
Abstract
Obscurin and its homolog, striated muscle preferentially expressed gene (SPEG), constitute a unique group of proteins abundantly expressed in striated muscles that contain two tandemly arranged MLCK-like kinases. The physiological significance of the dual kinase motifs is largely understudied; however, a collection of recent studies characterizing their binding interactions, putative targets, and disease-linked mutations have begun to shed light on their potential roles in muscle pathophysiology. Specifically, obscurin kinase 1 is proposed to regulate cardiomyocyte adhesion via phosphorylating N-cadherin, whereas SPEG kinases 1 and 2 regulate Ca2+ cycling by phosphorylating junctophilin-2 and the sarcoendoplasmic Ca2+ ATPase 2 (SERCA2). Herein, we review what is currently known regarding the potential substrates, physiological roles, and disease associations of obscurin and SPEG tandem kinase domains and provide future directions that have yet to be investigated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myosin light chain kinases (MLCKs) comprise a multifaceted family of Ca2+/calmodulin-dependent serine/threonine (Ser/Thr) kinases that play essential roles in a variety of cell types (Khapchaev and Shirinsky 2016). Ubiquitously expressed MLCKs participate in a number of important cellular processes such as cell adhesion and motility, proliferation, endo/exocytosis, and cytoskeletal stiffness by phosphorylating the regulatory light chain of myosin (Khapchaev and Shirinsky 2016). In a striated and smooth muscle, MLCKs primarily serve to regulate muscle contractility and the actomyosin cytoskeleton in response to cytosolic changes in Ca2+ (Kamm and Stull 2001).
Additional protein families that are expressed abundantly in muscle, such as titins, obscurins, and striated muscle preferentially expressed gene (SPEG), contain kinase domains that share significant homology to canonical MLCKs (Sutter et al. 2004; Gautel 2011; Russell et al. 2002). Obscurins, along with their invertebrate ortholog UNC-89 and vertebrate paralog SPEG, represent a unique subfamily of MLCK-like protein kinases that contain two tandem Ser/Thr kinase motifs (Sutter et al. 2004; Russell et al. 2002). As giant modular proteins that interact with a diverse set of proteins, obscurins, UNC-89, and SPEG serve established roles in myofibril assembly, the integration of the sarcomere with the surrounding subcellular and membrane structures, and Ca2+ cycling (Wang et al. 2018b; Quan et al. 2019). Importantly though, the regulation, enzymatic activities, and substrate specificities of their dual kinase domains have remained highly enigmatic.
Herein, we present an overview of what is known regarding the phylogenetic association, catalytic regulation, potential substrates and/or binding partners, and disease implications of the tandem kinase domains of obscurin/UNC-89 and SPEG in the heart. Given that these proteins are preferentially expressed in striated muscles, it has been postulated that the presence of the dual kinase motifs could confer a specialized purpose within muscle pathophysiology (Sutter et al. 2004).
Obscurin
Domain architecture and cellular distribution
The OBSCN gene, spanning more than 170 kb on human chromosome 1q42.13, encodes 117 exons that are alternatively spliced to give rise to the multitude of isoforms comprising the obscurin protein family, ranging in size between 50 and 870 kDa (Russell et al. 2002; Young et al. 2001; Fukuzawa et al. 2005; Ackermann et al. 2014; Bang et al. 2001). The prototypical obscurin isoform, obscurin A (~ 720 kDa), contains 65 immunoglobulin (Ig) and 2 fibronectin-III (FN-III) domains along with an array of COOH-terminal signaling domains, including a calmodulin-binding IQ motif, a Src homology-3 (SH3) domain, a Rho guanine nucleotide exchange factor (RhoGEF) domain, a pleckstrin homology (PH) domain, and a non-modular COOH-terminus that contains binding sites for ankyrins (Fukuzawa et al. 2005; Kontrogianni-Konstantopoulos et al. 2009). The largest obscurin isoform, obscurin B (~ 870 kDa), bears the same modular architecture as obscurin A but diverges COOH-terminal to Ig67, where it instead contains two Ser/Thr kinase domains referred to as kinase 1 and kinase 2 (Kontrogianni-Konstantopoulos et al. 2009; Fukuzawa et al. 2005) (Fig. 1). Kinase 1 is preceded by an additional Ig domain whereas kinase 2 is preceded by both an Ig and a FN-III domain, which is consistent with the domain arrangement of the tandem kinases present in the invertebrate obscurin ortholog UNC-89 and its closely related paralog SPEG (Fukuzawa et al. 2005; Sutter et al. 2004) (Fig. 1). Given this highly conserved organization and sequence similarity, it has been postulated that there could be significant overlap in the protein interactions, substrate specificity, targeting and functions of these three tandem kinase proteins (Sutter et al. 2004).

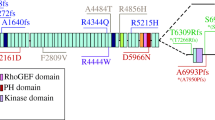
Schematic representation of the modular domain architecture of giant obscurin B, obscurin double kinase, obscurin single kinase, UNC-89-B, SPEGα, and SPEGβ. The currently known pathogenic mutations localized in the vicinity of the kinase domains are also shown; mutations present in obscurin are denoted according to accession numbers NP_001092093 (underlined) and NP_001258152 (in parenthesis), while mutations in SPEG are denoted according to accession numbers NM_005876.4 (marked with +) or NM_005876 (marked with #). Mutations are depicted in different colors to indicated their association with different forms of cardiomyopathy, as follows: LVNC-associated mutations in green; DCM-associated mutations in purple; HCM-associated mutations in orange; and systolic heart failure with hereditary ataxia-associated mutations in blue; while mutations not associated with cardiomyopathy are shown in black
OBSCN also gives rise to two small obscurin kinase isoforms, termed double (~ 145 kDa) and single (~ 55 kDa) kinases (Russell et al. 2002; Borisov et al. 2008; Hu and Kontrogianni-Konstantopoulos 2013). The double kinase isoform contains partial kinase 1 and the entirety of kinase 2, whereas the single kinase isoform contains only kinase 2 and the Ig/FN-III module that precedes it (Russell et al. 2002; Borisov et al. 2008; Hu and Kontrogianni-Konstantopoulos 2013) (Fig. 1). Northern blotting and PCR experiments have indicated that the transcripts of both the double and single kinase obscurins are abundantly present in the heart; however, these variants have remained largely uncharacterized (Russell et al. 2002).
