that enabled to infer single-cell CNA profiles. They identified a range of 1–5 abnormal CNA clones in both components, DCIS and IDC, from each of the 10 analyzed patients, no additional CNA events were acquired during invasion. Interestingly, the prevalence of individual subclones did vary between the in situ and invasive components, but the level of clonal diversity was not significantly different [7••]. The authors also found a mean of 87.4% of non-synonymous somatic mutations in known breast cancer genes such as TP53 and PIK3CA, which were concordant in the in situ and invasive carcinoma components, suggesting that they were acquired in the ducts before the invasion. From these findings, they proposed a model of multiclonal progression in which genetic heterogeneity develops within the duct system prior to the invasion. The TSCS study provided evidence to suggest that the genomic heterogeneity is maintained during progression from DCIS to invasive carcinoma [7••]. Therefore, epigenetic or other non-genomic biologic factors are likely important for invasive progression [52] and should be addressed together with genomics in a broad study encompassing normal breast, pure DCIS and DCIS with invasive synchronous tissue.
Extrinsic Factors on DCIS Progression
Since the extracellular matrix (ECM), fibroblasts, myoepithelial cells (MECs), lymphatics, and vascular endothelium to inflammatory cells, all components of the tumor microenvironment play a role and have been revealed substantial changes by gene expression profiling during progression from DCIS to IDC [52]. The invasive growth has been reported to express several matrix metalloproteases (MMP2, MMP11, and MMP14), which are known effectors of cancer progression in several cancer types [53]. Epigenetics changes in the microenvironment of the tumor also play a role in this process. Surrounding stromal cells and MECs phenotypically aberrant lose their normal function and may create a more permissive environment for the transition from in situ to invasive carcinoma [54].
Methylation is the epigenetic alteration most studied in DCIS. Park et al. evaluated the changes in promoter CpG islands hypermethylation during progression from pre-invasive lesions to invasive breast cancer, and found six new methylation markers of breast cancer, namely DLEC1, GRIN2B, HOXA1, MT1G, SFRP4, and TMEFF2, in addition to APC, GSTP1, HOXA10, IGF2, RARB, RASSF1A, RUNX3, SCGB3A1 (HIN-1), and SFRP1 [55]. The promoter CpG island methylation changed significantly in pre-invasive lesions, and was similar in DCIS and IDC, suggesting that CpG island methylation of tumor-related genes is an early event in breast cancer development [55].
Comparing the DNA methylome of 569 breast tissue samples, including 50 from cancer-free women and 84 from matched normal-cancer pairs, revealed dramatic changes between normal, cancer-free tissue, and normal adjacent tissue with widespread DNA methylation field effects [56]. The methylation patterns in the adjacent normal tissue were found strongly enriched at genetic regulator elements as CTCF and RAD21, proteins critical to chromatin looping suggesting that chromatin remodeling in adjacent normal tissue is critical to DCIS progression. The authors proposed a model where clonal epigenetic reprogramming towards reduced differentiation in normal tissue is an essential step in breast carcinogenesis [56]. Although the described epigenetic alterations in the tumor progression, much remains to be clarified about the mechanisms and the physiopathological role of the methylation in the whole process of invasion.
