
Abstract
To improve the performance of battery cathode materials that consist of carbonaceous organic material, carbon coatings on lithium iron phosphate (LiFePO4/C) materials were synthesized by different carbon sources. LiFePO4/C was synthesized by a combination method of sol–gel and gas-phase diffused permeation. LiFeO4/C materials were prepared by coating different carbon contents. High-performance composite materials were prepared by combining carbon with element doped by two modified methods. The elements of Fe and C came from Fe3+ and sucrose, glucose, citric acid. Thermogravimetry–differential thermal analysis (TG-DTA), X-ray diffractometer (XRD), scanning electron microscope (SEM), cycle voltammetry (CV), and charge–discharge test were used to characterize and test the surface morphology, structure, and electrochemical performance. The results show that LiFePO4/C synthesized with sucrose has higher specific discharge capacity than the other materials. The specific discharge capacity of this material is 84.27 mAh·g−1. The capacity retention could attain 94 % of the initial discharge capacity after 30 cycles, showing good electrochemical performance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Since LiFePO4 was reported [1], it has received much attention and research. Compared with similar cathode materials such as LiCoO2 which is short of resources, expensive, and toxic, as an interesting cathode material for lithium ion batteries, LiFePO4 cathode materials have advantages of excellent thermal stability, low cost, non-toxic, friendly to environment, etc. It becomes one of the most potential cathode materials for its high theoretical capacity which is up to 170 mAh·g−1 [1–4]. LiFePO4/C is olivine-structured, orthorhombic, and its space group is orthorhombic system (Pnma [5]). The way to prepare LiFePO4 is solid-phase method [6, 7], liquid-phase method [8–10], and microwave synthesis [11]. Its structural features make it have good cycle stability, but prevent the diffusion and electronic motion of lithium ion [12], leading to low electronic conductivity and diffusion rate of lithium ion. In order to improve the diffusion rate of lithium ion and the electronic conductivity of cathode material, most of the research is mainly carried out by coating the conductive material, doping ion, and decreasing particle size to improve the electrochemical properties of materials [13].
The solid-phase method is limited for the uneven phase, irregular crystals, large sizes, long period, and so on. Coating the surface of LiFePO4 can improve the conductivity among the particles and reduce the particle size to achieve the purpose of improving the electrochemical performance of LiFePO4. Studies have shown that coating the carbon surface can improve the high current discharge performance of the samples [14, 15]. Therefore, the LiFePO4 was modified by coating method [16], during which solid-phase coating [17] and gel coating [18] were taken.
Gas-phase diffusion permeation method originates from multi-component rare earth infiltration method which is the technology based on the basic standpoint of physical chemistry, using special structural characteristics and chemical properties of rare earth elements to modify metal surface. The earliest multi-element rare earth penetration method focused on rare earth permeating treatment [19] of pure 20 Steel and 08 Steel surfaces. In recent years, multi-component rare earth infiltration method has expanded to the polyoxometalates diffusion, processing many new functional materials. Based on the results of multi-element rare earth penetration [20], carbon coating was combined with gas-phase diffusion. Dispersive carbon was used as an electronic transport pathway to improve the electrical conductivity and the electrochemical performance. Through preparation of carbon-coated LiFePO4/C, it was aimed to enhance the conductivity of the material, increase the surface area of the material, and make the material and the electrolyte contact fully to improve the internal particle removal/block performance of Li+ and significantly improve the charge and discharge capacity as well as cycle performance [21, 22] of the material.
2 Experimental
2.1 Reagents and instruments
Ferric nitrate (Fe(NO3)3·9H2O) (analytically pure), lithium nitrate (LiNO3) (analytically pure), mono-ammonium phosphate [MAP] (NH4H2PO4) (analytically pure), sucrose (analytically pure), glucose (analytically pure), citric acid (analytically pure), and methanol (analytically pure) were purchased from Sinopharm Chemical Reagent Ltd., and lithium tablets (Beijing Research Institute of Nonferrous Metals), acetylene black (Shanghai Haotian Chemical Co., Ltd.), and polyvinylidene fluoride PVDF (industrial grade, Shanghai Hersbit Chemical Co., Ltd.) were also purchased.
