
Abstract
A series of nanosized CeO2–Fe2O3 mixed-oxide nanocomposites with different Ce4+/Fe3+ molar ratios were successfully prepared by a co-precipitation technique. The surface area increased with Fe2O3 content increasing up to 60 wt% in the composite. However, with further increase in Fe2O3 content, the surface area began to decrease. The reduction processes of the CeO2–Fe2O3 nanocomposites were studied in a hydrogen atmosphere at 300–600 °C. The reduction rates increased by increasing both the temperature and Fe2O3 content in the nanocomposites. The microstructure of the reduced composites at 500 °C illustrated the presence of a considerable number of macro- and micro-pores. The activation energy values were calculated which were in the range of 3.56–5.37 kJ·mol−1 at the initial stages (up to 35% reduction) and 5.21–10.2 kJ·mol−1 at the final stages (up to 80% reduction) of reduction. The rate-controlling mechanisms in both the initial and final reduction stages were determined, and the initial reaction stage was controlled by combined gaseous diffusion and interfacial chemical reaction mechanisms for all the composites except for pure CeO2, which was controlled by a chemical reaction mechanism. The final reaction stage was controlled by a gaseous diffusion mechanism for some composites, while for the others it was controlled by combined gaseous diffusion and interfacial chemical reaction mechanisms. The hydrogen sorption properties of the nanocomposites were studied by pressure composition isotherms using a volumetric method. Hydrogen storage in the nanocomposites increased by increasing the temperature because of the formation of oxygen vacancies which enhance atomic H adsorption and function as strong adsorption sites forming more metal hydride covalent bonds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In the last years, binary oxide composite systems containing the iron oxide and cerium oxide have attracted the attention of the scientific community due to their widespread applications in catalysis [1,2,3,4], water splitting [5], photocatalysis [6,7,8,9], supercapacitors [8] and sensors for toxic gases [1, 2, 10,11,12]. Moreover, metal oxide mixtures have been demonstrated to be an effective strategy for a stepwise course of reaction, where reduction of metal oxide can accelerate the reduction rate of the other such as in Fe2O3/NiO system. The formed metallic nickel accelerates the reduction rate of iron oxide [13].
Gas–solid reactions are of great importance in material processing operations, and the physical properties of the solids play an important role in the manner in which these reactions proceed with a great influence on the overall kinetics. Many researchers [14,15,16] investigated the reduction process of Fe2O3 which has potential applications [17, 18] because of its environmental friendliness, easy handling, low cost and non-toxicity [19]. The reduction of iron oxides was studied with different types of reducing gases such as H2 [20, 21], CO [22], CO–H2 mixtures [23] and CH4–H2 gas mixtures [24]. It was found that adding rare earth metals not only improved the quality in metallurgy, but also enhanced the properties of the product, thus opening new opportunities in numerous applications [25].
CeO2 is an essential rare earth metal oxide, which has distinct redox properties and high electrical conductivity [26], driving it to be applied in a wide range of applications [8, 27,28,29,30], although CeO2 has some disadvantages, such as poor thermal stability and high sintering tendency, which can decrease its catalytic behaviour [26]. Therefore, researchers doped it with a series of metal oxides, i.e. CuO [31], TiO2 [32], Al2O3 [33] and CoOx [34], to promote its structural stability and activity. Doping Fe2O3 with CeO2 increases the oxygen vacancy (OV) concentration to achieve charge balance [35] and enhances the catalytic property [36]. Moreover, Ce4+ induced the formation of structural defects in iron oxide, resulting in a smaller grain size and higher surface area [36]. Also, doping Fe3+ with Ce4+ reduces the sintering temperatures and facilitates ceramic densification [37]. Many studies have reported the behaviour of CeO2 combined with Fe2O3 [38]; Janoš et al. [39] successfully prepared a new type of magnetically separable composite consisting of maghemite grains and a nanocrystalline cerium oxide surface layer (CeO2/γ-Fe2O3), which functioned as a reactive sorbent to decompose dangerous organophosphate compounds. Arena et al. [40] studied the effects of Ce and Fe addition on the textural, structural and redox properties of MnOx systems. Additionally, CeO2-supported Fe2O3 has been investigated as a catalyst for ethanol conversion [41].
Various efforts have focused on the efficient development of hydrogen storage materials. Materials used for hydrogen storage must have unique properties, such as an easy refuelling capability, compact form, easy and affordable production [42]. Unconventional techniques have been used to improve hydrogen storage efficiency through the application of nanoscale materials, supporting active materials and redox-active metal oxides [43,44,45]. Moreover, Prabhukhot et al. [43] devoted considerable attention to studying solid-state materials as hydrogen storage materials for mobile applications due to their lightweight and compact form. Transition metal hydrides are promising for hydrogen storage [43], and furthermore, oxygen-defective metal oxides have been shown to exhibit excellent properties for storage applications [46]. The modification of CeO2 by Fe2O3 results in a significant enhancement of the oxygen storage capacity of the mixed oxides due to the formation of CeFeO3 after redox treatments [47]. Enhancement of the hydrogen storage and production activity by the cerium addition for the Fe–Ce mixed oxides was studied by Zhu et al. [5], and it was found that the maximum hydrogen storage amount was up to 1.51 wt% for the Fe/Ce composite with a 30% substitution of Ce for Fe at 750 °C. Various CeO2–Fe2O3 composites have been used as oxygen storage materials in the chemical looping process for CO2 conversion [48]. And one of the most interesting studies is the modification of CeO2 by Fe2O3 which provides better oxygen storage capacity by forming an intergranular interface between the two oxides, which enhances the low-temperature reducibility of Fe2O3 [49].