The Role of Myoepithelial Cells
In addition to the basement membrane (BM), normal breast ducts have a MEC layer between the epithelium and the BM. When DCIS cells acquire the invasive phenotype, they infiltrate through both the MEC and BM layers. Normal MECs and MEC layer integrity are seen in benign lesions, and they are reported to have natural tumor suppressor functions as the maintenance of the BM and the epithelial polarity, including expression of tumor suppressor proteins (p63, p73, WT-1, maspin, and laminin 1). During progression from in situ to invasive disease, interactions between intraductal malignant cells and MECs, which should exert tumor-suppressive effects, eventually allow the progression to invasive disease [57,58,59]. There is evidence that MECs lining ducts colonized by DCIS are phenotypically aberrant, lacking some of their differentiation markers and displaying up-regulation of genes related to angiogenesis and invasion [60]. Accordingly, normal MECs were shown to suppress tumor growth and invasion in the absence of detectable genomic alterations in the tumor epithelial cells. The elimination of mediators of myoepithelial differentiation-related pathways led to MEC loss and progression to invasive disease [59]. Interestingly, it has been noted that the sensitivity of some MEC markers using immunohistochemistry is lower in DCIS-associated MECs than in normal MECs, reflecting the presumable genetic alterations in these cells [61]. Rakha et al. described that MECs isolated from DCIS show gene expression and epigenetic differences compared to MECs isolated from normal breast tissue [62]. Hu et al. demonstrated the role of MECs and fibroblasts in the progression of DCIS to IDC using a mouse model of DCIS. They highlighted the importance of p63, sonic hedgehog signaling, TGFβ, and cell adhesion in the myoepithelial cell differentiation, without which DCIS had an enhanced propensity for progression [59]. Other studies have implicated cell-specific markers in the DCIS, namely CXCL14 in myoepithelial cells and CXCL12 in fibroblasts [63]. Exploring such differences further is clearly clinically important due to their potential as biomarkers of invasive progression.
The Role of Immune Cells
The immune system plays a role in tumor progression, probably by sculpting the immunogenic phenotype of tumors as they develop. The recognition that immunity plays a dual role in the complex interactions between tumors and the host prompted a refinement of the cancer immune-surveillance that is a critical step in tumor evolution [64, 65].
While the tumor infiltrating lymphocyte (TIL) score was not found to be associated with recurrence in breast cancer, there was a trend for recurrent lesions to have fewer TILs than the primary tumors. The loss of TILs in recurrent lesions may reflect suppression of anti-tumor adaptive immune responses during recurrence [52].
The studies analyzing leukocytes and the prognostic value of TILs in breast cancer have focused on invasive tumors, rather than DCIS. DCIS and premalignant lesions have been relatively neglected in the evaluation of lymphocyte role in the progression of DCIS to invasive disease. In the genomic characterization of DCIS, gene expression signatures implying the presence of activated T cells in a subset of tumors were identified [66]. Also, regulatory T cells (Treg) seemed to be increased during tumor progression, suggesting that this could be a marker of risk progression [67]. These two findings are not supported by other studies, which showed that a subset of CD8+ T cells (GZMB+CD8+ and Ki67+CD8+) were very common within DCIS and decreased in local invasive disease, implying that CD8+ cells could be potentially better biomarkers than Tregs for predicting invasive progression [65].
It has been reported that in triple-negative breast cancers (TNBCs), the invasive components have a higher fraction of T cells than DCIS of triple-negative phenotype, implying that more T cells are intermingling with cancer cells in TNBCs but not in HER2-positive breast cancers [65]. Moreover, the opposite was found by the same authors in HER2-positive DCIS, where higher numbers of T cells associated with HER2-positive DCIS compared with HER2-negative DCIS as well as higher numbers of T cells associated with high-grade compared with low-grade DCIS were observed. These findings suggest that HER2-positive DCIS and high-grade DCIS may be associated with a more immunogenic environment [52, 65].
The expression of proteins relevant to the use of immunotherapy for breast cancer patients has been investigated in DCIS. PD-L1, whose expression can be detected in immune cells, in particular CD68-positive cells, tumor cells have been found to display varying expression patterns in breast cancers. In a study regarding the immune escape in DCIS progression, the analysis of each T cell subtypes in DCIS and in synchronous DCIS-IDC by single-cell sequencing associated with functional studies may help to clarify the role of these cells in DCIS progression.
Conclusions
The understanding of the molecular processes involved in the DCIS progression to invasive breast cancer has been fundamentally transformed in the last decade; however, our ability to predict the behavior of individual cases of DCIS remains limited. The surgical treatment with or without sentinel lymph node biopsy followed by radiotherapy and endocrine therapy is still based on clinicopathological and immunohistochemical parameters.
