
Abstract
Neurons are classified into several morphological types according to the locations of their somata and the branching patterns of their axons and dendrites. Recent studies suggest that these morphological features are related to their physiological properties, including firing characteristics, responses to neuromodulators, and wiring patterns. Therefore, rapid morphological identification of electrophysiologically recorded neurons promises to advance our understanding of neuronal circuits. One of the most common anatomical cell identification methods is neuronal reconstruction with biocytin delivered through whole-cell patch-clamp pipettes. However, conventional reconstruction methods usually take longer than 24 h and limit the throughput of electrophysiological experiments. Here, we developed a quick, simple cell reconstruction method by optimizing the tissue clearing protocol ScaleSQ. We found that adding 200 mM NaCl almost entirely prevented tissue swelling without compromising optical clearing ability. This solution, termed IsoScaleSQ, allowed us to increase the transparency of the gray matter of 500-µm-thick slices within 30 min, meaning that the total time required to reconstruct whole-cell recorded neurons was reduced to 1 h. This novel method will improve the efficacy and effectiveness of electrophysiological experiments linked to cell morphology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurons show diversity in the locations of their somata and their subcellular morphologies (Larkman and Mason 1990; Markram et al. 2004; Klausberger and Somogyi 2008). These morphological features of neurons are associated with their electrophysiological properties. For example, hippocampal pyramidal cells located in the deep layer of the CA1 subarea exhibit higher firing rates and are more likely to have place fields than those located in the superficial layer (Mizuseki et al. 2011). In addition to being categorized according to their location, CA1 pyramidal cells are also divided into two subgroups based on dendritic morphology. These two types of pyramidal cells are modulated in opposite directions by acetylcholine (Graves et al. 2012). A large-scale connectivity analysis coupled with morphological identification of neurons revealed that the intrinsic cortical microcircuit in the visual cortex is governed by only a few types of connectivity motifs (Jiang et al. 2015). However, these pieces of information have not been fully integrated into a generalized model, so more post hoc morphological characterization of electrophysiologically recorded neurons are still required.
One of the major methods used for this purpose is neuronal reconstruction with biocytin delivered during whole-cell patch-clamp recordings (Margrie et al. 2002). The whole-cell patch-clamp technique is a powerful method that can monitor the subthreshold membrane potential of a single neuron with a high signal-to-noise (SN) ratio and high temporal resolution (Neher 1992). Using an intrapipette solution containing biocytin, one can reconstruct the fine-scale morphology of the recorded cells post hoc via visualization with avidin conjugates (Horikawa and Armstrong 1988). However, the current protocol requires as long as 24 h to complete fixation and labeling, precluding the user from confirming the result on the day of the recording. In addition, light scattering of brain tissues prevents the user from observing the labeled neurons when they are located deep in the tissues.
To facilitate studies associating the electrophysiological properties of neurons with their cellular morphology, we focused on optical clearing techniques (Chung et al. 2013; Ke et al. 2013; Yang et al. 2014; Hama et al. 2015; Ku et al. 2016; Pan et al. 2016; Kubota et al. 2017) for the following two reasons: (1) some optical clearing techniques permeabilize the treated samples, facilitating the penetration of proteins into the tissues (Chung et al. 2013; Yang et al. 2014), and (2) suppressing the background scattering of light allows the user to achieve sufficiently high SN ratios to observe the subcellular structures of neurons in deeper tissues, which may render post hoc sectioning of the tissue unnecessary.
In the present study, we focused on ScaleSQ, a quick clearing protocol for thick slices, because ScaleSQ consists of shorter procedures and preserves more intact membrane structures than other clearing protocols (Hama et al. 2015). This latter feature is of particular importance because biocytin is a small molecule and may leak out of the cell when the membrane structure is damaged. However, one drawback of ScaleSQ is the swelling of sample tissues (Hama et al. 2015). Sample swelling increases the risk of distortion of neurons and neurites, an occurrence that is undesirable for accurately evaluating the neuronal morphology. To reduce the swelling, we added NaCl to the clearing solution; this trick is used in another clearing protocol, CUBIC1A (Susaki and Ueda 2016), to alleviate excess swelling. With other refinements in the protocol, we optimized ScaleSQ for the simple and quick reconstruction of whole-cell recorded neurons. We refer to the modified ScaleSQ herein as IsoScaleSQ.