The current study focuses on studying the reduction behaviour of CeO2–Fe2O3 nanocomposites with different Ce4+/Fe3+ molar ratios at 300–600 °C in a H2 atmosphere. The morphological changes and kinetics data obtained from the isothermal reduction process to elucidate the reduction mechanism were reported. Additionally, the effect of the various Ce/Fe molar ratios on the hydrogen storage capacity at different temperatures was investigated.
2 Experimental
2.1 Preparation of CeO2–Fe2O3 samples
Nanocomposites of CeO2–Fe2O3 samples with different Ce4+/Fe3+ molar ratios were prepared by a co-precipitation method [50]. A mixed aqueous solution of cerium nitrate (Ce(NO3)3·6H2O) and ferric nitrate (Fe(NO3)3·9H2O) at the given Ce4+/Fe3+ molar ratio (Table 1) was heated in a thermostatic water bath at 70 °C. Then, a 33% ammonia solution was gradually added under continuous stirring. After complete precipitation, the formed precipitate was digested for 1–2 h at room temperature, followed by filtration and washing with hot distilled water several times. The product was then dried at 100 °C for 15 h. The obtained material was ground to obtain a fine homogenous powder. Subsequently, the fine powder was calcined in a muffle furnace at 700 °C for 1 h with a heating rate of 10 °C·min−1, then was left to gradually cool until safe to handle. The prepared samples were labelled as CeO2, 0.8CeO2-0.2Fe2O3, 0.6CeO2-0.4Fe2O3, 0.4CeO2-0.6Fe2O3, 0.2CeO2-0.8Fe2O3 and Fe2O3 according to the Ce4+/Fe3+ molar ratio.
2.2 Characterization of prepared samples
The phases and crystallinities of the prepared samples were characterized by X-ray diffractometer (XRD, D8 Advance, Bruker, Germany) with Cu Kα radiation (wavelength of λ = 0.154045 nm) at an accelerating voltage of 40 kV and current of 40 mA. The average crystallite sizes were calculated using Scherrer’s formula as follows:
where D is the crystallite size, λ is the X-ray wavelength, β is the diffraction peak broadening, β1 is the instrumental broadening and θ is the diffraction angle. The morphologies of the nanoparticles were observed using field emission scanning electron microscopy (FESEM, Gemini, Zeiss Ultra 55) and transmission electron microscopy (TEM, JEOL JEM-1230).
Zeta size distribution measurements were performed on a Malvern (Malvern Instruments Ltd). The specific surface area was calculated from N2 adsorption–desorption isotherms measured by a TriStar II 3020 instrument (Micromeritics, USA) using the Brunauer–Emmett–Teller (BET) equation. The pore-size distributions were obtained using the Barrett–Joyner–Halenda (BJH) method.
2.3 Reduction experiments
The prepared composite samples with equal weights of ~ 1.5 g were pressed in a cylindrical mould at 1471 N·cm−2 and calcined at 700 °C for 1 h in a muffle furnace. The compacts were used for isothermal reduction experiments using a thermogravimetric balance (PRECISA-SWISS) equipped with a vertical tube furnace. The reduction process was performed in the presence of a gas purification system to obtain 99.99% pure hydrogen at 300–600 °C. For each reduction experiment, the furnace was heated to the required reduction temperature, and then, the sample was weighed and placed in a platinum wire basket. The sample was then gradually introduced into the furnace to avoid thermal shock cracking and positioned in the middle of the constant hot zone of the furnace. First, nitrogen at a flow rate of 60 ml·min−1 was introduced. (Preliminary reduction experiments showed that the most suitable hydrogen flow rate required to ensure an adequate supply of gas and overcome the gas boundary layer diffusion resistance, thus avoiding gas starvation, was ~ 60 ml·min−1.) Then, after the sample was soaked for 2 min at the reduction temperature, the N2 gas flow was stopped, and the reducing gas (H2) at a flow rate of 60 ml·min−1 was introduced. The weight loss resulting from oxygen removal from the sample was recorded at specific time intervals. At the end of the experiment, the basket with the reduced sample compact was removed and dropped into a conical flask containing acetone to prevent pyrophoricity of the reduced sample. The structural changes accompanying the compact reduction were examined by FESEM.
2.4 Hydrogen storage experiments
Hydrogen content determination was performed by measuring pressure composition isotherms (PCIs) using a volumetric method (direct uptake of gas by the material) using an AMC PCI-HP 1200 equipment. The sorption measurements were performed at different temperatures. The temperature was controlled with a precision of ± 0.1 °C. The accuracy of the hydrogen content measurement was ± 0.04 wt%. To minimize contamination from air, the composite powders were degassed at 200 °C for 1 h under dynamic vacuum before the tests. PCIs were traced under the above-mentioned conditions.