Initial studies demonstrated that giant obscurins localize to both the M-bands and Z-disks of sarcomeres, with predominant localization to the M-bands (Bang et al. 2001; Young et al. 2001; Kontrogianni-Konstantopoulos et al. 2003). Unlike other giant sarcomeric proteins which are integrated longitudinally within the myofibrillar cytoskeleton, giant obscurins wrap around the periphery of myofibrils where they link proteins of the sarcomere with the surrounding subcellular and membrane structures (Kontrogianni-Konstantopoulos et al. 2003; Wang et al. 2018b; Kontrogianni-Konstantopoulos et al. 2009). Previous work from our group has delineated the precise distribution of kinase-bearing obscurin isoforms in the heart utilizing antibodies targeting different epitopes within kinase 2 or its preceding Ig/FN-III domains (Hu and Kontrogianni-Konstantopoulos 2013). Specifically, immunostaining of mouse myocardial sections and adult rat cardiomyocytes indicated that obscurin kinases localize at M-bands and Z-disks as well as the sarcolemma, the intercalated disc, and the nucleus (Hu and Kontrogianni-Konstantopoulos 2013). Whether the different obscurin kinase isoforms preferentially distribute to select subcellular compartments is not yet fully known due to the lack of unique epitopes among them; however, a ~ 70 kDa isoform proposed to correspond to the single kinase was shown to localize extracellularly where it possibly undergoes glycosylation and interacts with the β1-subunit of the N+/K+ ATPase (NKAβ1) (Hu and Kontrogianni-Konstantopoulos 2013).
Obscurin kinase domains phylogeny
Obscurin kinases 1 and 2 are members of the MLCK family (Russell et al. 2002; Sutter et al. 2004) (Fig. 2). Detailed phylogenetic analysis has established their close relation to UNC-89 and SPEG, which also contain dual MLCK-like kinase activity (Sutter et al. 2004) (Fig. 3), as well as other proteins which contain single MLCK-like domains, such as titin and its invertebrate orthologs and the death-associated protein kinases (DAPK) trio and duet (Russell et al. 2002; Sutter et al. 2004; Gautel 2011). Canonically, MLCK-like kinases are regulated by Ca2+/calmodulin (CaM) where binding of CaM to the autoinhibitory region of the regulatory domain confers activation (Kamm and Stull 2001; Stull et al. 1998). It has not yet been experimentally determined whether obscurin kinases 1 and 2 are regulated via CaM; however, the presence of a putative CaM-binding regulatory motif immediately downstream of the catalytic portion of kinase 1 highly supports this possibility (Fukuzawa et al. 2005; Hu and Kontrogianni-Konstantopoulos 2013). Interestingly though, CaM-dependent activation is not an entirely conserved feature among all MLCK-like family members. Titin kinase, for instance, sharing ~ 28 and ~ 27% homology to kinases 1 and 2 respectively (Fukuzawa et al. 2005), undergoes an alternative activation mechanism mediated by mechanical unfolding of the autoinhibitory region in response to stretch and subsequent phosphorylation of an inhibitory tyrosine residue (Tyr170) which exposes the catalytic aspartate (Gautel 2011; Grater et al. 2005; Mayans et al. 1998; Puchner et al. 2008). Intriguingly, both titin kinase and obscurin kinase 2 share a unique arginine residue in proximity to the catalytic aspartate (Fig. 2) that participates in the autoinhibitory mechanism mediated by Tyr170 in titin kinase (Fukuzawa et al. 2005). Therefore, it is possible that obscurin kinases 1 and 2 are regulated distinctly, and thus possess different physiological functions. In support of this notion, obscurin kinases 1 and 2 are only 30% identical to each other (Fukuzawa et al. 2005) (Fig. 2).

Sequence alignment of the kinase domains of human cardiac MLCK (UniProt Q32MK0; aa 515-770), human obscurin kinase 1 (UniProt Q5VST9; aa 6468-6721) and kinase 2 (UniProt Q5VST9; aa 7672-7924), C. elegans UNC-89 PK1 (UniProt O01761; aa 6592-6878) and PK2 (O01761, aa 7285-8035), Drosophila Unc89 PK1 (UniProt A8DYP0; aa 3186-3440) and PK2 (UniProt A8DYP0; aa 3897-4151), and human SPEGα/β kinase 1 (Uniprot Q15772; aa 1601-1854) and kinase 2 (Uniprot Q15772; aa 2966-3218). Domains were aligned using the CLUSTALW software (http://www.ebi.ac.uk/clustalw/). Residues identical in all kinases are denoted with an asterisk (*), residues conserved in closely related kinases with similar properties are marked with colon (:), and residues shared by kinases with weakly similar properties are indicated with a dot (.), while gaps in residue alignment are represented by dashes (-). The highly conserved lysine (K) residue within the ATP-binding site is denoted with an arrow, while the catalytic aspartate (D) is marked with an arrowhead; the arginine (R) at position + 2 from the catalytic aspartate (D) present in obscurin kinase 2 (similar to titin-MLCK) is underlined

Phylogenetic tree showing the evolutionary relationships between the kinase domains of human cardiac MLCK, human obscurin B, human SPEGα/β, C. elegans UNC-89, and D. melanogaster Unc-89. The phylogenetic tree was produced using the neighbor-joining algorithm. The evolutionary distances were computed using the Poisson correction method. Bootstrap values are shown on the branches, based on 2000 bootstrap replications. The calculated distance values are displayed in parenthesis. The optimal tree amino acids were aligned using MEGA X software, as in (Kumar et al. 2018). Note that the human obscurin kinases 1 and 2 domains are most closely related to the human SPEGα/β kinases 1 and 2 domains, respectively, consistent with the notion that SPEG arose from OBSCN via a gene duplication event
Putative substrates of obscurin kinases
Recent findings from our group have demonstrated that both obscurin kinases 1 and 2 are enzymatically active and undergo autophosphorylation in vitro (Hu and Kontrogianni-Konstantopoulos 2013). Yeast two-hybrid screening experiments utilizing kinases 1 or 2 as bait identified several putative binding partners and/or substrates from an adult human cardiac muscle cDNA library. In particular, N-cadherin, galectin, the immunoglobulin superfamily with leucine repeats protein, and titin were identified as potential ligands of kinase 1, whereas NKAβ1, galectin, fibronectin-1, γ-filamin, and four and a half LIM domain-2 protein (FHL2) were identified as potential ligands of kinase 2 (Hu and Kontrogianni-Konstantopoulos 2013). With the exception of N-cadherin and NKAβ1 (described in more detail below), these potential binding interactions have not been further confirmed or characterized. Given the identification of major components of the cytoskeleton (i.e., titin, γ-filamin, and FHL2) or extracellular matrix (i.e., fibronectin-1 and galectin) as potential ligands and/or substrates, it is tempting to speculate that the obscurin kinases may be essential in regulating cytoskeletal stability and/or remodeling, mechanotransduction, or cell-cell or cell-extracellular matrix communication.
