
Abstract
The purpose of this study was to investigate the role of Lactobacillus rhamnosus GG (LGG) probiotics in radiation enteritis using in vivo mice. A total of 40 mice were randomly assigned to four groups: control, probiotics, radiotherapy (RT), and RT + probiotics. For the group of probiotics, 0.2 mL of solution that contained 1.0 × 108 colony-forming units (CFU) of LGG was used and orally administered daily until sacrifice. For RT, a single dose of 14 Gy was administered using a 6 mega-voltage photon beam to the abdominopelvic area. Mice were sacrifice at day 4 (S1) and day 7 (S2) after RT. Their jejunum, colon, and stool were collected. A multiplex cytokine assay and 16 s ribosomal RNA amplicon sequencing were then performed. Regarding cytokine concentrations in tissues, pro-inflammatory cytokines, such as tumor necrosis factor-α, interleukin-6 and monocyte chemotactic protein-1, showed significantly decreased protein levels in colon tissues of the RT + probiotics group than in the RT alone group (all p < 0.05). As for comparing microbial abundance through alpha-diversity and beta-diversity, no significant differences were observed between the RT + probiotics and RT alone groups, except for an increase in alpha-diversity in the stool of the RT + probiotics group. Upon analysis of differential microbes based on treatment, the dominance of anti-inflammatory-related microbes, such as Porphyromonadaceae, Bacteroides acidifaciens, and Ruminococcus, was observed in the jejunum, colon, and stool of the RT + probiotics group. With regard to predicted metabolic pathway abundances, the pathways associated with anti-inflammatory processes, such as biosynthesis of pyrimidine nucleotides, peptidoglycans, tryptophan, adenosylcobalamin, and propionate, were differentially identified in the RT + probiotics group compared to the RT alone group. Protective effects of probiotics on radiation enteritis were potentially derived from dominant anti-inflammation-related microbes and metabolites.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radiation enteritis is a common treatment-related toxicity after external beam radiotherapy (RT) in the abdominal or pelvic area. It simply refers to an inflammatory process at the level of small and large intestines as a response to high-energy radiation exposure [1]. Its symptoms include diarrhea, nausea, abdominal pain, and vomiting. More than half of patients who receive pelvic RT experience acute bowel toxicity. Many of them develop subsequent late bowel toxicity with permanent intestinal histological changes [2, 3]. Bowel habit change induced by radiation exposure of the small intestine is considered a major limiting factor of delivering the highest tumoricidal radiation dose for gastrointestinal tract malignancies [4]. Therefore, preventing radiation enteritis and relieving patient’s symptoms have long been concerns for radiation oncologists.
The human gut harbors a complex and abundant aggregation of microbes that are collectively referred to as gut microbiota involved in host’s physiological functions including nutrition, immune development, and host defense [5]. Irradiation to a healthy gut can alter the composition and function of the gut microbiota, which is referred to as dysbiosis that can lead to aggravation of radiation-induced tissue damage by proinflammatory cytokines [6]. To alleviate this process, modulation of gut microbiome using probiotics has been suggested [7]. Probiotics contain live microorganisms such as Lactobacillus species with beneficial effects on the host by restoring bowel microbiota and reinforcing the intestinal barrier capacity [8]. Some studies have shown that probiotics can prevent radiation enteritis [9]. Experimental animal studies have also demonstrated that probiotics play a positive role in mucosal integrity [10]. Two randomized trials have tested the use of probiotics during pelvic radiotherapy in 214 patients [11, 12] and demonstrated less frequent diarrhea in the probiotics arm. However, the mechanism of how probiotics can prevent or relieve radiation enteritis remains unclear.
We designed this animal experiment to assess protective effect of probiotics on radiation enteritis and to investigate the mechanism involved in such effect. Given the potential effect of probiotics on the gut microbial community, we hypothesized that the mechanism involved in the effect of probiotics could be associated with microbial composition and metabolic pathways in terms of balance between anti-inflammatory and pro-inflammatory signals. Thus, in the current study, we compared gut microbiota and metabolites between mice receiving RT alone and those receiving RT + probiotics using 16 s rRNA amplicon sequencing metagenome methods.
Materials and Methods
Mouse Modeling for Radiation Enteritis
A total of 40 6-week-old C57BL/6 male mice weighing 20 to 25 g were prepared for this study. All mice were randomly assigned to four groups: control, probiotics, RT, and RT + probiotics. Each group was randomly stratified into groups with a sacrifice time of 4 days after irradiation (S1) and 7 days after irradiation (S2). Both RT + probiotics and probiotics groups received 0.2 mL of solution that contained 1.0 × 108 colony-forming units (CFU) of Lactobacillus rhamnosus GG (LGG) daily by using feeding canula until sacrifice. The LGG concentration was established based on the results of Ciorba et al. [13]. For both RT and RT + probiotics groups, on the third day of the study, mice were irradiated with a single dose of 14 Gy using a 6 mega-voltage photon beam. The control and probiotics groups did not receive RT. Radiation was administered to the abdominopelvic area at 2 cm depth through an anterior–posterior field using a linear accelerator (Linac 6 EX, Varian Medical System, Inc., Palo Alto, CA, USA). Dose rate of the irradiation was set at 1.04 Gy per minute. During the radiation delivery process, one mouse in the S2-RT alone group died due to asphyxia by a fixation tool. Detail process of experiment is described in Supplementary Fig. 1. At 4 days (S1) and 7 days (S2) after irradiation, mice were sacrificed. Their entire intestinal tissue samples were harvested. Fecal samples were collected at three time points (day 0, day 7, and day 10) generally before sacrifice. We confirmed the development of radiation enteritis in both RT and RT + probiotics groups by histomorphological comparison with non-irradiated groups (Supplementary Fig. 2 and 3).