3 Results and discussion
3.1 Characterization of prepared samples
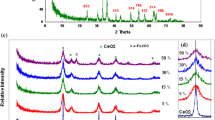
XRD patterns of the synthesized CeO2–Fe2O3 nanocomposite samples are shown in Fig. 1. The peaks of pure CeO2 were observed at 2θ values of 28.5°, 33.06°, 47.49°, 56.37°, 59.04° and 69.21°, corresponding to the (111), (200), (220), (311), (222) and (400) reflection planes of the cubic fluorite structure (assigned by comparing to the ICDD standard No. 75-0076). By gradually increasing Fe3+ content, the diffraction peak intensities corresponding to ceria gradually decreased and completely disappeared in the pure Fe2O3 sample, which showed diffraction peaks at 2θ values of 24.12°, 33.13°, 35.6°, 40.8°, 49.4° and 54.07°, corresponding to (012), (104), (110), (113), (024) and (116) reflection planes of the hexagonal structure (assigned by comparing to the ICDD standard No. 89-0598). The crystallite sizes were calculated using Scherrer’s formula, as tabulated in Table 1. The pure CeO2 nanoparticles were found to have an average crystallite size of 27.6 nm. By increasing Fe3+ molar ratio (0.8Fe:0.2Ce), the crystallite size of the ceria particles gradually decreased to 8.3 nm. The decreased ceria crystallite size is due to the interactions with Fe2O3, which hinders ceria grain growth and induces the formation of smaller ceria crystallites [51]. In addition, the average crystallite size of pure Fe2O3 was found to be 85.7 nm, and by increasing the ceria content in the CeO2–Fe2O3 nanocomposite, the average Fe2O3 nanoparticle crystallite size gradually decreased to 17.7 nm (molar ratio of 0.8Ce:0.2 Fe, Table 1).

XRD patterns of CeO2–Fe2O3 samples with different mole ratios
The lattice parameter for CeO2 cubic structure was calculated using Eq. (2):
The lattice parameter for Fe2O3 hexagonal structure was calculated using Eq. (3):
where d is the lattice spacing, a and c are the lattice parameters and h, k and l are the Miller indices. The lattice parameters for CeO2 and Fe2O3 are reported in Table 1. For Sample 2 (0.8CeO2–0.2Fe2O3), the calculated a value was 0.538 nm, which is slightly lower than that of 0.541 nm for Sample 1 (pure CeO2). Mixing Fe2O3 and CeO2 precursors led to a shift of the (111) diffraction plane to a higher diffraction angle in the final product (Fig. 1). The decreased unit cell parameter in the composite may be attributed to the smaller size of the Fe3+ cation radius (0.064 nm) compared to that of Ce4+ cation (0.087 nm) [41] or to OV formed [52]. For Fe2O3 lattice parameter, increased a and c values were observed in the nanocomposites with Fe contents increasing. The a value for iron oxide increased from 0.503 to 0.504 nm, while the c value increased from 1.373 to 1.391 nm (Table 1); this increase may be attributed to the reduction of Ce4+ (0.097 nm) to Ce3+ (0.423 nm) [52].
The nanocomposite colour changes from yellow to dark reddish brown with an increasing Fe2O3 content (Fig. S1 in supporting information). TEM images of the prepared samples are shown in Fig. 2a–c. Figure 2a shows that the ceria nanoparticles were slightly agglomerated, and the measured particle size was ~ 27 nm, which is in a good agreement with XRD results. In the composite sample, the number of particles clearly decreased and the particles condensed together with grain boundaries between them and particle sizes increased up to 29 nm, which support XRD results, indicating that the combined samples have an intermediate size between the pure oxides (Fig. 2b). However, in the pure iron oxide sample (Fig. 2c), the prepared nanoparticles were well dispersed possessing a clear hexagonal structure (inset in Fig. 2c) with particle sizes up to 100 nm. The size distribution diagram of the prepared composites is shown in Fig. S2, indicating that the particle size interval of the prepared samples was 570 nm for pure Fe2O3, 258 nm for pure CeO2 and in the range of 150–220 nm for all the other composites, which means that mixing of oxides decreases the average particle size of the composite, in accordance with TEM results.

TEM images of synthesized samples: a CeO2, b 0.6CeO2–0.4Fe2O3 and c Fe2O3 samples
The surface morphologies of the prepared samples were evaluated by SEM. The surface morphology changes of the pure and doped samples are shown in Fig. 3a–f. Pure CeO2 possesses large uniform grains and few pores (Fig. 3a). By incorporating Fe2O3 into CeO2 in Sample 2 (0.8CeO2–0.2Fe2O3), the surface morphology changes to large pores with clear grains (Fig. 3b). However, for Samples 3, 4 and pure Fe2O3, the enhancement of slight surface roughness is observed. Moreover, porosity and grain size increased due to the decreasing CeO2 mole ratio. CeO2 has been reported to help improving grains by reducing their size and distribution (Fig. 3c, d) [26]. The morphology of Sample 5 features large pores, high surface roughness and large grains (Fig. 3e). Nanosized Fe2O3 (Sample 6) possesses large grains, smooth surface and intermediate-sized pores (Fig. 3f).