Deletion analysis indicated that both intracellular and extracellular portions of N-cadherin are required for binding to the catalytic region of kinase 1 (Hu and Kontrogianni-Konstantopoulos 2013). Moreover, proximity ligation assay (PLA) and immunofluorescent microscopy confirmed that kinase-bearing obscurins and N-cadherin localize in proximity at the intercalated disc and the sarcolemma. In line with their direct binding and co-localization, these studies further demonstrated that kinase 1 can specifically phosphorylate the cytoplasmic domain of N-cadherin in vitro. Importantly, N-cadherin is the only confirmed substrate of an obscurin kinase to date. Although the exact phosphorylation site and physiological impact of this modification has yet to be elucidated, this implicates obscurin as a mediator of cardiomyocyte adhesion via the kinase 1 activity.
The binding interaction between obscurin kinase 2 and NKAβ1 generated novel insights on the possible roles of obscurin kinases in the heart, since the minimal interacting region that is necessary and sufficient to support binding to the catalytic subunit of kinase 2 resides within the extracellular domain of NKAβ1 (Hu and Kontrogianni-Konstantopoulos 2013). Prior to these findings, obscurins had not been shown to localize extracellularly. Experiments utilizing nonpermeabilized cardiomyocytes identified a small (~ 70 kDa) obscurin kinase variant proposed to encode the single obscurin kinase isoform that localizes extracellularly where it possibly undergoes glycosylation (Hu and Kontrogianni-Konstantopoulos 2013). Consistent with a possible interaction between obscurin kinase 2 and NKAβ1, obscurin kinase-bearing proteins and NKAβ1 localize in proximity at the sarcolemma and the intercalated disc in cardiomyocytes. However, unlike the phosphorylation event mediated by kinase 1 on N-cadherin, in vitro kinase assays could not confirm NKAβ1 as a substrate of kinase 2 (Hu and Kontrogianni-Konstantopoulos 2013). It remains to be clarified whether this lack of phosphorylation on NKAβ1 is due to limitations of the in vitro expression system used, or if NKAβ1 is not actually a substrate (but possibly a binding partner) of obscurin kinase 2.
Disease implications of obscurin kinases
To date, approximately ten missense, one splicing, and seven frameshift mutations have been identified in OBSCN that are linked to various forms of cardiomyopathy in humans, including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and left ventricular non-compaction (LVNC) (Grogan and Kontrogianni-Konstantopoulos 2019). Importantly, six of these mutations localize at the COOH-terminus of obscurin B where they reside within close proximity to kinases 1 and/or 2 (Fig. 1). Their close association to one or both kinases raises the possibility for pathogenic mechanisms related to the altered targeting, regulation, or enzymatic activity of obscurin kinases.
In particular, whole exome sequencing analysis performed on a cohort of 74 Chinese patients in 2015 discovered six novel OBSCN variants linked to HCM (Xu et al. 2015). Of these six variants, one missense mutation, G7500R, was identified within Ig69 (Fig. 1). Given that Ig69 is a component of the Ig/FN-III module immediately preceding kinase 2, the presence of G7500R could potentially impact the targeting and/or substrate specificity of the dual kinase domains. In the following year, Rowland and colleagues identified four frameshift mutations in OBSCN from a cohort of 10 LVNC and 325 DCM patients that all localize to the COOH-terminal region of obscurin B (Rowland et al. 2016) (Fig. 1). Although these mutations remain uncharacterized, their positioning immediately upstream of one or both kinases could lead to the loss of kinases 1 and/or 2 expressions due to frameshifts. These findings were particularly intriguing since three of these mutations (T7266Rfs*53; S7947Pfs*82; 25367-1 G>C) were associated with LVNC while the fourth mutation (A7950Pfs*79) was identified in a patient with DCM. Given that LVNC is a relatively rare form of cardiomyopathy and only represented ~ 3% of the tested cohort, OBSCN mutations that cluster around dual kinase domains could be more prevalent in the development of LVNC compared to HCM or DCM. Lastly, a missense mutation W7910R localized to kinase 2 was recently identified in a patient with chronic systolic heart failure and hereditary ataxia (Catalano et al. 2018) (Fig. 1). This represents the only known mutation residing directly within an obscurin kinase domain, potentially affecting the binding affinity to its substrates and/or enzymatic activity. Given that biochemical and functional analyses of these disease-linked OBSCN mutations residing in the vicinity of the dual kinase domains are considerably lacking at this time, their pathophysiological impact and molecular manifestations remain speculative.