Quantitative Analysis of Pro-Inflammatory Cytokine
For tissue cytokine measurements, frozen jejunum and colon tissue samples were subjected to the following processes: 1) thawing, 2) cutting, and 3) homogenization. Tumor Necrosis Factor-α (TNF-α), Interleukin-6 (IL-6), Monocyte Chemotactic Protein-1 (MCP-1), Interleukin-12p40 (IL-12p40), Interleukin-1α (IL-1α), Interleukin-1β (IL-1β), and Interleukin-10 (IL-10) protein levels were then quantitatively measured with a multiplex assay with a BD™ Cytometric Bead Array inflammatory cytokine kit (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s protocol. This kit uses the sensitivity of amplified fluorescence detection by flow cytometry to measure soluble analytes in a particle-based immunoassay. It enables multiplex analysis of complex biological samples on a flow cytometer. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed using a cDNA Synthesis kit (Fermentas, Glen Burnie, MD, USA). For Myeloperoxidase (MPO) assay, a Myeloperoxidase Peroxidation Fluorometric Assay Kit (Cayman chemical, Ann Arbor, Michigan) was used according to the manufacturer’s protocol. This kit can detect MPO peroxidase activity in both crude cell lysates and purified enzyme preparations based on a fluorescence-based method.
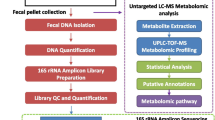
DNA Extraction, 16 s Ribosomal RNA Gene Amplicon Sequencing, and Bioinformatic Analysis
DNA was extracted from each jejunum, colon, and stool sample with a Maxwell® RSC PureFood GMO and Authentication kit and the test was prepared according to the protocol of 16S ribosomal RNA gene amplicons for the Illumina MiSeq System. Targeting 16 s ribosomal RNA V3-V4 region, MiSeq platform was used for sequencing. Using QIIME2 pipeline (version 2020.2), sequenced data were preprocessed including demultiplexing with Deblur, alignment with Mafft, and construction a phylogeny with fasttree2. Taxonomy was then assigned with a sklearn naïve Bayes taxonomy classifier based on the Greengenes 13_8 99% OTUs reference sequences.
Alpha diversity and beta diversity were analyzed with Chao index and a non-metric multidimensional scaling (NMDS) method, respectively. To explore differential microbes within and between groups, linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed based on the Huttenhower lab Galaxy web platform (http://huttenhower.sph.harvard.edu/galaxy). With respect to LEfSe, per-sample normalization of the sum of values to 1 M was performed. Input parameters were as follows: alpha value for the factorial Kruskal–Wallis test among classes = 0.05, alpha value for the pairwise Wilcoxon test between subclasses = 0.05, threshold on the logarithmic LDA score for discriminative features = 2.0, and all-against-all strategy for multi-class analysis.
To predict functional abundances, QIIME2 plugin (q2-picrust2) for phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt2) was facilitated based on MetaCyc metabolic pathway database. Predicted MetaCyc pathway abundances were compared between groups using ‘limma’ version 3.46 R package program after data transformation with ‘voom’ version 3.46. To make comparison metabolic pathways (N = 359) both within and between subjects, we firstly called ‘duplicateCorrelation’ function and estimated the correlation between measurement made on individual mouse. Next, we called the ‘voomWithQualityWeights’ function and obtained sample quality weights. The linear model was then fitted to compute empirical Bayes statistics. All statistically significant differential pathways were collected and presented as a heatmap where abundances were scaled, centered, and log transformed. In a heatmap, mice were bi-clustered by the k-means method according to the pattern of MetaCyc pathway abundances. We then compared the mean of predicted pathway abundances between groups stratified by anatomic sites and clusters.
Statistics
T-test or one-way analysis of variance (ANOVA) was used to perform statistical comparisons between samples. Chao index was compared between groups with ordinary one-way ANOVA. Using ‘vegan’ version 2.5–7 R package program, analysis of similarities (ANOSIM) was performed to test statistical difference for NMDS values between groups. Two-way ANOVA was performed for multiple comparison. Q value was then estimated with Benjamini and Hochberg correction method. Differential metabolic pathways with P-value < 0.05 were regarded as statistically significant. Cumulative bar graph, LDA bar graph, differential X–Y graph, and one-way/two-way ANOVA analyses were prepared or performed using Prism version 9.1.1 program.
Results
Tissue Cytokine Concentration
Seven inflammatory cytokines and chemokines (TNF-α, IL-12p40, IL-1α, IL-1β, IL-6, IL-10, and MCP-1 protein) were measured in jejunum and colon tissue specimens of 39 mice (the distribution was stated above). Results are summarized in Fig. 1A-H. Overall, TNF-α, IL-6, and MCP-1 protein levels were higher in both jejunum and colon tissues of irradiated mice than in non-irradiated ones at both S1 and S2. When results of RT alone and RT + probiotics groups were compared, the addition of probiotics significantly reduced TNF-α, IL-6, and MCP-1 levels compared to RT alone in S2-colon tissues (all p < 0.05). On the other hand, IL-12p40, and IL-1α, IL-1β, and IL-10 levels did not show significant difference between groups. Results of MPO assay showed no significant difference in MPO activity among the four groups (Fig. 1H). Repeated freezing and thawing of tissues for MPO assay might have reduced enzymatic activation. In summary, probiotic supplements reduced TNF-α, IL-6, and MCP-1 protein levels in colonic lysates of irradiated mice, especially at S2.