Methods
Preparation of the IsoScaleSQ solution
The IsoScaleSQ solution was prepared using the following reagents: 9.1 M urea (Nacalai Tesque, 35904-45), 22.5% (v/w) d-sorbitol (Wako Pure Chemical Industries, 198-03755), 200 mM sodium chloride (Nacalai Tesque, 31319-45), and 2% (v/w) Triton X-100 (Nacalai Tesque, 35501-15). The reagents were dissolved in water and stirred until well mixed.
Animal ethics
ICR mice (Japan SLC, Shizuoka, Japan) and Arc-dVenus transgenic mice (line D) (Eguchi and Yamaguchi 2009) were used in these experiments. Animal experiments were performed with the approval of the animal experiment ethics committee at the University of Tokyo (approval number: P29-9) and according to the University of Tokyo guidelines for the care and use of laboratory animals. These experimental protocols were carried out in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions (Ministry of Education, Culture, Sports, Science and Technology, notice no. 71 of 2006), the Standards for Breeding, Housing, and Pain Alleviation for Experimental Animals (Ministry of the Environment, notice no. 88 of 2006), and the Guidelines on Methods of Animal Disposal (Prime Minister’s Office, notice no. 40 of 1995). All animals were housed under a 12/12-h dark–light cycle (light from 07:00 to 19:00) at 22 ± 1 °C with ad libitum food and water.
Slice preparation for in vitro electrophysiology
Acute slices were prepared from the medial to the ventral part of the hippocampal formation. Postnatal 21- to 28-day-old male ICR mice were deeply anesthetized with isoflurane and decapitated. The brains were rapidly removed and horizontally sliced (400 µm thick) using a vibratome and an ice-cold oxygenated (95% O2, 5% CO2) cutting solution consisting of 222.1 mM sucrose, 27 mM NaHCO3, 1.4 mM NaH2PO4, 2.5 mM KCl, 1 mM CaCl2, 7 mM MgSO4, and 0.5 mM ascorbic acid. One to three slices per mouse were used in the experiments. Slices were incubated at 35 °C for 30 min and were maintained for at least 30 min at room temperature in a submerged chamber filled with oxygenated artificial cerebrospinal fluid (ACSF) containing 127 mM NaCl, 3.5 mM KCl, 1.24 mM NaH2PO4, 1.3 mM MgSO4, 2.4 mM CaCl2, 26 mM NaHCO3, and 10 mM d-glucose.
In vitro electrophysiology
Whole-cell current-clamp recordings were obtained from CA1 pyramidal cells, which were targeted using infrared differential interference contrast microscopy (IR-DIC). Patch pipettes (3–8 MΩ) were filled with a potassium-based solution consisting of 135 mM potassium gluconate, 4 mM KCl, 10 mM HEPES, 10 mM creatine phosphate, 4 mM Mg-ATP, 0.3 mM Na2-GTP, 0.3 mM EGTA, and 0.2% biocytin. Recordings were performed for at least 15 min in a submerged chamber perfused at 8 ml/min with oxygenated ACSF at 33–35 °C. Pyramidal cells were identified by their regular spiking properties and by post hoc histological analysis.
Surgery for in vivo electrophysiology
Whole-cell recordings were obtained from postnatal 28- to 40-day-old male ICR mice as previously described (Ishikawa et al. 2014; Funayama et al. 2015). The mice were anesthetized with urethane (2.25 g/kg, intraperitoneal [i.p.]). Anesthesia was confirmed by the lack of paw withdrawal, whisker movement, and eye-blink reflexes. The skin was subsequently removed from the head, and the animal was implanted with a metal head-holding plate. A craniotomy (2.5 × 2.0 mm2) was performed, with its center at 2.0 mm posterior to the bregma and 2.5 mm ventrolateral to the sagittal suture, and the neocortex above the hippocampus was aspirated (Kuga et al. 2011; Sakaguchi et al. 2012). The exposed hippocampal window was covered with 1.7% agar at a thickness of 1.5 mm.