In addition to the known mutations, previous studies have also reported upregulation of obscurin kinase transcripts and/or protein in various animal models of cardiac disease. In particular, Borisov and colleagues reported up to an 18-fold increase in transcripts encoding kinase 1 approximately 48 h after the induction of myocardial hypertrophy via aortic constriction in mice (Borisov et al. 2003). Consistent with this, a mouse model carrying the HCM-linked R4344Q mutation in Ig58 exhibited an increase in the expression of giant obscurin B in response to chronic pressure overload (Hu et al. 2017). Lastly, recent work from our lab utilizing a constitutive deletion mouse model (Obscn-ΔIg58/59) expressing obscurin lacking the Ig58/59 module exhibited an upregulation of obscurin B prior to, and during, progressive ventricular remodeling from HCM to DCM through aging (Grogan et al. 2019). These studies collectively signify that the upregulation of obscurin kinases could play a role in the hypertrophic response, provide a form of molecular compensation during stress, or serve as a marker of early manifestation of cardiac disease. Therefore, elucidating the precise roles and signaling pathways regulated by obscurin kinases 1 and 2 in the heart is essential.
Invertebrate obscurin homolog UNC-89
As there is still a lot to learn regarding the substrates, signaling events, and physiological functions of mammalian obscurin kinases 1 and 2, considering the regulation, established binding partners and purported roles of obscurin’s closely related invertebrate ortholog, UNC-89, has been proven highly informative. Similar to obscurin, UNC-89 is a giant modular protein composed of tandem Ig and FN-III domains along with an NH2-terminal signaling region consisting of an SH3 domain, a RhoGEF domain, a PH domain, and KSP motif (Benian et al. 1996). The largest isoforms, UNC-89-B and -F (750-900 kDa), contain two COOH-terminal Ser/Thr kinase domains termed PK1 and PK2 that are preceded by an Ig domain or Ig/FN-III module, respectively (Small et al. 2004; Ferrara et al. 2005) (Fig. 1). The unc-89 gene also gives rise to two smaller kinase isoforms, UNC-89-C and -D (~ 156 kDa), that both encompass partial PK1 through PK2, but differ in their NH2-termini where they contain a segment of 8 (UNC-89-C) or 11 (UNC-89-D) unique amino acids (Small et al. 2004).
The dual kinases of UNC-89 are also members of the MLCK protein family (Small et al. 2004) (Fig. 2). Based on homology modeling to other known kinases and members of the DAPK subfamily, PK2 is predicted to be catalytically active, whereas PK1 is considered to be enzymatically inactive due to the lack of several critical amino acids within the active site that are important for ATP or Mg2+ binding (Small et al. 2004). The potential activities of PK1 or 2 have not been experimentally proven yet, and there are no currently known substrates for either kinase, although Gieseler and colleagues mentioned (as unpublished data) that PK2 can phosphorylate several model peptide substrates, indicating that it is enzymatically active (Gieseler et al. 2017). Nevertheless, the identification of several proteins that bind to the kinase domains has provided insight on their potential cellular functions. In particular, a series of studies demonstrated that small C-terminal domain phosphatase-like 1 (SCPL-1), a novel protein phosphatase, and LIM-9, the nematode ortholog of FHL2, form a ternary complex with UNC-89 in C. elegans, where either one or both of the tandem kinases bind to SCPL-1 or LIM-9 directly (Xiong et al. 2009; Qadota et al. 2008). It was proposed that this complex could facilitate UNC-89 dimerization, or serve to link the M-band to the extracellular matrix via LIM-9’s indirect association with integrins (Xiong et al. 2009). Given that FHL2 was also identified as a putative binding partner of obscurin kinase 2 (Hu and Kontrogianni-Konstantopoulos 2013), it is tempting to speculate that these complexes could also exist in vertebrates. Lastly, PK1 binds Ball, an active Ser/Thr kinase, and both PK1 and PK2 support binding to the multiple ankyrin repeats single KH domain protein (MASK) in Drosophila (Katzemich et al. 2015). Experiments utilizing siRNA-mediated knockdown of Ball or MASK in indirect flight muscles (IFM) resulted in major sarcomeric disorganization, suggesting a potential role for these proteins in sarcomeric stability. Given that UNC-89 was properly localized to the M-bands in Ball and MASK knockdown IFMs, whereas the reciprocal knockdown of UNC-89 resulted in the improper localization of both Ball and Mask, it was proposed that UNC-89 kinases serve as signaling scaffolds for proteins involved in the assembly and maintenance of the sarcomere (Katzemich et al. 2015).
Importantly, the distance between the two kinase domains is highly conserved between vertebrate obscurin and nematode UNC-89 (Sutter et al. 2004). The interkinase region, consisting of a ~ 700 amino acid segment that lacks secondary structure, was recently demonstrated via single molecular force spectroscopy to exhibit properties of a random coil similar to the elastic spring elements found in titin (Qadota et al. 2020). Given that deletion of this region led to disorganized A-bands in C. elegans, it was proposed to serve important roles in sarcomeric assembly and stability of the thick filament (Qadota et al. 2020). It will therefore be important to also consider the potential roles of the interkinase region within vertebrate obscurins as well.
Striated Preferentially Expressed Gene
Domain architecture and cellular distribution
The SPEG gene encodes four distinct tissue-specific isoforms, APEG, BPEG, SPEGα and SPEGβ, which are generated by alternative splicing and two separate transcription initiation sites (Hsieh et al. 1999; Hsieh et al. 2000). APEG and BPEG are preferentially expressed in aortic/vascular smooth muscle and brain respectively, whereas SPEGα and β are specific to striated muscles where they are co-expressed (Hsieh et al. 2000; Hsieh et al. 1999). SPEGα (~ 250 kDa) and β (~ 355 kDa) represent the largest of the isoforms and are unique among the other members of their protein family in containing two Ser/Thr kinase domains at their COOH-terminus (Hsieh et al. 2000) (Fig. 1). In addition to the kinase domains, referred to herein as kinases 1 (internal kinase) and 2 (COOH-terminal kinase), are a total of 9 Ig domains and 2 FN-III domains that make up the SPEGβ isoform. SPEGα is identical to SPEGβ, but is transcribed by a separate intronic promoter and lacks the first two NH2-terminal Ig domains of SPEGβ (Hsieh et al. 2000; Agrawal et al. 2014). Importantly, the tandem kinases of SPEGα/β contain the same arrangement of preceding Ig and Ig/FN-III modules as described above for obscurin and UNC-89, alluding to their high homology and potentially conserved functions (Sutter et al. 2004).