Comparison of cytokines between control, probiotics, RT alone, and RT + probiotics groups. TNF-α (A), IL-6 (B), MCP-1 (C) protein, IL-1α (D), IL-12p40 (E), IL-1β (F), IL-10 (G), and MPO activity (H) were compared according to each anatomic site and time point. Abbreviations: RT, radiation therapy, S1, four days after RT; S2, seven days after RT; C, control; P, probiotics; R, RT; B, RT + probiotics; TNF, Tumor Necrosis Factor; IL, Interleukin; MCP, Monocyte Chemotactic Protein; MPO, myeloperoxidase; Asterisk represents p-value < 0.05 by ordinary one-way analysis of variance
Comparison of Microbes Abundance and Diversity between RT and RT + Probiotics Group
We examined alpha- and beta-diversity according to anatomic site, time (days after RT), and treatment to assess microbial abundance and diversity. As illustrated in Fig. 2A, there was a higher (p < 0.05) alpha diversity in the stool at S2 of the RT + probiotics group than that of the RT alone group. However, a lower (p < 0.05) alpha diversity in the jejunum of the RT + probiotics group at S1 was observed. There was no significant difference between timepoints (S1 and S2) at each anatomic site. In terms of beta-diversity, there were significant differences according to anatomic site at S1 (Fig. 2B, p < 0.05) and S2 (Fig. 2C, p < 0.05) regardless of treatment. However, we did not observe any significant difference in beta-diversity according to treatment at S1 (Fig. 2D, p = 0.465) and S2 (Fig. 3E, p = 0.990). Location within intestinal tract seemed to have great impact on the community diversity, suggesting that probiotics did not disturb the ecology of distinct normal microbial flora.

Comparison of diversity between RT and RT + probiotics (Both) groups. A. Alpha diversity in terms of Chao index (A) and beta-diversity (B-E) were compared according to each anatomic site and time point. In Figures (B) and (C), the blue, green, and red circles represent the distribution of beta-diversity in stool, jejunum, and colon, respectively. In Figures (D) and (E), the red and blue circles represent the distribution of beta-diversity in RT alone and RT + probiotics (Both) groups. Abbreviations: RT, radiation therapy, S1, four days after RT; S2, seven days after RT; S, stool; J, jejunum; C, colon. Asterisk represents p-value < 0.05 by ordinary one-way analysis of variance

Bar graphs showing relative abundances. Taxa at family level in stool (A), jejunum (B), and colon (C) were compared according to anatomic site and time point. Taxa at genus level in stool (D), jejunum (E), and colon (F) were compared according to anatomic site and time point. Colored taxa represent significantly decrease or increase across time points between RT and RT + probiotics (Both) groups. Two-way ANOVA was performed for multiple comparison. Q value was then estimated with Benjamini and Hochberg correction method. Abbreviations: RT, radiation therapy, S1, four days after RT; S2, seven days after RT
After profiling taxonomy, we calculated relative fractions at family and genus levels, stratifying groups according to time (S1 and S2) and treatment (RT + probiotics and RT). In groupwise comparison, we found several significantly changed microbes (all False Discovery Rate [FDR] < 0.05) by treatment and time in each stool, jejunum, and colon. Detail list of microbes at family and genus levels is shown in Table 1. At the family level, Desulfovibrionaceae, Helicobacteraceae, Lachnospiraceae, Prevotellaceae, and S24-7 in stool (Fig. 3A), Desulfovibrionaceae, Lachnospiraceae, and S24-7 in jejunum (Fig. 3B), and Desulfovibrionaceae, Helicobacteraceae, Pasteurellaceae, and Prevotellaceae in colon (Fig. 3C) showed changes in abundance. At the genus level, abundances of Bacteroides, Flexispira, and Mucispirillium in stool (Fig. 3D), Haemophilus, Lactobacillus, and Prevotella in jejunum (Fig. 3E), and Bacteroides, Flexispira, and Prevotella in colon (Fig. 3F) were changed.
Regardless of timepoint (S1 or S2) since RT, we found some consistently increased and decreased microbes according to site and treatment (Table 1). In colon, the RT + probiotics group showed more Bacteroides, less Prevotella, and less Clostridales compared to the RT alone group. In stool, Prevotella and S24-7 were significantly enriched in the RT + probiotics group than in the RT alone group. Flexispira and Oscillospira were found significantly more in stool from the RT alone group than in the RT + probiotics group. However, in jejunum, we could not identify consistently decreasing or increasing microbes at S2 time point (7 days after RT). It is known that Haemophilus, Desulfovibrionaceae, Helicobacteraceae, Flexispira, F16, and Mucispirillum are associated with inflammatory bowel disease or status. Meanwhile, Prevotella, S24-7, Clostridiales, Lactobacillus, Bacteroides, Oscillospira, Lachnospiraceae, and Ruminococcus were associated with anti-inflammatory or normal bowel status. For example, Clostridiales seems to increase over time early to late days after RT. During acute inflammation in a dextran sodium sulfate-colitis mouse model [14], decrease of Clostridiales was observed, which was correlated with a reduction of transcripts related to butyrate formation. Likewise, anti-inflammation-related microbes appeared in the colon/stool of the RT + probiotics group at 7 days after RT, and in the colon of the RT alone group at 7 days after RT as well. Meanwhile, inflammation-related microbes appeared in the stool of the RT alone group at 7 days after RT. Thus, at 7 days after RT, colon and stool might be appropriate samples for comparing effects of RT and RT + probiotics in terms of relative microbial abundances.