In vivo electrophysiology
Patch-clamp recordings were obtained from neurons in the CA1 stratum pyramidale (AP: − 2.0 mm; ML: 2.0 mm; DV: 1.1–1.3 mm) using borosilicate glass electrodes (4–7 MΩ). Pyramidal cells were identified by their regular spiking properties and by post hoc histological analysis. For current-clamp recordings, patch pipettes were filled with a potassium-based solution consisting of (in mM) 135 potassium gluconate, 4 KCl, 10 HEPES, 10 creatine phosphate, 4 Mg-ATP, 0.3 Na2-GTP, 0.3 EGTA, and 0.2% biocytin.
Histology
For the visualization of patch-clamped neurons, the slices were fixed in 10% paraformaldehyde with 0.1% Hoechst 33342 (Thermo Fisher Scientific; H1399) or 1% acridine orange (Sigma–Aldrich; A6014) at 4 °C for 30 min (in vitro), or the brains were coronally sectioned at a thickness of 500 µm using a brain matrix (Brain Science Idea. Co., Ltd., Osaka, Japan; MBS-S0.5C). The sections were fixed in 10% paraformaldehyde with 0.1% Hoechst 33342 or 1% acridine orange at 4 °C for 30 min (in vivo). After the solution was washed out, the sections were incubated in IsoScaleSQ with 2 µg/ml streptavidin-Alexa Fluor 594 conjugate (Thermo Fisher Scientific; S11227) for 30 min at 37 °C. Fluorescent images were acquired using a confocal microscope (FV1200; Olympus, Tokyo, Japan) and were subsequently merged.
Behavioral procedure and dVenus visualization
Arc-dVenus transgenic mice were transferred from their home cages to an environmentally enriched space and were allowed to explore it for 5 h (enriched environment group). Then the brains were removed and coronally sectioned at a thickness of 500 µm using a vibratome, and the sections were fixed in 10% paraformaldehyde with 0.1% Hoechst 33342 at 4 °C for 30 min. After the solution was washed out, the sections were incubated in IsoScaleSQ for 30 min at 37 °C. Fluorescent images of layers 2/3 and 5 of the primary visual cortex were acquired using a confocal microscope and were subsequently merged.
Measurement of transmittances
The transmittances of regions of interest (ROIs) were calculated using the following formula: (brightness of ROI − brightness of ROI (not clearing))/(brightness of transmitted light − brightness of ROI (not clearing)). Grids within a ROI were excluded from analysis.
Statistical analysis
A paired t test was used to compare areas of the hippocampus before and after swelling in reagents (ScaleSQ(5), ScaleSQ(2), and IsoScaleSQ) for 30 min (Fig. 1b). Two-way analysis of variance (ANOVA) was used with the Bonferroni correction to compare transmittance between two groups (ScaleSQ(2) and IsoScaleSQ) among twelve time points (Fig. 2c) and to compare transmittance among three regions (hippocampus, cortex, thalamus) from five groups defined by different slice thicknesses (100 µm, 300 µm, 500 µm, 700 µm, 900 µm) (Fig. 3b–d). One-way ANOVA was used with the Bonferroni correction was used to compare transmittance among the five slice-thickness groups (100 µm, 300 µm, 500 µm, 700 µm, 900 µm) (Fig. 3b–d). Student’s t test was used to compare the tortuosities of neurons visualized using the conventional method and our IsoScaleSQ method (Fig. 5d). Differences were considered to be significant at P < 0.05. Data are reported as the mean ± standard error (SEM).