Sample gas-phase diffusion experiment was carried out in gas-phase diffusion furnace (XMT-101). High-temperature thermal gravimetric (TG, ZRY-2P, Shanghai Precision and Scientific Instrument Co. Ltd.) analyzer and high temperature difference thermal analyzer (DTA, CRY-2P) were used to confirm the vapor diffusion temperature of the material; X-ray diffractometer (XRD, D/max-2000, Japan Science Company) was used to do the material phase analysis; scanning electron microscope (SEM, S-4800, Japan Hitachi company) was used to analyze surface topography of the material; battery assembly was carried out in the STX-type vacuum glove box; charge and discharge tests were completed in highly accurate test system of battery performance (BTS-5 V/2 mA, Shenzhen Xinweier Electronic Co. Ltd.) and electrochemical workstation (CHI 604C, Shanghai Chenhua Company); data acquisition and process were completed by Xinwei data processing software.
2.2 Experimental process
2.2.1 Sample preparation
LiFePO4/C composite cathode materials were prepared by sol–gel and gas-phase diffusion permeation method: the stoichiometric ratio of LiNO3. H2O, Fe (NO3)3·9H2O, and NH4H2PO4 was 1:1:1. They were dissolved in the solution that mixed water with ethanol, either without carbon source or adding soluble carbon source (glucose, citric acid, and sucrose). Then, they were mixed evenly with a magnetic stirrer, continuing at 80 °C for about 80 min until the sol appeared, then dried under 80 °C, and the precursor of LiFePO4/C was obtained. The diffusion experiments of LiFePO4/C precursor were carried out in a small crucible gas-phase permeability furnace that we made ourselves, and the temperature was controlled by precise temperature controller (XMT-101).
Diffusion experiment [23] was divided into three steps: first, the gas-phase permeability furnace was heated to a preset temperature of 700 °C [24] for 30 min, using methanol as the diffusion agent, making the diffusion furnace in a reducing atmosphere; second, the LiFePO4/C precursor was put into the furnace, regulating dripping speed of permeability agent to about 45 drops per minute; finally, LiFePO4/C composite cathode materials were cooled down to room temperature after 7 h diffusion and LiFePO4/C composite cathode material was obtained.
2.2.2 Battery assembly
Battery assembly process was conducted in a glove box filled with argon gas. Active substance acetylene black and polyvinylidene fluoride (PVDF) were weighed with the ratio of 75:15:10 (mass ratio). The mixed substances were dissolved in N-methyl pyrrolidone (NMP) and were evenly applied to the aluminum foil. After dried in a vacuum drying box under 120 °C, it was punched into card, tablet, and detect battery. The electrode was lithium metal wafer with a diameter of 12 mm, a thickness of 100 μm. The diaphragm was polypropylene with a diameter of 16 mm, a thickness of 16 μm; the electrolyte consisted of ethylene carbonate (EC) and dimethyl carbonate (DMC), the volume ratio of which was EC:DMC = 1:1 and the concentration of LiPF6 was 1 mol·L−1.
2.2.3 Cycle voltammetry (CV) characterization
The assembled simulation battery prepared by LiFePO4/C material mixed 70 % sucrose was scanned at voltage range of 2.5–4.3 V to test CV curve of the sample with scanning rate of 0.1 mV·s−1. In the experiment, electrochemical workstation (CHI604C, Shanghai Chen Hua Company) was used. Test condition was two-electrode system. Auxiliary electrode and reference electrode were both lithium. The electrode was active sample electrode.
3 Results and discussion
3.1 Determination of gas-phase diffusion permeation temperature
LiFePO4/C precursor without carbon source was tested by thermogravimetry–differential thermal analysis (TG–DTA) from room temperature to 800 °C, and the heating rate was 5 °C·min−1. The results were shown in Fig. 1. It can be seen from the TG curve that the sample was at great loss from 80 to 600 °C. The loss mainly occurred in three stages: the first stage was from room temperature to 178 °C, mainly removing the adsorbed water in the gel; the second stage was from 178 to 346 °C, which was mainly because NH4 +, NO −3 and other small molecules decomposed; the third stage was from 346 to 605 °C, and it was the main process of LiFePO4/C crystal type changes. No weight loss occurred after 600 °C, showing that the sample no longer had decomposition reaction after that temperature. According to the results of TG–DTA test, the diffusion temperature of LiFePO4/C precursor is 700 °C.