Early studies identified SPEGα/β at the Z-disks of sarcomeres in both the atria and ventricles of immunostained mouse hearts (Hsieh et al. 2000). Similar to obscurins, they were also revealed to exhibit a reticular distribution surrounding myofibrils and co-localizing with proteins of the sarcoplasmic reticulum (SR) (Agrawal et al. 2014). Moreover, a series of recent studies identified SPEGα/β at the cardiac diads as well, where they interact with junctophilin-2 (JPH2), the ryanodine receptor 2 (RyR2) (Quick et al. 2017), and the sarcoendoplasmic reticulum Ca2+ ATPase 2 (SERCA2) (Quan et al. 2019), suggesting their involvement in Ca2+ cycling and excitation-contraction coupling.
SPEG kinase domains phylogeny
Given the conserved exon structure between obscurin and SPEG tandem kinases, and the lack of an invertebrate SPEG ortholog, it is generally believed that SPEG arose from OBSCN by a gene duplication event (Sutter et al. 2004) (Fig. 3). The most striking difference between SPEG and obscurin, however, is that the exon immediately preceding SPEG kinase 2 contains an additional ~ 500 nucleotides, leading to increased separation between SPEG kinase 2 and the preceding Ig/FN-III module (Sutter et al. 2004). Although the distance between the NH2-terminal kinase 1 and its preceding Ig domain remains conserved, this suggests potential divergence in the binding interaction profiles of obscurin and SPEG kinases (Sutter et al. 2004).
Similar to obscurin kinases 1 and 2, SPEG kinases also show significant homology to members of the MLCK family (Hsieh et al. 2000) (Fig. 2). In particular, SPEG kinase 1 shows ~ 37% sequence identity to other MLCK-like kinases, including smooth muscle MLCK, titin and its invertebrate orthologs, and DAPKs (Hsieh et al. 2000). It also contains 14 of the 15 canonically conserved amino acid residues present in most protein kinases, providing evidence that it is catalytically active (Hsieh et al. 2000). Accordingly, in vitro kinase assays demonstrated that SPEG kinase 1 is an active kinase that undergoes autophosphorylation (Hsieh et al. 2000). In contrast, SPEG kinase 2 only shares ~ 24% sequence identity with the other MLCK-like proteins due to divergence within its ATP-binding region (Hsieh et al. 2000) (Fig. 3). Hsieh and colleagues therefore initially predicted that SPEG kinase 2 would be catalytically inactive (Hsieh et al. 2000); though recent evidence that SERCA2a is a substrate of SPEG kinase 2 (described in detail below) has suggested otherwise (Quan et al. 2019).
Putative substrates of SPEG kinases
Unlike obscurin and UNC-89, both kinase domains of SPEG have confirmed substrates where the physiological functions of these phosphorylation events have been characterized in relative detail. In an attempt to identify novel proteins of the junctional membrane complex (JMC), Quick and colleagues performed immunoprecipitation experiments followed by proteomics analysis and identified SPEG as a binding partner of both RyR2 and JPH2 in the heart (Quick et al. 2017). Although the minimal interacting domains of these protein complexes were not determined, these findings suggested that RyR2 and/or JPH2 could potentially serve as substrates of SPEG kinases. Using an inducible, cardiac-specific SPEG knockout mouse model, which develops ventricular dilation and contractile dysfunction leading to premature death, it was further demonstrated that SPEG deficiency is associated with enhanced SR Ca2+ leak by RyR2, and severely disrupted t-tubules consequent of reduced phosphorylation of JPH2. Importantly, t-tubule disarray preceded the onset of heart failure in SPEG-deficient mice, suggesting that the loss of phosphorylation of JPH2 was a direct effect of the loss of SPEG and causal in the development of disease (Quick et al. 2017). The exact phosphorylation site has not been confirmed yet; however, additional studies validated JPH2 as a direct substrate of SPEG kinase 1 in vitro (Quan et al. 2019).
In contrast to JPH2, the effect of SPEG knockout on RyR2 function is less clear at this time since the canonical RyR2 phosphorylation sites (Ser2808, Ser2814) were unaffected in SPEG-deficient mice (Quick et al. 2017). Moreover, unlike the impact on t-tubule organization, the observed alterations in diastolic Ca2+ handling did not occur prior to the onset of disease following SPEG knockout. It therefore remains unknown whether RyR2 function is directly regulated by SPEG or if these pathologies develop as a downstream consequence of cardiomyopathy.
Recent studies also employing immunoprecipitation and proteomics techniques identified SERCA2a as a binding partner of SPEG kinase 2 (Quan et al. 2019). In vitro kinase assays demonstrated that kinase 2, but not kinase 1, is capable of phosphorylating SERCA2a specifically on Thr484. This novel SERCA2a phosphorylation site was shown to increase SERCA2a oligomerization in vitro, which was associated with enhanced Ca2+ reuptake into the SR. Utilizing the inducible, cardiac-specific SPEG knockout mouse, Quan and colleagues revealed that loss of SPEG resulted in decreased SERCA2a phosphorylation on Thr484, providing evidence for this regulation in vivo. In addition, reduced Thr484 phosphorylation in SPEG-deficient mice preceded the development of ventricular dilation and dysfunction, suggesting that the loss of SERCA2a phosphorylation by SPEG contributes to disease development (Quan et al. 2019).
Collectively, these studies demonstrate that SPEG kinases serve important roles in JMC integrity, Ca2+ cycling, and excitation-contraction coupling through phosphorylation of major Ca2+ handling proteins.