No tissue swelling by ScaleSQ in the presence of NaCl. a Bright-field macroscopic images were acquired 0 min and 30 min after continuous treatment of brain hemisphere slices with ScaleSQ(5), ScaleSQ(2), or IsoScaleSQ. The grid interval is 2 mm. b The sectional areas of the hippocampus were measured 0 min and after 30 min of treatment with optical clearing reagents. Tissues swelled in ScaleSQ(5) and ScaleSQ(2), but not in IsoScaleSQ [n = 10 slices from two mice (ScaleSQ(5)), n = 11 slices from two mice (ScaleSQ(2)), n = 16 slices from four mice (IsoScaleSQ), ***P < 0.001, paired t test]. c Bright-field macroscopic images were acquired 0 min and 30 min after the treatment of brain hemisphere slices with IsoScaleSQ containing 300 mM or 400 mM NaCl. d The sectional areas of the hippocampus were measured 30 min after treatment with optical clearing reagents. n = 9 slices from two mice (300 mM), n = 9 slices from two mice (400 mM), #P < 0.001 versus 0 min, paired t test

Clearing efficiency of IsoScaleSQ. a, b Time-lapse imaging of ScaleSQ(2) and IsoScaleSQ-treated 500-µm-thick slices. The grid interval is 2 mm. c Time-dependent changes in the transparency of the slices were quantified in the hippocampus. The transparency increased rapidly within 10 min and reached a plateau after 30 min. Error bars are SEMs of nine slices from two mice (ScaleSQ(2)) and 16 slices from four mice (IsoScaleSQ)

Transparency of IsoScaleSQ-treated slices at different thicknesses. a Bright-field microscopic images were taken before and 30 min after IsoScaleSQ treatment of brain slices at thicknesses of 100, 300, 500, 700, and 900 µm. The grid interval is 2 mm. b–d The transmittances were quantified in the hippocampus (b), the neocortex (c), and the thalamus (d). n = 10 slices from four mice (100 µm), 12 slices from two mice (300 µm), 16 slices from four mice (500 µm), 12 slices from two mice (700 µm), 10 slices from two mice (900 µm) each. *P < 0.05 and ***P < 0.001 versus 100 µm. Two-way ANOVA followed by the Bonferroni correction
Results
The optical clearing procedure using ScaleSQ is reported to swell biological tissues, but the swelling has not been quantified. We applied ScaleSQ(5) or ScaleSQ(2) to 500-µm-thick coronal slices of the brain hemisphere at 37 °C for 30 min and measured their sectional areas (Fig. 1a). ScaleSQ(5) and ScaleSQ(2) differed in their detergent concentrations; the former contained 5% Triton X-100 and the latter contained 2%. ScaleSQ(2) was employed because a lower concentration of the detergent was expected to suppress the swelling. Treatment with ScaleSQ(5) and ScaleSQ(2) increased the tissue areas by 37.6 ± 3.8% (Fig. 1b, mean ± SEM of 10 slices from two mice, P = 8.5 × 10−5, t9 = 6.73, paired t test) and 31.7 ± 3.7% (n = 11 slices from two mice, P = 8.4 × 10−4, t8 = 5.18), respectively. Thus, both solutions induced tissue swelling. To suppress the swelling, we incorporated a trick used in CUBIC1A (Susaki and Ueda 2016). Thus, we added NaCl to ScaleSQ(2) at a concentration of 200 mM NaCl. The change in the sectional area caused by the treatment of this solution was 0.8 ± 1.5% (n = 16 slices from four mice), and there was no significant difference between the areas before and after the treatment (Fig. 1b; P = 0.91, t15 = 0.12, paired t test). Thus, we concluded that 200 mM NaCl-added ScaleSQ(2) induced no apparent swelling. We refer to this new solution herein as IsoScaleSQ. Incidentally, further increases in the concentration of NaCl caused tissue shrinkage at 30 min (Fig. 1c, d; 300 mM: 96.9 ± 3.4%, n = 9 slices from two mice; 400 mM: 85.4 ± 2.4%, n = 9 slices from two mice). Therefore, 200 mM NaCl was an optimal concentration.
The original procedure for ScaleSQ clearing consists of two steps: a clearing process and a refractive index matching process with a high sugar content solution (ScaleS4(0)) (Hama et al. 2015). However, we found that clearing with IsoScaleSQ alone was sufficient to make the gray matter of a 500-µm-thick slice transparent. Therefore, we skipped the refractive index matching process and were thus able to simplify and shorten the entire clearing procedure.
We next optimized the incubation time for IsoScaleSQ clearing. We measured time-dependent changes in light transmittance of the hippocampus in slices treated with ScaleSQ(2) or IsoScaleSQ at 37 °C (Fig. 2a, b). The light transmittance of ScaleSQ(2)-treated samples increased up to 63.9 ± 3.1% within 10 min (mean ± SEM of 9 slices from two mice) and thereafter increased slowly, reaching 67.6 ± 2.6% at 30 min and 74.7 ± 2.4% at 60 min (Fig. 2c; n = 9 slices from two mice). IsoScaleSQ treatment produced similar results. The light transmittance reached 65.4 ± 2.9%, 70.0 ± 1.1%, and 73.7 ± 1.6% at 10, 30, and 60 min, respectively (Fig. 2c; n = 16 slices from four mice). Thus, the addition of NaCl did not affect the clearing properties of IsoScaleSQ (P = 0.13, F1, 276 = 14.0, two-way ANOVA with the Bonferroni correction). We adopted a 30-min incubation time in the subsequent experiments.