SEM images of prepared samples: a CeO2, b 0.8CeO2–0.2Fe2O3, c 0.6CeO2–0.4Fe2O3, d 0.4CeO2–0.6Fe2O3, e 0.2CeO2–0.8Fe2O3 and f Fe2O3
The textural properties of the nanosized CeO2–Fe2O3 composites are represented in terms of surface area, pore volume and pore-size distribution using the BJH method (Fig. 4a–c and Table 2). The nitrogen adsorption–desorption isotherms of the prepared samples are shown in Fig. 4a. The adsorption isotherms exhibit type IV shapes according to IUPAC classification. Small closed adsorption–desorption hysteresis loops with relative pressures above 0.4 were observed, which is suggested to originate from capillary condensation in the mesopores [53]. The BET surface area of pure CeO2 was 22.81 m2·g−1, with a corresponding pore volume of 0.02 cm3·g−1. The average pore width was found to be 5.25 nm, indicating that the sample is mesoporous. The incorporation of mole ratio 20% Fe2O3 led to an increased surface area to 32.99 m2·g−1, which can be ascribed to structural defects formed in iron oxide resulting in a small grain size and high surface area [26, 36]. The corresponding pore volume was 0.05 cm3·g−1, and the average pore width was 6.1 nm (Table 2), indicating that the sample is mesoporous (Fig. 2b). Figure 4c shows that the surface area of the mixed oxides increased with Fe2O3 content increasing in composite up to 60 mol%, after which the surface area began to decrease. This decrease may be attributed to the reduction of Ce4+ to Ce3+, which can reduce the structural defects and thus reduce the grain size and surface area. However, for pure Fe2O3, the surface area was 3.73 m2·g−1, which may be attributed to the large crystallite size of pure Fe2O3 (Table 1) and large grains, as observed from SEM (Fig. 3e). The corresponding pore volume and pore width of pure Fe2O3 were 0.003 cm3·g−1 and 5.069 nm, respectively (Table 2). As clearly observed in Fig. 4c, all BET surface areas, pore volumes and porous surface areas displayed the same trends with Fe2O3 content varying, which was attributed to each of these properties being inversely proportional to the grain size.

a Nitrogen adsorption–desorption isotherms, b pore-size distribution and c relation between surface area, accumulative volume of pores and accumulative surface area of pores against Fe2O3 content for the prepared CeO2–Fe2O3
3.2 Reduction of prepared samples
The CeO2–Fe2O3 compacts were isothermally reduced under a dynamic H2 flow at different temperatures ranging from 300 to 600 °C. The reduction process was followed by measuring the sample weight loss at varying reduction times. The reduction per cent as a function of temperature for each prepared sample is shown in Fig. 5a–f. The reduction profiles reveal that a two-stage reduction process occurs. For the initial stage, the surface (up to 35% reduction) is characterized by a fast reduction rate, while for the final stage (up to 80% reduction extent), the surface and the bulk are characterized by a slower reduction rate, which is in agreement with the basic principles of chemical thermodynamics and kinetics. With temperature increasing, an increasing reduction rate was observed, indicating that the reduction process was endothermic.

Effect of temperature on reduction extent of prepared samples: a CeO2, b 0.8CeO2−0.2Fe2O3, c 0.6CeO2−0.4Fe2O3, d 0.4CeO2−0.6Fe2O3, e 0.2CeO2−0.8Fe2O3 and f Fe2O3
The reduction rate increased with Fe2O3 content increasing (Fig. S3a–d) due to the lower energy required to break Fe–O bonds than that is required to release oxygen atoms from cerium. Moreover, OVs formed in the reducing environment [46], which can accelerate the reduction process. At 300 °C, pure CeO2 did not completely reduce, reaching an 82% reduction degree within 33 min. However, at 400 °C, the reduction degree reached 99.5% after 32 min. Moreover, upon raising the reduction temperature to 500 °C and 600 °C, the CeO2 compacts were completely reduced within 20 and 19 min, respectively (Fig. 5a). The reduction behaviour of the composite samples shows the same general features, as observed in Fig. 5b-e. The reduction rate increased with Fe2O3 content increasing. For example, Sample 3 (0.6CeO2−0.4Fe2O3) reached 99% reduction within 33.5 min at 300 °C, while at 400 °C, 500 °C and 600 °C, the sample was completely reduced within 31.0, 17.5 and 12.8 min, respectively (Fig. 5c). For Sample 6, pure Fe2O3 (Fig. 5f), complete reduction (100%) was observed at 300 °C, 400 °C, 500 °C and 600 °C within 33, 17, 11 and 10 min, respectively.
3.3 Reduction-rate-controlling mechanism
To illustrate the rate-controlling mechanisms at both the initial (Fig. S4a-f) and final stages (Fig. S5a–f) of reduction, the apparent activation energy (Ea) of reduction was calculated from the Arrhenius equation:
where kr is the reaction rate constant, k0 is the frequency factor, Rg is the gas constant and T is the absolute temperature. kr can be derived from a rate equation of the form:
where p is the pressure of the reducing gas and n is the reaction order.
The correlated logarithm of the reduction rate (dr/dt) for the CeO2–Fe2O3 compacts with the reciprocal of the absolute temperature (1/T) is shown in Fig. 6a, b. From the obtained results, the Ea values were calculated at the initial and final stages, as listed in Table 3. The Ea values help elucidating the type of mechanism. Moreover, mechanisms based on the magnitude of the Ea are not decisive but indicative [54].