Disease implications of SPEG kinases
Early studies interrogating the precise roles of SPEG revealed the presence of a severe cardiac phenotype in SPEG-deficient mice manifested as significant dilation of the ventricles and atria accompanied by decreased contractility and cardiac function occurring in the developing heart by 18.5 days post-coitum and leading to prominent neonatal mortality (Liu et al. 2009). Additional studies found that SPEG mRNA levels are significantly reduced in mouse models of heart failure induced by isoproterenol or transaortic constriction (Quan et al. 2019). Lastly, an 83% reduction of SPEG transcripts were reported in human heart failure patients compared to non-failing hearts (Quick et al. 2017). Together, these findings implicate the loss of SPEG, and likely SPEG kinases, in the development of cardiomyopathy. Accordingly, a total of 13 mutations have been identified in SPEG to date that are all recessively inherited. Notably, seven of these mutations are found in patients with DCM or LVNC. Although only three of the currently known mutations localize directly within a kinase domain, most are predicted to cause significant truncations of SPEG where either one or both kinase domains are lost (Fig. 1).
Whole exome sequencing analysis on a cohort of 29 patients with centronuclear myopathy (CNM) identified three unrelated patients carrying mutations in SPEG, two of which also exhibited DCM (Agrawal et al. 2014). In particular, a total of five mutations that affected both SPEGα and β isoforms were found in either a homozygous or compound heterozygous state. The first patient carried a homozygous truncating mutation (Q2233*) that localized to the interkinase region. Given that this patient did not undergo a cardiac evaluation before dying at 3 weeks of age, it is not confirmed whether this mutation affected the heart. A second patient was compound heterozygous for a frameshift (T1237Sfs*46) and a nonsense (R1426*) mutation in Ig6 and Ig8 respectively. This patient developed a dilated left ventricle with severe cardiac dysfunction by 2 months of age. The third patient, carrying a frameshift mutation in Ig4 (A972Nfs*79) and a missense mutation in FN-III11 (G2757V), developed dilated cardiomyopathy by 1 month. Notably, skeletal muscle biopsies obtained from patients 2 and 3 demonstrated a significant reduction in both SPEGα and β, suggesting pathogenic mechanisms that are mediated by the loss of function of both isoforms (Agrawal et al. 2014).
Recently, two novel homozygous SPEG mutations were discovered in additional patients with CNM (Wang et al. 2017). The first patient, who did not develop cardiomyopathy, carried a frameshift mutation (T544Nfs*48) localized between Ig1 and Ig2 that introduced a premature stop codon. Importantly, this mutation is localized upstream of the SPEGα transcription initiation site and is predicted to only affect SPEGβ, suggesting that a functional SPEGα isoform can compensate for the loss of SPEGβ in the heart. The second CNM patient, who presented with ventricular dilation and reduced ejection fraction, carried a truncating mutation (R3196*) localized within kinase 2 that is predicted to cause partial loss of the kinase in both SPEGα and β. Similarly, another study by the same group reported the presence of a homozygous truncating mutation (Y2373*) that resulted in loss of the COOH-terminal Ig/FN-III and kinase 2 module in a CNM patient with LVNC (Wang et al. 2018a).
Lastly, two recent studies identified an additional five SPEG mutations that affect either one or both SPEG isoforms. Specifically, a truncating mutation localized between Ig1 and Ig2 (K359fs) and another localized to Ig8 (R1467*) were identified in a patient with congenital myopathy that did not show any apparent signs of cardiac disease (Lornage et al. 2018). Another patient presenting with CNM but no cardiac pathology carried two compound heterozygous truncating mutations localized to Ig2 (L728Rfs*82) and kinase 2 (V2997Gfs*52), respectively (Qualls et al. 2019). Given that SPEGα lacks Ig1 and Ig2, each of these patients only carries one (heterozygous) mutation affecting both isoforms, which further suggests that the presence of a single wild-type SPEGα allele can compensate for the presence of truncated SPEGβ in the heart. Consistent with this assertion, another CNM patient that was homozygous for a truncating mutation (V3062del) localized within Kin2 and present in both SPEGα and β, developed DCM (Qualls et al. 2019).
Taken together, these findings suggest that mutations truncating both SPEG isoforms and therefore resulting in the loss of kinases 1 and 2 are associated with the development of DCM and LVNC.
What we know and future perspectives
There is much left to discover regarding the potential roles of obscurin/UNC-89 and SPEG tandem kinases in the heart. Nonetheless, the currently identified binding partners, putative substrates, and disease associations have implicated them in a number of essential physiological processes, from cell adhesion, cytoskeletal and myofibrillar stability, to mechanotransduction, Ca2+ signaling, and excitation-contraction coupling. Despite having highly conserved domain architectures, the presence of unique substrates that are located in distinct cellular compartments suggest that obscurin and SPEG kinases serve specialized purposes. Accordingly, although both proteins appear to overlap at the Z-disk, SPEG’s unique positioning at the JMC and the distribution of obscurins primarily to the M-band could reflect that these tandem kinase proteins serve different roles depending on their localization. In addition, the varying length of the COOH-terminal Ig/FN-III module with kinase 2 further supports the notion that obscurin and SPEG kinases could form interactions with different protein complexes (Sutter et al. 2004).
It remains to be clarified how exactly the kinase domains of obscurin and SPEG are regulated. Canonically, MLCK-like kinases are activated via CaM binding to the regulatory domain which functions to relieve autoinhibition. However, other proteins containing MLCK-like domains that are regulated distinctly, such as titin kinase, signify that this regulatory mechanism is not entirely conserved. Whether or not obscurin and SPEG kinases 1 and 2 are regulated by CaM or through an alternative mechanism represents an important question that remains to be experimentally tested. Moreover, the recent discovery that the interkinase region of UNC-89 is an elastic random coil (Qadota et al. 2020) points to another form of potential regulation of kinase activity. Given that this region shares many physical properties with the elastic domains of titin (Qadota et al. 2020), it is conceivable that the interkinase region could mediate mechanical signals to kinases 1 and/or 2.