We next quantified the light transmittance as a function of the thickness of brain slices. The brains were coronally sliced at thicknesses of 100 µm (n = 10 slices from four mice), 300 µm (n = 12 slices from two mice), 500 µm (n = 16 slices from four mice), 700 µm (n = 12 slices from two mice), or 900 µm (n = 10 slices from two mice), and were fixed with PFA. The light transmittance of the hippocampus after incubation with IsoScaleSQ at 37 °C for 30 min was 78.5 ± 1.8%, 72.8 ± 1.3%, 70.0 ± 1.1%, 42.4 ± 1.7%, and 41.1 ± 1.3% at 100, 300, 500, 700, and 900 µm, respectively (Fig. 3a, b). There was a sudden decrease in light transmittance between 500 and 700 µm. In the neocortex, the light transmittance also decreased steeply between 500 and 700 µm (Fig. 3c). The transmittance of the thalamus was 59.8 ± 7.2%, 40.0 ± 2.2%, 40.8 ± 2.5%, 15.1 ± 1.4%, and 13.2 ± 0.7% at 100, 300, 500, 700, and 900 µm, respectively; these values were consistently lower than for two other regions (Fig. 3d; P = 2.0 × 10−16, F2, 173 = 298.6 vs. hippocampus, P = 3.6 × 10−15, F2, 173 = 298.6 vs. cortex, two-way ANOVA with the Bonferroni correction). In general, the white matter of the tissue was less well cleared using IsoScaleSQ. For example, the light transmittance was 73.9 ± 1.2% at the lobes and 35.9 ± 2.3% at the fissures of the cerebellum in IsoScaleSQ-treated 500-µm-thick slices (n = 12 slices from two mice). Thus, our IsoScaleSQ-based clearing protocol was applicable to the gray matter of up to 500-µm-thick slices.
Staining of the cell nuclei helps to identify the location of a given neuron in a brain region. Among the dyes used for nuclear staining, Hoechst 33342 and acridine orange are known to penetrate well into fixed tissues. Thus, we quantified the penetration of these dyes in 30 min. The neocortex was coronally sliced at thicknesses of 500 µm and 1000 µm and fixed with 10% PFA at 4 °C for 30 min. During fixation, 0.1% Hoechst 33342 and 1% acridine orange were added to the PFA solution. To evaluate the penetration of these dyes, we perpendicularly sectioned the slices and confocally imaged the cross-sections (Fig. 4a). Both dyes stained the nuclei near-uniformly across the Z-axis of 500-µm-thick slices (Fig. 4b). In 1000-µm-thick slices, acridine orange provided uniform tissue staining, but Hoechst 33342 did not strongly stain deep parts of the tissues (Fig. 4c). These results suggest again that the brain should be sliced at thicknesses 500 µm or thinner for IsoScaleSQ clearing.