Arrhenius plots for reduction at a initial stages and b final stages
The grain model by Szekely et al. [55] was used to analyze the experimental results. According to Szekely, for the reduction of metal oxides in a H2 atmosphere following a first-order reaction, the chemical reaction control formula is as follows:
where φ(X) is the interfacial chemical reaction control conversion factor and X is the decimal fractional reduction. Fg is the shape factor, which is 2 for cylinder-type compacts. On the other hand, the gaseous diffusion reaction control formula is:
where g(X) is the gaseous diffusion reaction control conversion factor. The combined interfacial chemical reaction and gaseous diffusion control formula is:
By applying the different equations derived from the gas–solid reaction model, the following results were obtained. For pure CeO2 (Sample 1), the reduction initial stage is controlled by a chemical reaction mechanism (Fig. 7a) due to the textural nature of the surface of Sample 1, which contains a porous surface (Fig. 4c) and a high surface area, pore volume and pore width. Pure CeO2 has a total surface area of 22.81 m2·g−1 and porous surface area of 17.67 m2·g−1 (Table 2), and therefore, H2 gas can easily diffuse through the oxide layer. However, in the final reduction stage, the mechanism is controlled by gaseous diffusion (Fig. S6a), which may be attributed to the pores being surrounded by the oxide layer, hindering gas diffusion through the pores. The combined gaseous diffusion and interfacial chemical reaction mechanisms control the initial reduction stage for composites 0.8CeO2–0.2Fe2O3 and 0.2CeO2–0.8Fe2O3 containing highly porous surfaces (Fig. 4c). These composites have very high surface area parameters (Table 2), indicating that the interfacial chemical reaction is the rate-controlling mechanism for Samples 2 (Fig. 7b) and 5 (Fig. 7e). On the other hand, the gaseous diffusion mechanism controls the final stage (Fig. S6b, e), where the pores are filled with a thick oxide layer, hindering H2 diffusion and allowing solid-state diffusion to serve as the prevailing mechanism in the final stage.

Application of different mechanisms at initial stages for reduced nanosized compacts at 300–600 °C: a CeO2, b 0.8CeO2−0.2Fe2O3, c 0.6CeO2−0.4Fe2O3, d 0.4CeO2−0.6Fe2O3, e 0.2CeO2−0.8Fe2O3 and f Fe2O3
For Samples 3, 4 and 6, the reduction mechanism is controlled by a combined gaseous diffusion and interfacial chemical reaction process for both the initial stage (Fig. 7c, d, f) and final stage (Fig. S6c, d, f). For Sample 3, this mechanistic control may be related to the high surface area, pore volume, porous surface area (Fig. 4c) and the uniform pore-size distribution (Fig. 4b), in addition to the low pore width, which hinders H2 gas penetrating the oxide film. However, Samples 4 and 6 have low surface areas (Table 2), which correspond to increased crystallite sizes from grain growth, allowing hydrogen gas to slowly diffuse and leading to a contribution from solid-state diffusion in the rate-controlling process.
Figure 8a shows the microstructure of the reduced CeO2 compact at 500 °C, illustrating the presence of numerous macro- and micro-pores. Therefore, the material will have no resistance to gaseous diffusion, and thus, only the chemical reaction would be rate-controlling in the initial stage. In the final stage, the dense CeO2 matrix resists gas diffusion, and therefore, the final stage is controlled by the gaseous diffusion mechanism. For Samples 2 and 5 (Fig. 8b, e), the presence of some pores facilitates gaseous diffusion, and the partial dense matrix hinders gaseous diffusion; thus, the controlling mechanism is a combination of gaseous diffusion and the interfacial chemical reaction in the initial stage. On the other hand, in the final stage, the gaseous diffusion resistance increases, and therefore, the gaseous diffusion mechanism controls reduction. For Samples 3, 4 and 6 (Fig. 8c, d, f), both the initial and final reduction stages can be described by a combined gaseous diffusion and interfacial chemical reaction mechanism, as the composite surfaces are characterized by open pores and dense matrices.

SEM images for pure and doped oxides reduced at 500 °C: a CeO2, b 0.8CeO2–0.2Fe2O3, c 0.6CeO2–0.4Fe2O3, d 0.4CeO2–0.6Fe2O3, e 0.2CeO2–0.8Fe2O3 and f Fe2O3
3.4 Hydrogen storage uptake of CeO2–Fe2O3 nanocomposites
The hydrogen sorption properties of the prepared CeO2–Fe2O3 nanocomposites were studied using volumetric methods, and the PCIs are displayed in Fig. 9a, b. A comparison between the PCIs for CeO2–Fe2O3 nanocomposites at 25 and 100 °C is shown. At both 25 and 100 °C, hydrogen adsorption increased at an increasing applied hydrogen pressure. At 25 °C (Fig. 9a), Sample 5 (0.2CeO2–0.8Fe2O3) displayed the highest hydrogen storage capacity (1.02 wt%), followed by Sample 2 (0.8CeO2–0.2Fe2O3) and Sample 1 (CeO2), resulting from their high surface areas (Table 2). On the other hand, metal hydride formation depended on the surface structure and morphology [43], while the reactivity of the CeO2 surface was closely related to OV formation [56]. OVs were mainly formed from doping cerium oxide with iron oxide, resulting in structural defects. Moreover, atomic H adsorption has been shown to increase on defect-free CeO2(111) as the lattice strain increases [56]. In addition to H bonding to Fe sites at OV-rich surface sites [57], tensile strain enhanced surface OV formation more than compressive strain [56]. Therefore, we concluded that OV formation enhanced atomic H adsorption, in agreement with the principle that OVs function as strong adsorption sites [58]. The OV concentration was greatly affected by the molar ratio of the doped metal oxides [59], which can explain the increased hydrogen storage capacity for Sample 5 (0.2CeO2–0.8Fe2O3), possessing the largest surface area. However, OVs rearranged upon raising the temperature to 100 °C (Fig. 9b), and OVs controlled the bonding site selectivity [57], where metal hydride formation was affected by the metal surface [43] and the rapid formation of OVs distorted the crystal lattice [60]. Therefore, at 100 °C, Sample 4 (0.4CeO2–0.6Fe2O3) possesses the maximum number of hydrogen adsorption sites and has the maximum hydrogen adsorption capacity (2.3 wt%), followed by pure CeO2. Notably, both Samples 3 (0.6CeO2–0.4Fe2O3) and 6 (pure Fe2O3) have the lowest hydrogen adsorption capacity at high and low temperatures, respectively. These low hydrogen adsorption capacities can be explained as follows: Sample 3 has a narrow pore width (4.858 nm), while Sample 6 has the smallest surface area (3.73 m2·g−1) and a small pore width (Fig. 4), thus decreasing the hydrogen adsorption process on their surfaces. In general, the samples show obvious changes in their hydrogen storage behaviour based on their OV concentrations at increased temperature. The evident increase in the maximum hydrogen storage capacity proved that the hydrogen uptake of the samples increased at increasing temperature.