Lastly, the exact physiological significance of having two closely associated kinase domains within the same protein molecule is largely unknown. One possibility is that kinases 1 and 2 regulate each other through direct phosphorylation events, or by indirect mechanisms such as modulating other kinases and phosphatases that act on the other. Alternatively, given their large size and distribution to multiple cellular compartments, the presence of tandem kinase domains could function to spatially integrate multiple signaling pathways together. Therefore, detailed investigations of their individual targets, physiological roles, and disease mechanisms are essential in order to shed light on these intriguing questions.
References
Ackermann MA, Shriver M, Perry NA, Hu LY, Kontrogianni-Konstantopoulos A (2014) Obscurins: Goliaths and Davids take over non-muscle tissues. PLoS One 9(2):e88162. https://doi.org/10.1371/journal.pone.0088162
Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC, Haliloglu G, Talim B, Yau KS, Allcock RJ, Laing NG, Perrella MA, Beggs AH (2014) SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet 95(2):218–226. https://doi.org/10.1016/j.ajhg.2014.07.004
Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S (2001) The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89(11):1065–1072. https://doi.org/10.1161/hh2301.100981
Benian GM, Tinley TL, Tang X, Borodovsky M (1996) The Caenorhabditis elegans gene unc-89, required fpr muscle M-line assembly, encodes a giant modular protein composed of Ig and signal transduction domains. J Cell Biol 132(5):835–848. https://doi.org/10.1083/jcb.132.5.835
Borisov AB, Raeker MO, Kontrogianni-Konstantopoulos A, Yang K, Kurnit DM, Bloch RJ, Russell MW (2003) Rapid response of cardiac obscurin gene cluster to aortic stenosis: differential activation of Rho-GEF and MLCK and involvement in hypertrophic growth. Biochem Biophys Res Commun 310(3):910–918. https://doi.org/10.1016/j.bbrc.2003.09.035
Borisov AB, Raeker MO, Russell MW (2008) Developmental expression and differential cellular localization of obscurin and obscurin-associated kinase in cardiac muscle cells. J Cell Biochem 103(5):1621–1635. https://doi.org/10.1002/jcb.21551
Catalano J, Paynton B, Kaniper S, Gerhard G, Alvarez R (2018) Identification of a novel obscurin protein variant in nonischemic cardiomyopathy. J Am Coll Cardiol 71(11)
Ferrara TM, Flaherty DB, Benian GM (2005) Titin/connectin-related proteins in C. elegans: a review and new findings. J Muscle Res Cell Motil 26(6-8):435–447. https://doi.org/10.1007/s10974-005-9027-4
Fukuzawa A, Idowu S, Gautel M (2005) Complete human gene structure of obscurin: implications for isoform generation by differential splicing. J Muscle Res Cell Motil 26(6-8):427–434. https://doi.org/10.1007/s10974-005-9025-6
Gautel M (2011) Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch 462(1):119–134. https://doi.org/10.1007/s00424-011-0946-1
Gieseler K, Qadota H, Benian GM (2017) Development, structure, and maintenance of C. elegans body wall muscle. WormBook 2017:1–59. https://doi.org/10.1895/wormbook.1.81.2
Grater F, Shen J, Jiang H, Gautel M, Grubmuller H (2005) Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys J 88(2):790–804. https://doi.org/10.1529/biophysj.104.052423
Grogan A, Kontrogianni-Konstantopoulos A (2019) Unraveling obscurins in heart disease. Pflugers Arch 471(5):735–743. https://doi.org/10.1007/s00424-018-2191-3
Grogan A, Hu L-YR, Ward C, Kontrogianni-Konstantopou A (2019) Loss of binding between giant obscurin and titin results in cardiac maladaptation. Biophys J 116:115a
Hsieh CM, Yet SF, Layne MD, Watanabe M, Hong AM, Perrella MA, Lee ME (1999) Genomic cloning and promoter analysis of aortic preferentially expressed gene-1. Identification of a vascular smooth muscle-specific promoter mediated by an E box motif. J Biol Chem 274(20):14344–14351. https://doi.org/10.1074/jbc.274.20.14344
Hsieh CM, Fukumoto S, Layne MD, Maemura K, Charles H, Patel A, Perrella MA, Lee ME (2000) Striated muscle preferentially expressed genes alpha and beta are two serine/threonine protein kinases derived from the same gene as the aortic preferentially expressed gene-1. J Biol Chem 275(47):36966–36973. https://doi.org/10.1074/jbc.M006028200
Hu LY, Kontrogianni-Konstantopoulos A (2013) The kinase domains of obscurin interact with intercellular adhesion proteins. FASEB J 27(5):2001–2012. https://doi.org/10.1096/fj.12-221317
Hu LR, Ackermann MA, Hecker PA, Prosser BL, King B, O'Connell KA, Grogan A, Meyer LC, Berndsen CE, Wright NT, Jonathan Lederer W, Kontrogianni-Konstantopoulos A (2017) Deregulated Ca(2+) cycling underlies the development of arrhythmia and heart disease due to mutant obscurin. Sci Adv 3(6):e1603081. https://doi.org/10.1126/sciadv.1603081
Kamm KE, Stull JT (2001) Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem 276(7):4527–4530. https://doi.org/10.1074/jbc.R000028200
Katzemich A, West RJ, Fukuzawa A, Sweeney ST, Gautel M, Sparrow J, Bullard B (2015) Binding partners of the kinase domains in Drosophila obscurin and their effect on the structure of the flight muscle. J Cell Sci 128(18):3386–3397. https://doi.org/10.1242/jcs.170639