TG–DTA curves of LiFePO4 precursor
The TG–DTA test of LiFePO4/C precursor with glucose as carbon source was carried out from room temperature to 800 °C, the heating rate was 5 °C·min−1 and the test results were shown in Fig. 2. According to the TG curve, the sample had obvious weight loss from 80 to 600 °C. Weight loss mainly occurred in three stages: the first stage was from room temperature to 178 °C, mainly removing the adsorbed water in the gel; the second stage was from 178 to 246 °C, which was mainly caused by glucose decomposition; the third stage was from 246 to 600 °C, and it was the main process of LiFePO4/C crystal type changes. No weight loss after 600 °C means that the sample no longer had decomposition reaction after that temperature. According to the results of TG–DTA test, the diffusion temperature of LiFePO4/C precursor with glucose as carbon source is 700 °C.

TG–DTA curves of LiFePO4/C precursor with glucose
The TG–DTA test of LiFePO4/C precursor with 50 % sucrose as carbon source was carried out from room temperature to 800 °C, the heating rate was 5 °C·min−1. The test results were shown in Fig. 3. According to the TG curve, the sample had obvious weight loss from 80 to 600 °C. Weight loss mainly occurred in three stages: the first stage was from room temperature to 125 °C, mainly resulting from removal of the adsorbed water in the gel; the second stage was from 125 to 330 °C, mainly caused by sucrose decomposition; the third stage was from 330 to 610 °C, which was the main process of LiFePO4/C crystal type changes. No weight loss after 610 °C shows that the sample no longer had decomposition reaction after that temperature. According to the test results of TG–DTA, the diffusion temperature of LiFePO4/C precursor with sucrose as carbon source is 700 °C.

TG–DTA curves of LiFePO4/C precursor with sucrose
The TG–DTA test of LiFePO4/C precursor with 50 % citric acid as carbon source was carried out from room temperature to 800 °C, and the heating rate was 5 °C·min−1. The test results were shown in Fig. 4. According to the TG curve, the sample had obvious weight loss from 80 to 600 °C. Weight loss mainly occurred in three stages: the first stage was from room temperature to 160 °C, mainly removing the adsorbed water in the gel; the second stage was from 160 to 350 °C, mainly caused by citric acid decomposition; the third stage was from 350 to 590 °C, which was the main process of LiFePO4/C crystal type changes. No weight loss after 600 °C shows that the sample no longer had decomposition reaction after that temperature. According to the results of TG–DTA test, the diffusion temperature of LiFePO4/C precursor with citric acid as carbon source is 700 °C.

TG–DTA curves of LiFePO4/C precursor with citric acid
Through the TG–DTA diagrams, it can be seen that different carbon source types determine different degrees of penetration.
3.2 XRD analysis
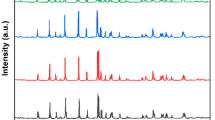
XRD patterns of the LiFePO4/C sample prepared without carbon source and the LiFePO4/C sample used glucose, citric acid, and sucrose as carbon source are shown in Fig. 5. Compared with the standard spectrum (JCPD40-1499), all diffraction peaks were indexed to olivine structure. The results show that LiFePO4/C prepared with different carbon sources have pure phase. No diffraction peaks appeared means that carbon exists in the form of amorphous carbon. Three peaks of all the samples were observed between 20° and 40°, among which LiFePO4/C samples used citric acid as carbon source are the strongest. Angles of strong diffraction peaks of samples prepared with these four carbon sources are almost consistent.

XRD patterns of LiFePO4/C synthesized by different carbon sources
3.3 SEM morphology analysis
The morphologies of LiFePO4/C samples with different carbon sources are shown in Fig. 6. From the chart, the grain size of the prepared sample is about 1 μm with carbon coated. Samples without carbon source among the precursors are hardened to a certain extent. However, samples prepared by adding different carbon sources (glucose, sucrose and citric acid) to precursors have even particles. LiFePO4/C sample with sucrose as the carbon source is the best among them as Fig. 6c shows [25].