Differential Microbes According to Treatment
Among the three treatment groups (RT + probiotics, probiotics, and RT), different microbes were found through group-pairwise comparisons. In jejunum at 4 days after RT (Fig. 4A), Phascolarctobacterium (LDA = 3.64, p = 0.008) and Streptococcus (LDA = 4.09, p = 0.008) were differentially enriched in RT alone and probiotics alone group, respectively. In the RT + probiotics group, Lactobacillales (LDA = 4.22, p = 0.006) and Lactobacillus (LDA = 3.77, p = 0.008) were differentially found compared to other treatment groups. These microbes were not observed as differential taxa in the jejunum at 7 days after RT (Fig. 4B).

Differential microbes among RT, probiotics, and RT + probiotics groups according to anatomic site and timepoint. By treatment groups, differential taxa in jejunum at S1 (A) and S2 (B) time points, and those in colon at S1 (C) and S2 (D) time points, and those in stool at S1 (E) and S2 (F) time points are plotted. Linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed and those with LDA score ≥ 3 were selected. Abbreviations: RT, radiation therapy, S1, four days after RT; S2, seven days after RT; LDA, linear discriminant analysis
In the colon at 4 days after RT, Desulfovibrionaceae (LDA = 4.74, p = 0.044) was profoundly more abundant in the RT alone group and Neisseria (LDA = 4.12, p = 0.042) was significantly more abundant in the probiotics alone group (Fig. 4C). Desulfovibrionaceae has sulfate-reducing capacities that might reduce disulfide bonds and break mucus barrier, potentially leading to inflammation [15]. YS2 (LDA = 4.43, p = 0.008) and Enterobacteriaceae (LDA = 4.26, p = 0.027) were differentially abundant in the RT + probiotics group compared to other groups. In colon at 7 days after RT, Prevotellaceae (LDA = 3.29, p = 0.032) was significantly more abundant in the RT alone group and Odoribacter (LDA = 4.43, p = 0.007) was significantly more abundant in the probiotics alone group (Fig. 4D). Reduced abundance of Odoribacter belonging to family Porphyromonadaceae has been found in inflammatory bowel disease [16]. In the RT + probiotics group, Porphyromonadaceae (LDA = 4.03, p = 0.011) was the most differentially enriched one (Fig. 4D). Porphyromonadaceae has a protective effect against inflammation-driven colorectal cancer in a murine model [17].
In stool, Neisseria subflava (LDA = 3.46, p = 0.018) was significantly more abundant in RT alone group at 4 days after RT and SR1 (LDA = 3.32, p = 0.012) was significantly more abundant in probiotics alone group at 4 days after RT (Fig. 4E). SR1 (also known as Absconditabacteria) and Neisseria subflava are considered as oral bacteria. At four days after RT, Bifidobacterium animalis (LDA = 2.89, p = 0.004), Enterobacteriaceae (LDA = 3.39, p = 0.009), and Lactobacillus (LDA = 3.80, p = 0.009) were significantly enriched in the RT + probiotics group than in other treatment groups (Fig. 4E). TM7-3 was consistently and differentially found in the stool in RT alone group at both 4 and 7 days after RT (Fig. 5E and F). TM7 is positively correlated with levels of inflammatory cytokines associated with inflammatory bowel disease [17]. At 7 days after RT in probiotics alone group, Peptococcus (LDA = 3.10, p = 0.022) and Bacteroides (LDA = 2.97, p = 0.046) were differentially enriched in the stool. Veillonella, Parvimonas, and Aggregatibacter segnis are known to be present in the oral microbiome of murine [18]. Abundance of Peptococcus is relatively decreased in patients with inflammatory bowel disease than in healthy controls [19]. Except Helicobacteraceae (LDA = 4.56, p = 0.029), anti-inflammation-related microbes were enriched in stool from RT + probiotics group, including Lactobacillus zeae (LDA = 3.50, p = 0.020), Bacteroides acidifaciens (LDA = 3.15, p = 0.045), and Ruminococcus (LDA = 3.28, p = 0.021).

Differential MetaCyc pathways and their patterns. For predicted MetaCyc pathway abundances, differential taxa between RT + probiotics and RT groups in jejunum (A), colon (B), and stool (C) are plotted. Patterns of all differential taxa are presented as a heatmap (D). Comparisons of mean predicted pathway abundance using k-means clustering (E, F) are also shown. Abbreviations: RT, radiation therapy, S1, four days after RT; S2, seven days after RT; KM, k-means clustering; FC, fold change
Collectively, in RT + probiotics group, these results indicated that probiotics-related microbes and anti-inflammation-related microbes are dominant in the jejunum, colon, and stool from early to later days after RT. However, unfavorable microbes such as Fusobacterium or Helicobacteraceae also appeared in the colon and stool during later days.
MetaCyc Metabolic Pathway
Predicted MetaCyc pathway abundances were calculated and compared between treatments (RT versus RT + probiotics) stratified by time points (S1 and S2). Minus log FC indicates enrichment of pathway in the RT + probiotics group.