The progression of DCIS to invasive disease is a complex biological phenomenon that involves considerable genomic and epigenetic alterations of the tumor and its microenvironment (Fig. 2), and the mechanisms of progression may vary from patient to patient (e.g., clonal selection vs multiclonal invasion). Despite these challenges, the implementation of cost-effective single-cell sequencing DNA, RNA, and ATAC sequencing approaches coupled with the constitution of international consortia of investigators tackling the biology of DCIS and the validation of biomarkers to predict their clinical behavior will likely result in the development of approaches for the management of DCIS that are biology-driven rather than based on empiricism.

Hypothetical models of invasion of ductal carcinoma in situ (DCIS). a Invasive ductal carcinoma can originate de novo or from non-obligate precursors, following an independent evolutionary pattern. b The evolutionary bottleneck progression postulates that, in a heterogeneous lesion with multiple subclones harboring private mutations, a single subclone is selected and with the support of the microenvironment escapes the basement membrane constituting the invasive carcinoma. c Multiple subclones with support of the microenvironment can escape the basement membrane and co-migrate to adjacent tissue constituting the invasive carcinoma. DCIS, ductal carcinoma in situ; IDC, invasive ductal carcinoma; Treg, regulatory T cells
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ. WHO classification of tumors of the breast. 4th Edition ed. IARC Press; 2012.
Solin LJ, Gray R, Baehner FL, Butler SM, Hughes LL, Yoshizawa C, et al. A multigene expression assay to predict local recurrence risk for ductal carcinoma in situ of the breast. J Natl Cancer Inst. 2013;105(10):701–10. https://doi.org/10.1093/jnci/djt067.
• Hernandez L, Wilkerson PM, Lambros MB, Campion-Flora A, Rodrigues DN, Gauthier A, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227(1):42–52. https://doi.org/10.1002/path.3990. They demonstrate that some DCIS and invasive breast carcinomas are composed of mosaics of neoplastic cells that harbor genetic alterations in addition to the founder genetic events, and the selection of populations with specific genomic alterations takes place in the progression from DCIS to invasive breast cancer.
Cowell CF, Weigelt B, Sakr RA, Ng CK, Hicks J, King TA, et al. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol. 2013;7(5):859–69. https://doi.org/10.1016/j.molonc.2013.07.005.
•• Martelotto LG, Baslan T, Kendall J, Geyer FC, Burke KA, Spraggon L, et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat Med. 2017;23(3):376–85. https://doi.org/10.1038/nm.4279. Applying single-cell sequencing to FFPE tumor material, they studied the progression from in situ to invasive breast cancer, which revealed that DCIS display intra-tumor genetic heterogeneity at diagnosis and that these lesions may progress to invasive breast cancer through a variety of evolutionary processes.
Casasent AK, Edgerton M, Navin NE. Genome evolution in ductal carcinoma in situ: invasion of the clones. J Pathol. 2017;241(2):208–18. https://doi.org/10.1002/path.4840.
•• Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell. 2018;172(1–2):205–17.e12. https://doi.org/10.1016/j.cell.2017.12.007. The authors developed a spatially resolved single-cell genome sequencing method. Studying DCIS-IDC, genomic evolution was reported to occur in the DCIS, and invasion was found to entail the co-migration of multiple clones into the adjacent tissues.
Allegra CJ, Aberle DR, Ganschow P, Hahn SM, Lee CN, Millon-Underwood S, et al. National Institutes of Health state-of-the-science conference statement: diagnosis and Management of Ductal Carcinoma in Situ September 22-24, 2009. J Natl Cancer Inst. 2010;102(3):161–9. https://doi.org/10.1093/jnci/djp485.
Wellings SR, Jensen HM. On the origin and progression of ductal carcinoma in the human breast. J Natl Cancer Inst. 1973;50(5):1111–8.
Sanders ME, Schuyler PA, Simpson JF, Page DL, Dupont WD. Continued observation of the natural history of low-grade ductal carcinoma in situ reaffirms proclivity for local recurrence even after more than 30 years of follow-up. Mod Pathol. 2015;28(5):662–9. https://doi.org/10.1038/modpathol.2014.141.