PCI curves of CeO2–Fe2O3 nanocomposite samples at a 25 °C and b 100 °C
4 Conclusion
In this study, we reported the preparation of pure and doped nanosized CeO2–Fe2O3 nanocomposites with different Ce4+/Fe3+ molar ratios by a co-precipitation technique. Doped composites featured a higher specific surface area and surface area of pores than pure oxides. Pure and mixed nanosized CeO2–Fe2O3 nanocomposites were used for isothermal reduction using a H2 atmosphere. The further temperature increase led to the increase in the reduction rates of the initial and final stages. The hydrogen storage features of synthetic nanocomposites were evaluated to be 1.02 wt% and 2.3 wt% at room temperature and 100 °C, respectively, which are beneficial for hydrogen storage application.
References
Bao H, Chen X, Fang J, Jiang Z, Huang W. Structure-activity relation of Fe2O3–CeO2 composite catalysts in CO oxidation. Catal Lett. 2008;125(1–2):160.
Wei Y. Syngas generation from methane using a chemical-looping concept: a review of oxygen carriers. J Chem. 2013. https://doi.org/10.1155/2013/294817.
Xiao S, Mei H, Han D, Dassios KG, Cheng L. Ultralight lamellar amorphous carbon foam nanostructured by SiC nanowires for tunable electromagnetic wave absorption. Carbon. 2017;122:718.
Mei H, Zhao X, Bai S, Li Q, Xia J, Bai H, Cheng L. Tuning SERS properties of pattern-based MWNTs-AuNPs substrates by adjustment of the pattern spacings. Carbon. 2018;136:38.
Zhu X, Sun L, Zheng Y, Wang H, Wei Y, Li K. CeO2 modified Fe2O3 for the chemical hydrogen storage and production via cyclic water splitting. Int J Hydrogen Energy. 2014;39(25):13381.
Channei D, Inceesungvorn B, Wetchakun N, Phanichphant S, Nakaruk A, Koshy P, Sorrell C. Photocatalytic activity under visible light of Fe-doped CeO2 nanoparticles synthesized by flame spray pyrolysis. Ceram Int. 2013;39(3):3129.
Channei D, Inceesungvorn B, Wetchakun N, Phanichphant S. Kinetics study of photocatalytic activity of flame-made unloaded and Fe-loaded CeO2 nanoparticles. Int J Photoenergy. 2013. https://doi.org/10.1155/2013/484831.
Arul NS, Mangalaraj D, Ramachandran R, Grace AN, Han JI. Fabrication of CeO2/Fe2O3 composite nanospindles for enhanced visible light driven photocatalysts and supercapacitor electrodes. J Mater Chem A. 2015;3(29):15248.
Mei H, Huang W, Hua C, Xu Y, Cheng L. A novel approach to strengthen naturally pored wood for highly efficient photodegradation. Carbon. 2018;139:378.
Kanan SM, El-Kadri OM, Abu-Yousef IA, Kanan MC. Semiconducting metal oxide based sensors for selective gas pollutant detection. Sensors. 2009;9(10):8158.
Neri G, Bonavita A, Rizzo G, Galvagno S, Capone S, Siciliano P. A study of the catalytic activity and sensitivity to different alcohols of CeO2–Fe2O3 thin films. Sens Actuators B Chem. 2005;111:78.
Liu C, Shan H, Liu L, Li S, Li H. High sensing properties of Ce-doped α-Fe2O3 nanotubes to acetone. Ceram Int. 2014;40(1):2395.
Al-Kelesh H, Halim KA, Nasr M. Synthesis of heavy tungsten alloys via powder reduction technique. J Mater Res. 2016;31(19):2977.
Kang KS, Kim CH, Cho WC, Bae KK, Woo SW, Park CS. Reduction characteristics of CuFe2O4 and Fe3O4 by methane; CuFe2O4 as an oxidant for two-step thermochemical methane reforming. Int J Hydrogen Energy. 2008;33(17):4560.
El-khalek NAA, Naser MI, Yassin KE, Al-Kelesh H. Studying the reduction behavior of eastern desert iron after benefication using falcon concentrator. J Ore Dress. 2014;16(31):11.