Nucleus labeling of thick slices using acridine orange and Hoechst 33342. a Schematic drawing of optical Z-sectioning (indicated by the hatched area) in a slice preparation. b Confocal Z-sections of a 500-µm-thick slice treated with Hoechst 33342 and acridine orange for 30 min. In the inset graphs, their normalized fluorescence intensities are plotted as a function of depth. c The same as b, but for a 1000-µm-thick slice. Acridine orange but not Hoechst 33342 permeated the slice. The same experiments were repeated in ten slices from each of two mice (500 µm) and two slices from one mouse (1000 µm), producing similar results. Scale bars represent 100 µm
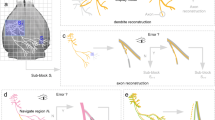
The cell reconstruction protocol conventionally involves overnight fixation and overnight histology (Fig. 5a, b). As a result, the entire procedure usually takes more than 24 h. Based on the optimizations described above, we aimed to establish a simple and quick reconstruction of in vitro patch-clamped neurons using IsoScaleSQ. After filling neurons with biocytin during recordings of > 15 min, we fixed the slices with 10% PFA at 4 °C for 30 min. This process was conducted simultaneously with Hoechst 33342 staining. The fixed tissues were briefly rinsed with PBS three times and were immersed in the IsoScaleSQ solution containing Alexa Fluor 594-conjugated streptavidin at 37 °C for 30 min (Fig. 5a, c). Fluorescence microscopic inspection revealed that the soma location was identified against Hoechst 33342 staining and that the soma and dendrites were finely reconstructed. To quantify the possible distortions, we calculated the tortuosity d/D of the dendrites of the reconstructed neurons, where D represents the straight-line distance between two ends of each dendrite segment and d represents the path length between them (Ke et al. 2013). The tortuosity did not statistically differ between two groups, suggesting no apparent distortions in subcellular morphology (Fig. 5d; the conventional method: 1.049 ± 0.003, n = 180 from two neurons; IsoScaleSQ method: 1.049 ± 0.003, n = 286 from five neurons. P = 0.87, t464 = 0.17, Student’s t test). These results indicate that this protocol is applicable for the post hoc morphological classification of recorded cells, despite its experimental ease. We also applied this protocol for the reconstruction of in vivo patch-clamped cells. In a conventional protocol, one must prepare thin sections using vibratomes, but in this study we cut unfixed whole-brain tissues into 500-µm-thick coronal slices using brain matrices and then visualized the cell using the same procedure described above, except for treatment with IsoScaleSQ and streptavidin for 1 h. Instead of 30 min of incubation, we selected 1 h of incubation so that streptavidin could penetrate deep inside the tissue; note that in vivo patch-clamped cells may be located in deeper parts of the tissues. As a result, in vivo patch-clamped cells were successfully visualized within a total procedure time of 1.5 h (n = 5, data not shown). The 1-h incubation with IsoScaleSQ caused slight swelling (114.3 ± 2.9%). Alternatively, users may employ 300 mM NaCl, leading to little swelling at 1 h of incubation (101.6 ± 4.0%, n = 9 slices from two mice).

Quick and simple reconstruction using IsoScaleSQ. a Experimental schedules for the post hoc visualization of the recorded cells. b, c In vitro whole-cell recorded cells are reconstructed in 500-µm-thick slices using the conventional method (b) and the IsoScaleSQ-based method (c). The cell nuclei are counterstained using Hoechst 33342. The same experiments were repeated in a total of two slices (conventional) and five slices (IsoScaleSQ) each, producing similar results. Scale bars are 100 µm. d Tortuosity of the dendrites of reconstructed neurons in each group. n = 180 from 2 neurons (conventional), 286 from 5 neurons (IsoScaleSQ), Student’s t test
ScaleSQ preserves the fluorescent proteins (Hama et al. 2015). To examine whether IsoScaleSQ quenches fluorescent proteins, we cleared brain slices prepared from Arc-dVenus transgenic mice in which the fluorescent protein dVenus is expressed in a neuronal-activity-dependent manner (Eguchi and Yamaguchi 2009). These mice were transferred from their home cage to an environmentally enriched space and were allowed to explore it for 5 h. The brains were then removed and cleared using the same protocol as used for in vivo whole-cell recorded neurons. IsoScaleSQ was unlikely to strongly quench fluorescent protein. We observed that dVenus was more abundantly expressed in layers 2/3 and 5 of the primary visual cortex compared to slices prepared from the home-cage control group (Fig. 6).