Page DL, Dupont WD, Rogers LW, Jensen RA, Schuyler PA. Continued local recurrence of carcinoma 15-25 years after a diagnosis of low grade ductal carcinoma in situ of the breast treated only by biopsy. Cancer. 1995;76(7):1197–200.
Collins LC, Tamimi RM, Baer HJ, Connolly JL, Colditz GA, Schnitt SJ. Outcome of patients with ductal carcinoma in situ untreated after diagnostic biopsy: results from the Nurses' health study. Cancer. 2005;103(9):1778–84. https://doi.org/10.1002/cncr.20979.
Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378(9805):1812–23. https://doi.org/10.1016/S0140-6736(11)61539-0.
Hanby AM. The pathology of breast cancer and the role of the histopathology laboratory. Clin Oncol (R Coll Radiol). 2005;17(4):234–9.
Maxwell AJ, Clements K, Hilton B, Dodwell DJ, Evans A, Kearins O, et al. Risk factors for the development of invasive cancer in unresected ductal carcinoma in situ. Eur J Surg Oncol. 2018;44(4):429–35. https://doi.org/10.1016/j.ejso.2017.12.007.
Bryan BB, Schnitt SJ, Collins LC. Ductal carcinoma in situ with basal-like phenotype: a possible precursor to invasive basal-like breast cancer. Mod Pathol. 2006;19(5):617–21. https://doi.org/10.1038/modpathol.3800570.
Meijnen P, Peterse JL, Antonini N, Rutgers EJ, van de Vijver MJ. Immunohistochemical categorisation of ductal carcinoma in situ of the breast. Br J Cancer. 2008;98(1):137–42. https://doi.org/10.1038/sj.bjc.6604112.
Tamimi RM, Baer HJ, Marotti J, Galan M, Galaburda L, Fu Y, et al. Comparison of molecular phenotypes of ductal carcinoma in situ and invasive breast cancer. Breast Cancer Res. 2008;10(4):R67. https://doi.org/10.1186/bcr2128.
Muggerud AA, Hallett M, Johnsen H, Kleivi K, Zhou W, Tahmasebpoor S, et al. Molecular diversity in ductal carcinoma in situ (DCIS) and early invasive breast cancer. Mol Oncol. 2010;4(4):357–68. https://doi.org/10.1016/j.molonc.2010.06.007.
Clark SE, Warwick J, Carpenter R, Bowen RL, Duffy SW, Jones JL. Molecular subtyping of DCIS: heterogeneity of breast cancer reflected in pre-invasive disease. Br J Cancer. 2011;104(1):120–7. https://doi.org/10.1038/sj.bjc.6606021.
Fisher ER, Dignam J, Tan-Chiu E, Costantino J, Fisher B, Paik S, et al. Pathologic findings from the National Surgical Adjuvant Breast Project (NSABP) eight-year update of protocol B-17: intraductal carcinoma. Cancer. 1999;86(3):429–38.
Fisher B, Land S, Mamounas E, Dignam J, Fisher ER, Wolmark N. Prevention of invasive breast cancer in women with ductal carcinoma in situ: an update of the National Surgical Adjuvant Breast and bowel project experience. Semin Oncol. 2001;28(4):400–18.
Bijker N, Meijnen P, Peterse JL, Bogaerts J, Van Hoorebeeck I, Julien JP, et al. Breast-conserving treatment with or without radiotherapy in ductal carcinoma-in-situ: ten-year results of European Organisation for Research and Treatment of Cancer randomized phase III trial 10853--a study by the EORTC breast Cancer cooperative group and EORTC radiotherapy group. J Clin Oncol. 2006;24(21):3381–7. https://doi.org/10.1200/JCO.2006.06.1366.
Han MS, Khan SA. Clinical trials for ductal carcinoma in situ of the breast. J Mammary Gland Biol Neoplasia. 2018;23:293–301. https://doi.org/10.1007/s10911-018-9413-3.