Halim KA. Isothermal reduction behavior of Fe2O3/MnO composite materials with solid carbon. Mater Sci Eng A. 2007;452:15.
Llusar M, Royo V, Badenes J, Tena M, Monrós G. Nanocomposite Fe2O3–SiO2 inclusion pigments from post-functionalized mesoporous silicas. J Eur Ceram Soc. 2009;29(16):3319.
Mohapatra M, Anand S. Synthesis and applications of nano-structured iron oxides/hydroxides—a review. Int J Eng Sci Technol. 2010;2(8):127.
Dharanipragada NA, Meledina M, Galvita VV, Poelman H, Turner S, Van Tendeloo G, Detavernier C, Marin GB. Deactivation study of Fe2O3–CeO2 during redox cycles for CO Production from CO2. Ind Eng Chem Res. 2016;55(20):5911.
Turkdogan E, Vinters J, Vinters J. Gaseous reduction of iron oxides: part I. Reduction of hematite in hydrogen. Metall Mater Trans B. 1971;2(11):3175.
Turkdogan E, Olsson R, Vinters J. Gaseous reduction of iron oxides: part II. Pore characteristics of iron reduced from hematite in hydrogen. Metall Mater Trans B. 1971;2(11):3189.
El-Geassy AA, Shehata KA, Ezz SY. Mechanism of iron-oxide reduction with hydrogen-carbonmonoxide mixtures. Trans Iron Steel Inst Jpn. 1977;17(11):629.
El-Geassy A. Gaseous reduction of Fe2O3 compacts at 600 to 1050 °C. J Mater Sci. 1986;21(11):3889.
Gold R, Sandall W, Cheplick P, MacRae D. Plasma reduction of iron oxide with H and natural gas at 100 kW and 1 MW. Ironmak Steelmak. 1977;4(1):10.
Pan F, Zhang J, Chen HL, Su YH, Kuo CL, Su YH, Chen SH, Lin KJ, Hsieh PH, Hwang WS. Effects of rare earth metals on steel microstructures. Materials. 2016;9(6):417.
Maisang W, Phuruangrat A, Thongtem S, Thongtem T. Photoluminescence and photonic absorbance of Ce2(MoO4)3 nanocrystal synthesized by microwave–hydrothermal/solvothermal method. Rare Met. 2018;37(10):868.
El Rouby W, Farghali A, Hamdedein A. Microwave synthesis of pure and doped cerium (IV) oxide (CeO2) nanoparticles for methylene blue degradation. Water Sci Technol. 2016;74(10):2325.
Farghali AA, El Rouby WM, Hamdedein A. Effect of hydrothermal conditions on microstructures of pure and doped CeO2 nanoparticles and their photo-catalytic activity: degradation mechanism and pathway of methylene blue dye. Res Chem Intermed. 2017;43(12):7171.
Yu XX, Sun J, Li ZT, Dai H, Fang HJ, Zhao JF, Yin DF. Solidification behavior and elimination of undissolved Al2CuMg phase during homogenization in Ce-modified Al–Zn–Mg–Cu alloy. Rare Met. 2018. https://doi.org/10.1007/s12598-018-1172-1.
Aboud AA, Al-Kelesh H, El Rouby WM, Farghali AA, Hamdedein A, Khedr MH. CO2 responses based on pure and doped CeO2 nano-pellets. J Mater Res Technol. 2018;7(1):14.
Cao S, Shi M, Wang H, Yu F, Weng X, Liu Y, Wu Z. A two-stage Ce/TiO2–Cu/CeO2 catalyst with separated catalytic functions for deep catalytic combustion of CH2Cl2. Chem Eng J. 2016;290:147.
Shi Z, Yang P, Tao F, Zhou R. New insight into the structure of CeO2–TiO2 mixed oxides and their excellent catalytic performances for 1,2-dichloroethane oxidation. Chem Eng J. 2016;295:99.
Aresta M, Dibenedetto A, Pastore C, Cuocci C, Aresta B, Cometa S, De Giglio E. Cerium (IV) oxide modification by inclusion of a hetero-atom: a strategy for producing efficient and robust nano-catalysts for methanol carboxylation. Catal Today. 2008;137(1):125.
Lin S, Su G, Zheng M, Ji D, Jia M, Liu Y. Synthesis of flower-like Co3O4–CeO2 composite oxide and its application to catalytic degradation of 1,2,4-trichlorobenzene. Appl Catal B. 2012;123:440.
Wang W, Zhu Q, Dai Q, Wang X. Fe doped CeO2 nanosheets for catalytic oxidation of 1,2-dichloroethane: effect of preparation method. Chem Eng J. 2017;307:1037.
Sahoo S, Mohapatra M, Pandey B, Verma H, Das R, Anand S. Preparation and characterization of α-Fe2O3–CeO2 composite. Mater Charact. 2009;60(5):425.
Brito PC, Santos DA, Duque JGS, Macêdo MA. Structural and magnetic study of Fe-doped CeO2. Physica B. 2010;405(7):1821.
Cardillo D, Konstantinov K, Devers T. The effects of cerium doping on the size, morphology, and optical properties of α-hematite nanoparticles for ultraviolet filtration. Mater Res Bull. 2013;48(11):4521.