In the jejunum at 4 days after RT (S1) (Fig. 5A), Pyrimidine Deoxyribonucleotides De Novo Biosynthesis Iii was more abundant in the RT alone group than in the RT + probiotics group (log FC = 1.07, p < 0.001). Several inhibitors of pyrimidine nucleotide biosynthesis are FDA-approved to treat rheumatoid arthritis, multiple sclerosis, acute leukemia, or lymphoproliferative disorders [20]. In Jejunum at 7 days after treatment (S2), the Petidoglycan biosynthesis pathway was less abundant in the RT alone group than in the RT + probiotics group (log FC = 4.90, p < 0.001). Petidoglycans derived from various Lactobacillus strains are known to have anti-inflammatory capacity [21]. Chorismate Biosynthesis Ii (Archaea) pathway was also more abundant in the RT + probiotics group (log FC = -2.71, p = 0.001). Chorismate is a precursor of tryptophan. It can be produced by Bifidobacteria [22]. Tryptophan is metabolized in the gut into several metabolites known to contribute its anti-inflammatory or radiation-mitigating effect [14]. Thus, in the jejunum, a metabolic effect by adding probiotics to RT is anticipated at ≥ 7 days after RT.
In the colon, significantly differential metabolic pathways in the RT alone group and RT + probiotics group were mostly found at S1 and S2 time points, respectively (Fig. 5B). At 4 days after RT, Adenosylcobalamin Biosynthesis Ii (Aerobic) metabolic pathway was profoundly more predicted in the RT alone group than in the RT + probiotics group (log FC = 6.95, p < 0.001). In contrast, at 7 days after RT, this pathway was enriched in the RT + probiotics group (log FC = -3.15, p = 0.016). Adenosylcobalamin (vitamin B12) is involved in anti-oxidative stress. It can be produced by several probiotic Lactobacilli and Bifidobacteria [23]. A metabolic benefit induced by the addition of probiotics seems to be observed in the colon at 7 days after intervention.
In the stool at 4 days after RT (Fig. 5C), Superpathway of Ornithine Degradation was more predicted in the RT alone group (log FC = 5.68, p < 0.001) and 2-Methylcitrate Cycle Ii (log FC = -3.00, p < 0.001) was more abundant in the RT + probiotics group. L-ornithine degradation is involved in bacterial-Induced IL-6 cytokine response in human microbiome [24]. 2-Methylcitrate Cycle Ii is one of propionate degradation pathways [25]. Several studies have found that propionate can reduce inflammation in the colon of mice [26, 27]. At 7 days after RT, Biotin Biosynthesis Ii was more active in the RT + probiotics group (log FC = -1.59, p = 0.002) and Glyoxylate Cycle was active in the RT alone group (log FC = 1.44, p = 0.011). B group vitamins can be produced by probiotics [28]. The glyoxylate cycle is a variation of the TCA cycle. An increase of the glyoxylate cycle indicates an increased energy metabolism in the RT alone group [29]. Consistent with findings for the jejunum and colon, we observed beneficial effect of probiotics in the stool at 7 days after treatment.
All statistically differential metabolic pathways between RT + probiotics and RT alone group were collected. Predicted abundances of these pathways are presented as an heatmap (Fig. 5D). After performing k-means clustering, we found two dominant clusters across anatomic site, time, and treatment. For unsupervised cluster 1 (KM = 1), the mean of predicted MetaCyc pathway abundances increased in the order probiotics alone, RT alone, and RT + probiotics in the jejunum and colon/stool (Fig. 5E). For another unsupervised cluster 2 (KM-2), the predicted abundance of pathways was increased in the jejunum than in the colon/stool. These pathways were associated with the biosynthesis of mycothiol, biotin, Heme B, polyamine, or L-Lysine fermentation to acetate and butanoate. Within jejunum, the mean of predicted MetaCyc pathway abundances membered into KM2 was different among treatment groups, with the RT + probiotics group showing the highest mean abundances (Fig. 5F). Collectively, these results indicate that probiotics and RT might promote the overall activity of metabolic pathways.
Discussion
In the present study, we could observe several meaningful findings based on a multiplex cytokine assay and 16 s rRNA amplicon sequencing of metagenome regarding the effect of probiotics on radiation enteritis. In terms of tissue cytokine concentration, major pro-inflammatory cytokines TNF-α, IL-6, and MCP-1 were significantly reduced in colon tissues at 7 days after RT after probiotics intake. In general, expression levels of pro-inflammatory cytokines and chemokines (IL-1, IL-6, and TNF-α) are upregulated in a murine colitis model, whereas anti-inflammatory cytokines such as IL-10 are downregulated. Other parameters to characterize mouse colitis include elevation of myeloperoxidase level which suggests neutrophil infiltration into the epithelium. In another aspect, dysbiosis after pelvic radiotherapy is also associated with enhanced expression of inflammatory cytokines such as TNF-α and IL-1β [30]. It has been demonstrated that probiotics can modulate inflammatory cytokine in patients with ulcerative colitis [31]. To understand the mechanism involved in the protective effect of probiotics on radiation enteritis, identifying changes in microbial environment modulated by probiotics is essential.
Regarding microbial diversity, we were able to observe increased alpha diversity in the stool of the RT + probiotics group than in that of the RT alone group. However, there were no significant differences between other samples. Gut dysbiosis commonly accompanies decreased diversity [32]. Reduced diversity is associated with active inflammatory bowel disease such as Crohn’s disease [33]. This might be due to a negative impact of probiotics on microbiome’s non-redundant contributions to the development of anti-inflammatory networks and inflammatory networks formed by diverse microbes [34]. Unexpected and reversed trend was observed in jejunum at an early time point. This might be because the jejunum at this time was spatiotemporally too early to benefit from probiotics. Beta-diversity was not changed after adding probiotics to RT. Given that distinct microbial flora variations existed among small intestine, colon, and fecal material [35], we found that administration of probiotics did not disturb the distinct microbial flora in each anatomic site.