Pinder SE, Duggan C, Ellis IO, Cuzick J, Forbes JF, Bishop H, et al. A new pathological system for grading DCIS with improved prediction of local recurrence: results from the UKCCCR/ANZ DCIS trial. Br J Cancer. 2010;103(1):94–100. https://doi.org/10.1038/sj.bjc.6605718.
Fisher B, Dignam J, Wolmark N, Wickerham DL, Fisher ER, Mamounas E, et al. Tamoxifen in treatment of intraductal breast cancer: National Surgical Adjuvant Breast and bowel project B-24 randomised controlled trial. Lancet. 1999;353(9169):1993–2000. https://doi.org/10.1016/S0140-6736(99)05036-9.
Allred DC, Anderson SJ, Paik S, Wickerham DL, Nagtegaal ID, Swain SM, et al. Adjuvant tamoxifen reduces subsequent breast cancer in women with estrogen receptor-positive ductal carcinoma in situ: a study based on NSABP protocol B-24. J Clin Oncol. 2012;30(12):1268–73. https://doi.org/10.1200/JCO.2010.34.0141.
Liang K, Lu Y, Jin W, Ang KK, Milas L, Fan Z. Sensitization of breast cancer cells to radiation by trastuzumab. Mol Cancer Ther. 2003;2(11):1113–20.
Siziopikou KP, Anderson SJ, Cobleigh MA, Julian TB, Arthur DW, Zheng P, et al. Preliminary results of centralized HER2 testing in ductal carcinoma in situ (DCIS): NSABP B-43. Breast Cancer Res Treat. 2013;142(2):415–21. https://doi.org/10.1007/s10549-013-2755-z.
Hughes LL, Wang M, Page DL, Gray R, Solin LJ, Davidson NE, et al. Local excision alone without irradiation for ductal carcinoma in situ of the breast: a trial of the eastern cooperative oncology group. J Clin Oncol. 2009;27(32):5319–24. https://doi.org/10.1200/JCO.2009.21.8560.
Solin LJ, Gray R, Hughes LL, Wood WC, Lowen MA, Badve SS, et al. Surgical excision without radiation for ductal carcinoma in situ of the breast: 12-year results from the ECOG-ACRIN E5194 study. J Clin Oncol. 2015;33(33):3938–44. https://doi.org/10.1200/JCO.2015.60.8588.
Bremer T, Whitworth PW, Patel R, Savala J, Barry T, Lyle S, et al. A Biological Signature for Breast Ductal Carcinoma. Clin Cancer Res. 2018;24(23):5895–901. https://doi.org/10.1158/1078-0432.CCR-18-0842.
Wärnberg F, Garmo H, Emdin S, Hedberg V, Adwall L, Sandelin K, et al. Effect of radiotherapy after breast-conserving surgery for ductal carcinoma in situ: 20 years follow-up in the randomized SweDCIS trial. J Clin Oncol. 2014;32(32):3613–8. https://doi.org/10.1200/JCO.2014.56.2595.
Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100(10):5974–9. https://doi.org/10.1073/pnas.0931261100.
•• Doebar SC, Krol NM, van Marion R, Brouwer RWW, van Ijcken WFJ, Martens JM, et al. Progression of ductal carcinoma in situ to invasive breast cancer: comparative genomic sequencing. Virchows Arch. 2018;474(2):247–51. https://doi.org/10.1007/s00428-018-2463-5. The authors found a high genomic concordance between synchronous DCIS and invasive breast cancers. The proportion of tumor cells harboring shared mutations between the DCIS and IDC was higher in the invasive component in a subset of patients. Their findings support the notion that progression of DCIS to invasive carcinoma could be driven by clonal selection.
•• Pang JB, Savas P, Fellowes AP, Mir Arnau G, Kader T, Vedururu R, et al. Breast ductal carcinoma in situ carry mutational driver events representative of invasive breast cancer. Mod Pathol. 2017;30(7):952–63. https://doi.org/10.1038/modpathol.2017.21. GATA3 mutations in DCIS were found to be a marker of less aggressive behavior. In contrast, TP53 mutations were associated with adverse tumor characteristics.