Fluorescence detection of green fluorescent proteins in IsoScaleSQ-treated tissues. a A representative confocal image of the V1 cortex in a slice prepared from an Arc-dVenus transgenic mouse housed in its home cage. The cell nuclei are counterstained using Hoechst 33342. Scale bar represents 200 µm. b The same as a, but for a slice prepared from an Arc-dVenus transgenic mouse that was allowed to explore an enriched environment for 5 h. The boxed region is magnified in the left inset, in which dVenus-positive cells are observed. The same experiments were repeated in a total of two mice (enriched environment) and one mouse (control) each, producing similar results. Scale bars represent 200 µm and 50 µm
Discussion
In this study, we modified ScaleSQ—a rapid clearing protocol—to reduce tissue swelling, and optimized the histological reconstruction of cells after whole-cell patch-clamp recording. We found that the addition of 200 mM NaCl to the ScaleSQ solution suppresses tissue swelling without compromising the clearing efficiency (at least in the gray matter of the brain) or inducing any apparent distortion of subcellular neuronal structures. Refractive index matching was dispensable for IsoScaleSQ to clear 500-µm-thick slices. As a result, IsoScaleSQ shortens the total time required for reconstruction down to 1 h for in vitro patch-clamped neurons and 1.5 h for in vivo patch-clamped neurons. Thus, this protocol is routinely applicable to the characterization of recorded neurons within the day of recording. Furthermore, we demonstrated that IsoScaleSQ does not quench fluorescent proteins.
The successful visualization achieved using IsoScaleSQ seems to be due, at least in part, to the membrane-preserving characteristics of ScaleSQ. It is necessary to ensure that biocytin remains inside the whole-cell recorded neurons. In fact, when we tried to visualize whole cell patch-clamped cells using the CUBIC1A protocol (Susaki and Ueda 2016), a protocol that does not preserve the cell membrane, we failed to visualize the cells in most cases. We occasionally observed some neurons under extreme conditions with a strong laser power and a long exposure time; however, even in those cases, we were not able to capture the fine morphological structures of the cells (data not shown). Stronger fixation with glutaraldehyde might suppress the possible leakage of biocytin, but the strong fixation usually slows the clearing step.
Swelling and shrinkage of tissues are frequent problems with clearing procedures (Ke et al. 2013; Chen et al. 2015; Hama et al. 2015; Ku et al. 2016; Pan et al. 2016; Kubota et al. 2017). These tissue volume changes are produced by multiple factors; in the cases of ScaleSQ and IsoScaleSQ, we speculate that tissue swelling depends on protein crosslinking (Ku et al. 2016) and osmotic pressure caused by ions that are immobilized within a tissue (Ricka and Tanaka 1984).
In general, the degree of swelling depends on the types of crosslinkers present within the tissue. For example, tissues that are strongly crosslinked by formaldehyde and acrylamide-bisacrylamide hydrogel are subject to only moderate swelling after SDS-based delipidation (Chung et al. 2013). Indeed, the omission of bisacrylamide from the hydrogel results in severe swelling (Yang et al. 2014). Thus, the degree of crosslinking is a major factor that influence on the tissue volume. It is also known that higher concentrations of acrylamide during crosslinking are associated with larger volumes of delipidated tissues, probably because acrylamide prevents protein crosslinking by quenching reactive methylols formed through the protein–formaldehyde reaction (Ku et al. 2016). This fact suggests that both the presence of crosslinker proteins and their structure affect the tissue volume. The swelling induced by ScaleSQ may be explained in a similar way. Urea, which accounts for 25% of the solution, is a strong denaturant and may induce tissue swelling by unfolding the structure of the crosslinkers.
Sodium polyacrylate swells when it is immersed in deionized water because the immobilized, dissociable carboxylic groups in the polymer increase the osmotic pressure (Ricka and Tanaka 1984). However, when sodium polyacrylate is immersed in solutions containing high concentrations of cations, the swelling is reduced because the immobilized carboxylic groups do not dissociate. In chemical terms, fixed biological samples are regarded as ionic hydrogels, such as sodium polyacrylate. Therefore, we consider that the suppression of tissue swelling by NaCl is mediated by its high concentration of sodium ions, which act as counterions against the carboxylates of amino acids.
The exact reason for the slight acceleration of clearing by IsoScaleSQ is unclear. It may involve the suppression of background light scattering and permeabilization of tissues, both of which are due to the high concentration of Triton X-100 and urea in the IsoScaleSQ solution. Within 30 min, IsoScaleSQ optically cleared the gray matter of 500-µm-thick brain slices. Therefore, our method is applicable not only to hippocampal neurons but also to cortical and cerebellar neurons. However, the clearing of white matter was insufficient for the morphological classification of neurons. Hence, we still recommend the conventional method for observing biocytin-reconstructed cells in the white matter.