Janoš P, Kuráň P, Pilařová V, Trögl J, Šťastný M, Pelant O, Henych J, Bakardjieva S, Životský O, Kormunda M. Magnetically separable reactive sorbent based on the CeO2/γ-Fe2O3 composite and its utilization for rapid degradation of the organophosphate pesticide parathion methyl and certain nerve agents. Chem Eng J. 2015;262:747.
Arena F, Gumina B, Lombardo AF, Espro C, Patti A, Spadaro L, Spiccia L. Nanostructured MnOx catalysts in the liquid phase selective oxidation of benzyl alcohol with oxygen: part I. Effects of Ce and Fe addition on structure and reactivity. Appl Catal B. 2015;162:260.
Gobara HM, Aboutaleb WA, Hashem KM, Hassan SA, Henein SA. A novel route for synthesis of α-Fe2O3–CeO2 nanocomposites for ethanol conversion. Mater Sci. 2017;52(1):550.
Broom DP. Hydrogen sorption properties of materials. Hydrogen Storage Mater. 2011. https://doi.org/10.1007/978-0-85729-221-6.
Prabhukhot Prachi R, Wagh Mahesh M, Gangal Aneesh C. A review on solid state hydrogen storage material. Adv Energy Power. 2016;4(2):11.
Lee DH, Cha KS, Lee YS, Kang KS, Park CS, Kim YH. Effects of CeO2 additive on redox characteristics of Fe-based mixed oxide mediums for storage and production of hydrogen. Int J Hydrogen Energy. 2009;34(3):1417.
Santos MC, Kesler O, Reddy ALM. Nanomaterials for energy conversion and storage. J Nanomater. 2012. https://doi.org/10.1155/2012/159249.
Wang G, Yang Y, Han D, Li Y. Oxygen defective metal oxides for energy conversion and storage. Nano Today. 2017;13:23.
Gu Z, Li K, Qing S, Zhu X, Wei Y, Li Y, Wang H. Enhanced reducibility and redox stability of Fe2O3 in the presence of CeO2 nanoparticles. RSC Adv. 2014;4(88):47191.
Galvita VV, Poelman H, Bliznuk V, Detavernier C, Marin GB. CeO2-Modified Fe2O3 for CO2 utilization via chemical looping. Ind Eng Chem Res. 2013;52(25):8416.
Li K, Haneda M, Gu Z, Wang H, Ozawa M. Modification of CeO2 on the redox property of Fe2O3. Mater Lett. 2013;93:129.
Farghali AA, Sayed SG. Synthesis, characterisation and photo-catalytic activity of CeO2/Fe2O3 nano-composite. Int J Nanopart. 2015;8(2):171.
Kongzhai L, Hua W, Yonggang W, Mingchun L. Preparation and characterization of Ce1−xFexO2 complex oxides and its catalytic activity for methane selective oxidation. J Rare Earths. 2008;26(2):245.
Kurian M, Kunjachan C. Effect of lattice distortion on physical properties and surface morphology of nanoceria framework with incorporation of iron/zirconium. Nano-Struct Nano-Objects. 2015;1:15.
Sohn S, Kim D. Modification of Langmuir isotherm in solution systems—definition and utilization of concentration dependent factor. Chemosphere. 2005;58(1):115.
Khedr MH. Isothermal reduction kinetics at 900–1100 °C of NiFe2O4 sintered at 1000–1200 °C. J Anal Appl Pyrol. 2005;73(1):123.
Szekely J, Evans JW, Sohn HY. Gas–Solid Reactions. New York: Academic Press; 1976.
Ma D, Lu Z, Tang Y, Li T, Tang Z, Yang Z. Effect of lattice strain on the oxygen vacancy formation and hydrogen adsorption at CeO2 (111) surface. Phys Lett A. 2014;378(34):2570.
Liu F, Chen C, Guo H, Saghayezhian M, Wang G, Chen L, Chen W, Zhang J, Plummer E. Unusual Fe–H bonding associated with oxygen vacancies at the (001) surface of Fe3O4. Surf Sci. 2017;655:25.
Zhang C, Geng X, Li J, Luo Y, Lu P. Role of oxygen vacancy in tuning of optical, electrical and NO2 sensing properties of ZnO1−x coatings at room temperature. Sens Actuators B Chem. 2017;248:886.
Tan X, Lan H, Xie H, Zhou G, Jiang Y. Role of surface oxygen species of mesoporous CeCu oxide catalyst in OVOCs catalytic combustion. J Environ Chem Eng. 2017;5(2):2068.
Hai G, Huang J, Cao L, Jie Y, Li J, Wang X, Zhang G. Influence of oxygen deficiency on the synthesis of tungsten oxide and the photocatalytic activity for the removal of organic dye. J Alloys Compd. 2017;690:239.
Acknowledgements
The authors gratefully acknowledge Prof. M. H. Khedr (Materials Science and Nanotechnology Department, Faculty of Postgraduate Studies for Advanced Sciences (PSAS), Beni-Suef University, Egypt) for his contributions. The authors also acknowledge Beni-Suef University for supporting this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sayed, S.G., El Rouby, W.M.A. & Farghali, A.A. Preparation and characterization of (CeO2)x–(Fe2O3)1−x nanocomposites: reduction kinetics and hydrogen storage. Rare Met. 39, 218–229 (2020). https://doi.org/10.1007/s12598-019-01244-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12598-019-01244-z