SEM images of LiFePO4/C synthesized by different carbon sources: a without carbon source, b glucose, c sucrose, and d citric acid
3.4 Charge and discharge test
In order to study the effects of different carbon sources on the electrochemical properties of LiFePO4/C samples, the LiFePO4/C samples of button-type battery which were prepared with different carbon sources which were tested by charging and discharging. The first charge and discharge curves and cycle performance curves of samples without carbon source and with different carbon sources under 0.5 °C are shown in Figs. 7 and 8. According to the Figures, the initial specific discharge capacity of LiFePO4/C samples used glucose as carbon source is 64.80 mAh·g−1 and the initial discharge efficiency is 69.8 %. As for LiFePO4/C samples used citric acid as carbon source, the initial specific discharge capacity is 15.0 mAh·g−1. The first specific discharge capacity of LiFePO4/C samples prepared with sucrose as carbon source is 84.27 mAh·g−1, and the initial discharge efficiency is 71.2 %, higher than the initial efficiency (69.8 %) of those used glucose as carbon source. The cycle performance is good, as well. After 30 cycles, the capacity retention rate is 94 %, so LiFePO4/C material is chosen as adding sucrose during the preparation of precursor.

First discharge curves of LiFePO4/C synthesized by different carbon sources

Cycle performance curves of LiFePO4/C synthesized by different carbon sources
3.5 CV analysis
The symmetry of CV curve and redox peak area are important references of material cycle performance and the reversible capacity size. The better the symmetry of material circulation is, the greater the performance and the bigger the reversible capacity. The better the uniform of redox peak size is, the better cycle performance of the material and the greater reversible capacity of the assembled simulation battery. The curves of samples added with 75 % sucrose are consistent with the peak of charge and discharge. The charge peak is at 3.6 V, discharge peak is at 3.25 V. The peak sharpness is better and the discharge peak area is consistent with charge peak area, showing the good reversible capacity and cycle performance of the material, as shown in Fig. 9.