Remarkably, recent developments in optical clearing techniques may permit near-complete transparentization of fixed tissues—a major goal of these methods. However, in regard to applying these techniques to electrophysiological experiments in the field of neuroscience, the preferred goal is not necessarily finished tissue transparency but rather procedural convenience and rapidity of the clearing procedure. Future aims for the development of clearing techniques should include more rapid clearing that transparentizes the white matter or thicker samples without changing the tissue size at the macroscopic or microscopic scale.
References
Chen F, Tillberg PW, Boyden ES (2015) Optical imaging. Expansion microscopy. Science 347:543–548
Chung K, Wallace J, Kim SY et al (2013) Structural and molecular interrogation of intact biological systems. Nature 497:332–337
Eguchi M, Yamaguchi S (2009) In vivo and in vitro visualization of gene expression dynamics over extensive areas of the brain. Neuroimage 44:1274–1283
Funayama K, Minamisawa G, Matsumoto N et al (2015) Neocortical rebound depolarization enhances visual perception. PLoS Biol 13:e1002231
Graves AR, Moore SJ, Bloss EB, Mensh BD, Kath WL, Spruston N (2012) Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron 76:776–789
Hama H, Hioki H, Namiki K et al (2015) ScaleS: an optical clearing palette for biological imaging. Nat Neurosci 18:1518–1529
Horikawa K, Armstrong WE (1988) A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J Neurosci Methods 25:1–11
Ishikawa D, Matsumoto N, Sakaguchi T, Matsuki N, Ikegaya Y (2014) Operant conditioning of synaptic and spiking activity patterns in single hippocampal neurons. J Neurosci 34:5044–5053
Jiang X, Shen S, Cadwell CR et al (2015) Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350:aac9462
Ke MT, Fujimoto S, Imai T (2013) SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci 16:1154–1161
Klausberger T, Somogyi P (2008) Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57
Ku T, Swaney J, Park JY et al (2016) Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat Biotechnol 34:973–981
Kubota SI, Takahashi K, Nishida J et al (2017) Whole-body profiling of cancer metastasis with single-cell resolution. Cell Rep 20:236–250
Kuga N, Sasaki T, Takahara Y, Matsuki N, Ikegaya Y (2011) Large-scale calcium waves traveling through astrocytic networks in vivo. J Neurosci 31:2607–2614
Larkman A, Mason A (1990) Correlations between morphology and electrophysiology of pyramidal neurons in slices of rat visual cortex. I. Establishment of cell classes. J Neurosci 10:1407–1414
Margrie TW, Brecht M, Sakmann B (2002) In vivo, low-resistance, whole-cell recordings from neurons in the anaesthetized and awake mammalian brain. Pflugers Arch 444:491–498
Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C (2004) Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5:793–807
Mizuseki K, Diba K, Pastalkova E, Buzsaki G (2011) Hippocampal CA1 pyramidal cells form functionally distinct sublayers. Nat Neurosci 14:1174–1181
Neher E (1992) Ion channels for communication between and within cells. Science 256:498–502
Pan C, Cai R, Quacquarelli FP et al (2016) Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nat Methods 13:859–867
Ricka J, Tanaka T (1984) Swelling of ionic gels: quantitative performance of the Donnan theory. Macromolecules 17:2916–2921
Sakaguchi T, Ishikawa D, Nomura H, Matsuki N, Ikegaya Y (2012) Normal learning ability of mice with a surgically exposed hippocampus. Neuroreport 23:457–461
Susaki EA, Ueda HR (2016) Whole-body and whole-organ clearing and imaging techniques with single-cell resolution: toward organism-level systems biology in mammals. Cell Chem Biol 23:137–157
Yang B, Treweek JB, Kulkarni RP et al (2014) Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell 158:945–958
Acknowledgements
This work was supported by JST ERATO (JPMJER1801), JSPS Grants-in-Aid for Scientific Research (18H05525), and the Human Frontier Science Program (RGP0019/2016). This work was conducted partly as a program at the International Research Center for Neurointelligence (WPI-IRCN) of The University of Tokyo Institutes for Advanced Study at The University of Tokyo.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sato, Y., Miyawaki, T., Ouchi, A. et al. Quick visualization of neurons in brain tissues using an optical clearing technique. Anat Sci Int 94, 199–208 (2019). https://doi.org/10.1007/s12565-018-00473-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12565-018-00473-z