Corzo C, Corominas JM, Tusquets I, Salido M, Bellet M, Fabregat X, et al. The MYC oncogene in breast cancer progression: from benign epithelium to invasive carcinoma. Cancer Genet Cytogenet. 2006;165(2):151–6. https://doi.org/10.1016/j.cancergencyto.2005.08.013.
Heselmeyer-Haddad K, Berroa Garcia LY, Bradley A, Ortiz-Melendez C, Lee WJ, Christensen R, et al. Single-cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol. 2012;181(5):1807–22. https://doi.org/10.1016/j.ajpath.2012.07.012.
Pan A, Zhou Y, Mu K, Liu Y, Sun F, Li P, et al. Detection of gene copy number alterations in DCIS and invasive breast cancer by QM-FISH. Am J Transl Res. 2016;8(11):4994–5004.
• Sakr RA, Weigelt B, Chandarlapaty S, Andrade VP, Guerini-Rocco E, Giri D, et al. PI3K pathway activation in high-grade ductal carcinoma in situ--implications for progression to invasive breast carcinoma. Clin Cancer Res. 2014;20(9):2326–37. https://doi.org/10.1158/1078-0432.CCR-13-2267. The authors found genetic alterations affecting the PI3K pathway in a subset of pure DCIS and DCIS adjacent to invasive breast cancer. A receptor subtype-matched comparison of pure DCIS and DCIS adjacent to invasive breast cancer revealed that PIK3CA hotspot mutations and pAKT expression were significantly more prevalent in ER-positive /HER2-negative DCIS adjacent to invasive carcinoma. They concluded that molecular alterations affecting the PI3K pathway may play a role in the progression from high-grade DCIS to invasive breast cancer in a subset of ER-positive/ HER2-negative lesions.
Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–33. https://doi.org/10.1038/ng.2762.
Burkhardt L, Grob TJ, Hermann I, Burandt E, Choschzick M, Jänicke F, et al. Gene amplification in ductal carcinoma in situ of the breast. Breast Cancer Res Treat. 2010;123(3):757–65. https://doi.org/10.1007/s10549-009-0675-8.
Rane SU, Mirza H, Grigoriadis A, Pinder SE. Selection and evolution in the genomic landscape of copy number alterations in ductal carcinoma in situ (DCIS) and its progression to invasive carcinoma of ductal/no special type: a meta-analysis. Breast Cancer Res Treat. 2015;153(1):101–21. https://doi.org/10.1007/s10549-015-3509-x.
Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–60. https://doi.org/10.1038/nature13600.
Robanus-Maandag EC, Bosch CA, Kristel PM, Hart AA, Faneyte IF, Nederlof PM, et al. Association of C-MYC amplification with progression from the in situ to the invasive stage in C-MYC-amplified breast carcinomas. J Pathol. 2003;201(1):75–82. https://doi.org/10.1002/path.1385.
Jang M, Kim E, Choi Y, Lee H, Kim Y, Kim J, et al. FGFR1 is amplified during the progression of in situ to invasive breast carcinoma. Breast Cancer Res. 2012;14(4):R115. https://doi.org/10.1186/bcr3239.
Kitamura M, Nakayama T, Mukaisho KI, Mori T, Umeda T, Moritani S, et al. Progression potential of ductal carcinoma in situ assessed by genomic copy number profiling. Pathobiology. 2018;86:1–10. https://doi.org/10.1159/000492833.
Doebar SC, Sieuwerts AM, de Weerd V, Stoop H, Martens JWM, van Deurzen CHM. Gene expression differences between ductal carcinoma in situ with and without progression to invasive breast Cancer. Am J Pathol. 2017;187(7):1648–55. https://doi.org/10.1016/j.ajpath.2017.03.012.
Newburger DE, Kashef-Haghighi D, Weng Z, Salari R, Sweeney RT, Brunner AL, et al. Genome evolution during progression to breast cancer. Genome Res. 2013;23(7):1097–108. https://doi.org/10.1101/gr.151670.112.
Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, et al. Lobular carcinomas. Clin Cancer Res. 2018;25:674–86. https://doi.org/10.1158/1078-0432.CCR-18-1103.
Abba MC, Gong T, Lu Y, Lee J, Zhong Y, Lacunza E, et al. A molecular portrait of high-grade ductal carcinoma in situ. Cancer Res. 2015;75(18):3980–90. https://doi.org/10.1158/0008-5472.CAN-15-0506.
Nelson AC, Machado HL, Schwertfeger KL. Breaking through to the other side: microenvironment contributions to DCIS initiation and progression. J Mammary Gland Biol Neoplasia. 2018;23(4):207–21. https://doi.org/10.1007/s10911-018-9409-z.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74. https://doi.org/10.1038/nrc745.
Hu M, Yao J, Cai L, Bachman KE, van den Brûle F, Velculescu V, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905. https://doi.org/10.1038/ng1596.
Park SY, Kwon HJ, Lee HE, Ryu HS, Kim SW, Kim JH, et al. Promoter CpG island hypermethylation during breast cancer progression. Virchows Arch. 2011;458(1):73–84. https://doi.org/10.1007/s00428-010-1013-6.
Teschendorff AE, Gao Y, Jones A, Ruebner M, Beckmann MW, Wachter DL, et al. DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun. 2016;7:10478. https://doi.org/10.1038/ncomms10478.
Gudjonsson T, Rønnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115(Pt 1:39–50.
Barsky SH, Karlin NJ. Myoepithelial cells: autocrine and paracrine suppressors of breast cancer progression. J Mammary Gland Biol Neoplasia. 2005;10(3):249–60. https://doi.org/10.1007/s10911-005-9585-5.
Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406. https://doi.org/10.1016/j.ccr.2008.03.007.
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M, Marchió C, Reis-Filho JS. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology. 2010;57(2):171–92. https://doi.org/10.1111/j.1365-2559.2010.03568.x.
Hilson JB, Schnitt SJ, Collins LC. Phenotypic alterations in ductal carcinoma in situ-associated myoepithelial cells: biologic and diagnostic implications. Am J Surg Pathol. 2009;33(2):227–32. https://doi.org/10.1097/PAS.0b013e318180431d.
Rakha EA, Miligy IM, Gorringe KL, Toss MS, Green AR, Fox SB, et al. Invasion in breast lesions: the role of the epithelial-stroma barrier. Histopathology. 2018;72(7):1075–83. https://doi.org/10.1111/his.13446.
Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32. https://doi.org/10.1016/j.ccr.2004.06.010.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. https://doi.org/10.1146/annurev.immunol.22.012703.104803.
Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune escape in breast Cancer during. Cancer Discov. 2017;7(10):1098–115. https://doi.org/10.1158/2159-8290.CD-17-0222.
Kristensen VN, Vaske CJ, Ursini-Siegel J, Van Loo P, Nordgard SH, Sachidanandam R, et al. Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc Natl Acad Sci U S A. 2012;109(8):2802–7. https://doi.org/10.1073/pnas.1108781108.
Lal A, Chan L, Devries S, Chin K, Scott GK, Benz CC, et al. FOXP3-positive regulatory T lymphocytes and epithelial FOXP3 expression in synchronous normal, ductal carcinoma in situ, and invasive cancer of the breast. Breast Cancer Res Treat. 2013;139(2):381–90. https://doi.org/10.1007/s10549-013-2556-4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jorge S. Reis-Filho reports personal fees from Belgian Volition, Goldman Sachs, Invicro, Paige.AI, Roche Diagnostics, Grail and Genentech outside the submitted work. Kimberly Dessources, Ana Paula Martins Sebastiao, Fresia Pareja and Britta Weigelt declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Translational Research
Rights and permissions
About this article

Cite this article
Dessources, K., Sebastiao, A.P.M., Pareja, F. et al. How Did We Get There? The Progression from Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Curr Breast Cancer Rep 11, 175–184 (2019). https://doi.org/10.1007/s12609-019-00318-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12609-019-00318-2