CV curve of LiFePO4/C synthesized by 70 wt% sucrose
4 Conclusion
Pure-phase LiFePO4/C materials were prepared by sol–gel and gas-phase diffusion permeation method. LiFePO4/C precursor material prepared with glucose, sucrose, and citric acid as carbon source was diffused in diffusion furnace under 700 °C, the diffusion time was 7 h. XRD, SEM, and charge–discharge test were used to analyze LiFePO4/C materials prepared with different carbon sources. The results show that LiFePO4/C material prepared with sucrose as carbon source has even particles with ~1 μm in diameter. The initial specific discharge capacity of the material is the highest at 84.27 mAh·g−1 and has good cycle performance. After 30 cycles, the capacity retention rate is 94 %, so the choice of carbon source for preparing LiFePO4/C materials is sucrose. Owing to small electronic conductivity of LiFePO4 and low speed of diffusion of lithium ion, the volume energy density is low, which severely affects the utility of the material. Therefore, using different methods to increase the conductivity of the LiFePO4 material is the main research direction.
References
Padhi AK, Nanjundaswamy KS, Goodenough JB. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J Electrochem Soc. 1997;144(4):1188.
Lin L, Ma XG, Guo Y, Wang Q, Xiao D. Study on preparation and electrochemical performance of LiFePO4 with microwave method. Res Appl Chem. 2010;22(5):570.
Jiang ZJ. Development and challenge of LiFePO4 cathode materials for Li-ion batteries. J Funct Mater. 2010;3(41):365.
Yang SF, Song YN, Peter Y, Zavalij M, Stanley W. Reactivity, stability and electrochemical behavior of lithium iron phosphates. Electrochem Commun. 2002;4(3):75.
Zhang SH, Dou QS. Research progress of material preparation method of cathode material for lithium-ion batteries lithium iron cathode. Xinjiang Nonferrous Metals. 2009;62(S1):134.
Yamada A, Chang SC, Hinokuma K. Optimized LiFePO4 for lithium battery cathodes. J Electrochem Soc. 2001;148(3):224.
Hu J, Feng ZS, Wang Y, Chen JJ, Wang XJ. Study on the preparation of composite LiFePO4/C cathode material mixing carbon doped by carbon thermal reduction method. Electron Compon Mater. 2013;32(11):23.
Tang ZY, Qiu RL, Teng GP. Study of LiFePO4 cathode material for lithium ion battery. Chem Ind Eng Prog. 2008;27(7):995.
Wang EX, Xu GR, Zheng HB, Zhang YX. An overview of coprecipitation method to synthesize LiFePO4. Guangdong Chem Ind. 2011;38(1):97.
Gao MY, Liu NQ, Li ZB, Li C. A gelatin-based sol–gel procedure to synthesize the LiFePO4/C nanocomposite for lithium ion batteries. Solid State Ion. 2014;34(6):8.
Zhang HF, Sun Z, Chen MJ. Preparation and electrochemical performance of LiFePO4 by microwave synthesis. Power Syst. 2012;36(12):1787.
Wang LL, Ma PH, Li FQ. The structure and the electrochemical reaction mechanism of LiFePO4 cathode material for lithium ion batteries. Chem Bull. 2008;1(9):17.
Zhong SK, Xu YB, Li YH, Zeng HH, Li W, Wang J. Synthesis and electrochemical performance of LiMnPO4/C composites cathode materials. Rare Met. 2012;31(5):474.
Hee PS, Hyoung KC. Preparation of Li[Ni1/3Co1/3Mn1/3]O2 powders for cathode material in secondary battery by solid-state method. Rare Met. 2006;25(6):186.
Zhou JX, Shen XQ, Jing MX, Zhan Y. Synthesis and electrochemical performances of spherical LiFePO4 cathode materials for Li-ion batteries. Rare Met. 2006;25(6):20.
Hamamoto K, Fukushima M, Mamiya M, Yoshizawa Y, Akimoto J, Suzuki T, Fujishiro Y. Morphology control and electrochemical properties of LiFePO4/C composite cathode for lithium ion batteries. Solid State Ion. 2012;225(14):562.
Kim JK, Choi JW, Cheruvally G, Ahn JH. A modified mechanical activation synthesis for carbon-coated LiFePO4 cathode in lithium batteries. Mater Lett. 2007;61(18):3822.
Cao YL, Yu LH, Li T, Yang HX. Synthesis and electrochemical characterization of carbon-coated nanocrystalline LiFePO4 prepared by polyacrylates-pyrolysis route. J Power Sources. 2007;172(2):1.
Neelima M, Amitava B, Alka G, Shobit O, Kantesh B. Progress in material selection for solid oxide fuel cell technology: a review. Prog Mater Sci. 2015;72(7):145.
Zhang YH, Cai Y, Li BW, Ren HP, Hou ZH, Zhao DL. Electrochemical hydrogen storage characteristics of the as-cast and annealed La0.8−x Pr x Mg0.2Ni3.35Al0.1Si0.05 (x = 0−0.4) electrode alloys. Rare Metal Mater Eng. 2013;42(10):1985.
Wang GX, Zhang XH, Han ES, Wang CX. Study on electrochemical performance of LiFePO4 cathode materials mixing different carbon sources. Inorg Salt Chem Ind. 2008;41(8):35.
Prosini PP, Carewska M, Scaccia S, Pasquali M. Long-term cyclability of nanostructured LiFePO4. Electrochim Acta. 2003;48(28):4205.
Xu J, Chen G, Xie CD. Synthesis and electrochemical performance of LiFePO4/C prepared by a new method. Solid State Commun. 2008;147(11–12):443.
Peng ZD, Cao YB, Zhou YL, Hu GR. Synthesis of LiFePO4 using FeSO4·7H2O byproduct from TiO2 production as raw materials. Rare Met. 2009;28(6):612.
Luo WB, Wen L, Luo HZ, Song RS, Zhai YC, Liu C, Li F. Carbon nanotube-modified LiFePO4 for high rate lithium ion batteries. New Carbon Mater. 2014;29(4):292.
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (No. 51274143).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, X., Jiang, Y.Z., Li, X.K. et al. Electrochemical property of LiFePO4/C composite cathode with different carbon sources. Rare Met. 37, 743–749 (2018). https://doi.org/10.1007/s12598-016-0781-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12598-016-0781-9