Several studies have revealed that radiation can change gut microbial compositions. Johnson et al. [36] have shown altered abundances of aerobic, anaerobic, Enterobacteriaceae, and Lactobacillus in an irradiated C57/B16 murine model. Kim et al. [37] have reported increases of bacterial members of genera Alistipes and Corynebacterium with decreases of bacteria in genus of Prevotella in irradiated mice. Meanwhile, probiotic bacteria can produce shot-chain fatty acids (SCFAs) [28]. They also can synthesize vitamin K and most water-soluble B vitamins [23]. SCFA can enhance barrier function of the colonic epithelium [38]. It has anti-inflammatory effects [39]. Thus, SCFA-producing microbes could mitigate RT-induced inflammation. In the current study, Odoribacter splanchnus was found in the probiotics group. It is known that Odoribacter splanchnus can produce acetate, propionate, and butyrate [40]. Ruminococcus was found in the RT + probiotics group. Ruminococcus bromii can produce butyrate in the colon by fermenting resistant starch [41]. We also observed several oral microbes such as SR2 and Neisseria subflava in stool samples. This might be due to a time lag between mouse defecation and stool obtainment by researchers. During the time lag, mice might have oral contact with stool. Nevertheless, oral SR-2 microbiome is associated with gut inflammation [17].
According to anatomic site and time points, we found consistently increased or decreased taxa between groups. Anti-inflammatory-related microbes’ dominance pattern in the RT + probiotics group than in the RT alone group was manifested at 7 days (S2) after RT compared with that at 4 days after RT (S1). We speculate that every ≥ 7 days administration of probiotics might be needed for anti-inflammatory microbial networks to be dominated. In more detail, we investigated differentially existed microbes in each anatomic site and each timepoint according to three intervention groups: RT, RT + probiotics, and probiotics. Probiotics-related microbes such as Lactobacillales and Lactobacillus were differentially found and started to appear from the early time point (S1) in the RT + probiotics group than in other groups. This result was expected considering direct administration of probiotics microbes. However, we also observed well-known unfavorable microbes such as Fusobacterium or Helicobacteraceae in the RT + probiotics group at a later time point (S2) after RT. This result can be interpreted with two points. Firstly, since anti-inflammatory microbes dominated at 7 days after RT, reduction of unfavorable microbes by probiotics might need prolonged administration such as more than 7 days. Rectosigmoid transit time should also be considered. Gerassy-Vainberg et al. [6] have demonstrated that microbiota change is significantly manifested at six weeks after irradiation. Secondly, there was a possibility of detecting 16 s RNA of dead or partially digested microbes [42]. Indeed, H. pylori in stool was likely to be dead or dying [43]. Thus, increased detection of Helicobacteraceae might reflect the status of beneficial effect such as elimination of unfavorable bacteria caused by probiotics [44].
In metagenome analysis, we found that predicted abundances of metabolic pathways were increased in the order of probiotic, RT alone, and RT + probiotics groups. Administration of probiotics can lead to increased production of short-chain fatty acid, frequency of defecation, and stool volume [45]. RT can increase metabolic activities since RT-induced injury can induce inflammation by recruiting immune cells and damage gut epithelial cells. In Crohn’s disease mouse model, inflammation is associated with increased metabolic functions [45]. Altogether, the RT + probiotics group demonstrated the most increase in the abundance of metabolic pathways than the RT alone or the probiotics alone group. We also observed differential MetaCyc pathways between jejunum and colon/stool samples, indicating the unique feature of metabolism by microbes in the jejunum. In the later time point (S2), we observed differentially abundant MetaCyc pathways associated with anti-inflammatory processes in the RT + probiotics group compared to the RT alone group. However, in the early timepoint (S1), inflammation-related MetaCyc pathway such as 2-Methylcitrate Cycle Ii was differentially found in the RT + probiotics group from stool samples. These results may support the need of prolonged administration of probiotics during RT. Since metagenome profile was less sensitive than functional omics [46], additional functional profiles such as metatranscriptomics, metaproteomics, and metabolomics might be required to reveal functional perturbation caused by probiotics.
The current study has several limitations in. First, radiation was delivered in a single fraction with a large fraction size (14 Gy), which might have different biological effects on intestinal mucosa and microbial environment compared to conventional multi-fractionation with smaller fraction sizes commonly used for human cancer patients. The appropriate duration of probiotics administration before irradiation remains unclear. Regrading analysis methodology, poor resolution of species level of taxa by 16 s rRNA amplicon sequencing could limit accurate and direct measurements of metabolic predictions. The gold standard of metagenomic analysis is the shot-gun sequencing method. However, coarse-grained predictions from metagenome have been frequently used to date [22]. Most importantly, since mice of current study did not bear tumor, how anti-inflammatory effects of probiotics affect the prognosis of cancer-bearing host was unclear. Previously, Jang et al. have demonstrated that gut microbiome is associated with tumor regression after radiotherapy for rectal cancer patients [47].
Conclusions
In conclusion, probiotics showed protective effects by controlling inflammatory process after radiation exposure. Among various environmental factors, the presence of dysbiosis induced by irradiation was observed. In this condition, probiotics are likely to play an important role by enhancing microbes and metabolic pathways that could benefit the host. Meanwhile, anti-inflammatory microbes and metabolic pathways were differentially found in jejunum, colon, and stool samples between the RT + probiotics group and the RT alone group. This effect is likely to be pronounced if prolonged duration is used for the administration of probiotics after RT. Further studies for humans are needed to determine optimal conditions for achieving the maximal benefit of probiotics during RT.
Data Availability
The data that support the findings of this study are openly available in figshare at https://doi.org/10.6084/m9.figshare.21101686, and within the article and its supplementary materials. The 16s ribosomal RNA raw sequencing data can be found at the BioProject database (ID: PRJNA934953).
Abbreviations
- RT:
-
Radiotherapy
- CFU:
-
Colony-forming units
- LGG:
-
Lactobacillus rhamnosus GG
- TNF-α:
-
Tumor Necrosis Factor-α
- IL-6:
-
Interleukin-6
- MCP-1:
-
Monocyte Chemotactic Protein-1
- IL-12p40:
-
Interleukin-12p40
- IL-1α:
-
Interleukin-1α
- IL-1β:
-
Interleukin-1β
- IL-10:
-
Interleukin-10
- MPO:
-
Myeloperoxidase
- NMDS:
-
Non-metric multidimensional scaling
- LDA:
-
Linear discriminant analysis
- ANOVA:
-
One-way analysis of variance
- FDR:
-
False Discovery Rate
- SCFA:
-
Short-chain fatty acids
References
Andreyev HJ, Vlavianos P, Blake P, Dearnaley D, Norman AR, Tait D (2005) Gastrointestinal symptoms after pelvic radiotherapy: role for the gastroenterologist? Int J Radiat Oncol Biol Phys 62(5):1464–1471
McGough C, Baldwin C, Frost G, Andreyev HJ (2004) Role of nutritional intervention in patients treated with radiotherapy for pelvic malignancy. Br J Cancer 90(12):2278–2287
Tharavichtikul E, Meungwong P, Chitapanarux T et al (2014) The association of rectal equivalent dose in 2 Gy fractions (EQD2) to late rectal toxicity in locally advanced cervical cancer patients who were evaluated by rectosigmoidoscopy in Faculty of Medicine, Chiang Mai University. Radiat Oncol J 32(2):57–62
Bismar MM, Sinicrope FA (2002) Radiation enteritis. Curr Gastroenterol Rep 4(5):361–365
O’Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep 7:688–693
Gerassy-Vainberg S, Blatt A, Danin-Poleg Y et al (2018) Radiation induces proinflammatory dysbiosis: transmission of inflammatory susceptibility by host cytokine induction. Gut 67:97–107
Zhao T-S, Xie L-W, Cai S et al (2021) Dysbiosis of Gut Microbiota Is Associated With the Progression of Radiation-Induced Intestinal Injury and Is Alleviated by Oral Compound Probiotics in Mouse Model. Front Cell Infect Microbiol 11:717636. https://doi.org/10.3389/fcimb.2021.717636
Ki Y, Kim W, Cho H, Ahn K, Choi Y, Kim D (2014) The effect of probiotics for preventing radiation-induced morphological changes in intestinal mucosa of rats. J Korean Med Sci 29(10):1372–1378
Delia P, Sansotta G, Donato V et al (2007) Use of probiotics for prevention of radiation-induced diarrhea. World J Gastroenterol 13(6):912–915
Demirer S, Aydintug S, Aslim B et al (2006) Effects of probiotics on radiation-induced intestinal injury in rats. Nutrition (Burbank, Los Angeles County, Calif) 22:179–186
Salminen E, Elomaa I, Minkkinen J, Vapaatalo H, Salminen S (1988) Preservation of intestinal integrity during radiotherapy using live Lactobacillus acidophilus cultures. Clin Radiol 39:435–437
Delia P, Sansotta G, Donato V et al (2002) Prophylaxis of diarrhoea in patients submitted to radiotherapeutic treatment on pelvic district: personal experience. Dig Liver Dis 34(Suppl 2):S84–S86
Ciorba MA et al (2012) Lactobacillus probiotic protects intestinal epithelium from radiation injury in a TLR-2/cyclo-oxygenase-2-dependent manner. Gut 61(6):829–838
Guo H, Chou WC, Lai Y et al (2020) Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science 370(6516):eaay9097
Ijssennagger N, van der Meer R, van Mil SWC (2016) Sulfide as a mucus barrier-breaker in inflammatory bowel disease? Trends Mol Med 22(3):190–199. https://doi.org/10.1016/j.molmed.2016.01.002
Morgan XC, Tickle TL, Sokol H et al (2012) Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13(9):R79
Zackular JP, Baxter NT, Iverson KD et al (2013) The gut microbiome modulates colon tumorigenesis. mBio 4(6):e00692-13
Fardini Y, Chung P, Dumm R, Joshi N, Han YW (2010) Transmission of diverse oral bacteria to murine placenta: Evidence for the oral microbiome as a potential source of intrauterine infection. Infect Immun 78(4):1789–1796. https://doi.org/10.1128/IAI.01395-09
Willing BP, Dicksved J, Halfvarson J et al (2010) A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139:1844–54.e1
Okesli A, Khosla C, Bassik MC (2017) Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr Opin Biotechnol 48:127–134. https://doi.org/10.1016/j.copbio.2017.03.010
Matsumoto S, Hara T, Nagaoka M et al (2009) A component of polysaccharide peptidoglycan complex on Lactobacillus induced an improvement of murine model of inflammatory bowel disease and colitis-associated cancer. Immunology 128:e170–e180
Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK (2014) Specialized metabolites from the microbiome in health and disease. Cell Metab 20(5):719–730. https://doi.org/10.1016/j.cmet.2014.10.016
Averina OV, Poluektova EU, Marsova MV, Danilenko VN (2021) Biomarkers and utility of the antioxidant potential of probiotic lactobacilli and bifidobacteria as representatives of the human gut microbiota. Biomedicines 9:1340
Schirmer M, Smeekens SP, Vlamakis H et al (2016) Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167:1125–36.e8
Zheng C, Yu Z, Du C et al (2020) 2-Methylcitrate cycle: a well-regulated controller of Bacillus sporulation. Environ Microbiol 22(3):1125–1140. https://doi.org/10.1111/1462-2920.14901
Tong LC, Wang Y, Wang ZB et al (2016) Propionate ameliorates dextran sodium sulfate-induced colitis by improving intestinal barrier function and reducing inflammation and oxidative stress. Front Pharmacol 15(7):253. https://doi.org/10.3389/fphar.2016.00253
Agus A, Richard D, Faïs T et al (2021) Propionate catabolism by CD-associated adherent-invasive E. coli counteracts its anti-inflammatory effect. Gut Microbes 13:1–18
LeBlanc JG, Chain F, Martín R, Bermúdez-Humarán LG, Courau S, Langella P (2017) Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb Cell Fact 16:1–10
Wang K, Xu X, Maimaiti A et al (2021) Gut microbiota disorder caused by diterpenoids extracted from Euphorbia pekinensis aggravates intestinal mucosal damage. Pharmacol Res Perspect 9(5):e00765. https://doi.org/10.1002/prp2.765
Wang Z, Wang Q, Wang X et al (2019) Gut microbial dysbiosis is associated with development and progression of radiation enteritis during pelvic radiotherapy. J Cell Mol Med 23(5):3747–3756. https://doi.org/10.1111/jcmm.14289
Bai AP, Ouyang Q, Xiao XR, Li SF (2006) Probiotics modulate inflammatory cytokine secretion from inflamed mucosa in active ulcerative colitis. Int J Clin Pract 60(3):284–288. https://doi.org/10.1111/j.1368-5031.2006.00833.x
Ott SJ, Musfeldt M, Wenderoth DF et al (2004) Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53:685–693
Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI (2014) Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 6(220):220ra11. https://doi.org/10.1126/scitranslmed.3008051
Petersen C, Round JL (2014) Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol 16(7):1024–1033. https://doi.org/10.1111/cmi.12308
Holzapfel WH, Haberer P, Snel J, Schillinger U, Huis In’T Veld JHJ (1998) Overview of gut flora and probiotics. Int J Food Microbiol 41(2):85–101. https://doi.org/10.1016/s0168-1605(98)00044-0
Johnson LB, Riaz AA, Adawi D et al (2004) Radiation enteropathy and leucocyte-endothelial cell reactions in a refined small bowel model. BMC Surg 4:10
Kim YS, Kim J, Park S-J (2015) High-throughput 16S rRNA gene sequencing reveals alterations of mouse intestinal microbiota after radiotherapy. Anaerobe 33:1–7
Suzuki T, Yoshida S, Hara H (2008) Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Br J Nutr 100(2):297–305
Lomax AR, Calder PC (2009) Prebiotics, immune function, infection and inflammation: A review of the evidence. Br J Nutr 101(5):633–658. https://doi.org/10.1017/S0007114508055608
Göker M, Gronow S, Zeytun A et al (2011) Complete genome sequence of odoribacter splanchnicus type strain (1651/6 T). Stand Genomic Sci 4(2):200–209. https://doi.org/10.4056/sigs.1714269
Ze X, Duncan SH, Louis P, Flint HJ (2012) Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J 6:1535–1543
Weaver LT (1999) Helicobacter pylori in the faeces? QJM 92:361–364
Dolan B, Burkitt-Gray L, Shovelin S et al (2018) The use of stool specimens reveals Helicobacter pylori strain diversity in a cohort of adolescents and their family members in a developed country. Int J Med Microbiol 308(2):247–255. https://doi.org/10.1016/j.ijmm.2017.11.005
Hamilton-Miller JMT (2003) The role of probiotics in the treatment and prevention of Helicobacter pylori infection. Int J Antimicrob Agents 22(4):360–366. https://doi.org/10.1016/s0924-8579(03)00153-5
Plaza-Diaz J, Ruiz-Ojeda FJ, Gil-Campos M, Gil A (2019) Mechanisms of action of probiotics. Adv Nutr 10(suppl_1):S49–S66. https://doi.org/10.1093/advances/nmy063
Heintz-Buschart A, Wilmes P (2018) Human gut microbiome: function matters. Trends Microbiol 26(7):563–574. https://doi.org/10.1016/j.tim.2017.11.002
Jang B-S, Chang JH, Chie EK et al (2020) Gut microbiome composition is associated with a pathologic response after preoperative chemoradiation in patients with rectal cancer. Int J Radiat Oncol Biol Phys 107(4):736–746. https://doi.org/10.1016/j.ijrobp.2020.04.015
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant Number: 2019R1A2C1002071), the New Faculty Startup Fund, 3020200090 from SNUH Research Fund, and funded by the Seoul Metropolitan Government-Seoul National University Boramae Medical Center (Grant Number 03-2017-19).
Author information
Authors and Affiliations
Contributions
SL and BJ were involved in the conception design, analysis and interpretation of the data, and drafting of the manuscript. YN and SL collected and analyzed the data and assisted in manuscript drafting. SH collected and analyzed the data. JC and HK developed the conception design, analyzed and interpreted the data, and revised the manuscript critically for intellectual content.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
The Institutional animal care and use committee of Seoul National University Hospital approved the experimental protocol (IACUC NO. 18–0096).
Consent for Publication
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, S.U., Jang, BS., Na, Y.R. et al. Effect of Lactobacillus Rhamnosus GG for Regulation of Inflammatory Response in Radiation-Induced Enteritis. Probiotics & Antimicro. Prot. 16, 636–648 (2024). https://doi.org/10.1007/s12602-023-10071-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12602-023-10071-9