Khapchaev AY, Shirinsky VP (2016) Myosin light chain kinase MYLK1: anatomy, interactions, functions, and regulation. Biochemistry (Mosc) 81(13):1676–1697. https://doi.org/10.1134/S000629791613006X
Kontrogianni-Konstantopoulos A, Jones EM, Van Rossum DB, Bloch RJ (2003) Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol Biol Cell 14(3):1138–1148. https://doi.org/10.1091/mbc.e02-07-0411
Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ (2009) Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol Rev 89(4):1217–1267. https://doi.org/10.1152/physrev.00017.2009
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549. https://doi.org/10.1093/molbev/msy096
Liu X, Ramjiganesh T, Chen YH, Chung SW, Hall SR, Schissel SL, Padera RF Jr, Liao R, Ackerman KG, Kajstura J, Leri A, Anversa P, Yet SF, Layne MD, Perrella MA (2009) Disruption of striated preferentially expressed gene locus leads to dilated cardiomyopathy in mice. Circulation 119(2):261–268. https://doi.org/10.1161/CIRCULATIONAHA.108.799536
Lornage X, Sabouraud P, Lannes B, Gaillard D, Schneider R, Deleuze JF, Boland A, Thompson J, Bohm J, Biancalana V, Laporte J (2018) Novel SPEG mutations in congenital myopathy without centralized nuclei. J Neuromuscul Dis 5(2):257–260. https://doi.org/10.3233/JND-170265
Mayans O, van der Ven PF, Wilm M, Mues A, Young P, Furst DO, Wilmanns M, Gautel M (1998) Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature 395(6705):863–869. https://doi.org/10.1038/27603
Puchner EM, Alexandrovich A, Kho AL, Hensen U, Schafer LV, Brandmeier B, Grater F, Grubmuller H, Gaub HE, Gautel M (2008) Mechanoenzymatics of titin kinase. Proc Natl Acad Sci U S A 105(36):13385–13390. https://doi.org/10.1073/pnas.0805034105
Qadota H, McGaha LA, Mercer KB, Stark TJ, Ferrara TM, Benian GM (2008) A novel protein phosphatase is a binding partner for the protein kinase domains of UNC-89 (Obscurin) in Caenorhabditis elegans. Mol Biol Cell 19(6):2424–2432. https://doi.org/10.1091/mbc.E08-01-0053
Qadota H, Moody JC, Lesanpezeshki L, Moncrief T, Kitzler D, Bhat PD, Vanapalli SA, Oberhauser AF, Benian GM (2020) A region of UNC-89 (obscurin) lying between two protein kinase domains is a highly elastic spring required for proper sarcomere assembly. bioRxiv:2020.2004.2003.023374. https://doi.org/10.1101/2020.04.03.023374
Qualls AE, Donkervoort S, Herkert JC, D'Gama AM, Bharucha-Goebel D, Collins J, Chao KR, Foley AR, Schoots MH, Jongbloed JDH, Bonnemann CG, Agrawal PB (2019) Novel SPEG mutations in congenital myopathies: genotype-phenotype correlations. Muscle Nerve 59(3):357–362. https://doi.org/10.1002/mus.26378
Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, Ouyang K, Wang HY, Chen S (2019) SPEG controls calcium reuptake into the sarcoplasmic reticulum through regulating SERCA2a by its second kinase-domain. Circ Res 124(5):712–726. https://doi.org/10.1161/CIRCRESAHA.118.313916
Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, Showell J, McCauley MD, Scholten A, Wehrens XH (2017) SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ Res 120(1):110–119. https://doi.org/10.1161/CIRCRESAHA.116.309977
Rowland TJ, Graw SL, Sweet ME, Gigli M, Taylor MR, Mestroni L (2016) Obscurin variants in patients with left ventricular noncompaction. J Am Coll Cardiol 68(20):2237–2238. https://doi.org/10.1016/j.jacc.2016.08.052
Russell MW, Raeker MO, Korytkowski KA, Sonneman KJ (2002) Identification, tissue expression and chromosomal localization of human Obscurin-MLCK, a member of the titin and Dbl families of myosin light chain kinases. Gene 282(1-2):237–246. https://doi.org/10.1016/s0378-1119(01)00795-8
Small TM, Gernert KM, Flaherty DB, Mercer KB, Borodovsky M, Benian GM (2004) Three new isoforms of Caenorhabditis elegans UNC-89 containing MLCK-like protein kinase domains. J Mol Biol 342(1):91–108. https://doi.org/10.1016/j.jmb.2004.07.006
Stull JT, Lin PJ, Krueger JK, Trewhella J, Zhi G (1998) Myosin light chain kinase: functional domains and structural motifs. Acta Physiol Scand 164(4):471–482. https://doi.org/10.1111/j.1365-201x.1998.tb10699.x
Sutter SB, Raeker MO, Borisov AB, Russell MW (2004) Orthologous relationship of obscurin and Unc-89: phylogeny of a novel family of tandem myosin light chain kinases. Dev Genes Evol 214(7):352–359. https://doi.org/10.1007/s00427-004-0413-5
Wang H, Castiglioni C, Kacar Bayram A, Fattori F, Pekuz S, Araneda D, Per H, Erazo R, Gumus H, Zorludemir S, Becker K, Ortega X, Bevilacqua JA, Bertini E, Cirak S (2017) Insights from genotype-phenotype correlations by novel SPEG mutations causing centronuclear myopathy. Neuromuscul Disord 27(9):836–842. https://doi.org/10.1016/j.nmd.2017.05.014
Wang H, Schanzer A, Kampschulte B, Daimaguler HS, Logeswaran T, Schlierbach H, Petzinger J, Ehrhardt H, Hahn A, Cirak S (2018a) A novel SPEG mutation causes non-compaction cardiomyopathy and neuropathy in a floppy infant with centronuclear myopathy. Acta Neuropathol Commun 6(1):83. https://doi.org/10.1186/s40478-018-0589-y
Wang L, Geist J, Grogan A, Hu LR, Kontrogianni-Konstantopoulos A (2018b) Thick filament protein network, functions, and disease association. Compr Physiol 8(2):631–709. https://doi.org/10.1002/cphy.c170023
Xiong G, Qadota H, Mercer KB, McGaha LA, Oberhauser AF, Benian GM (2009) A LIM-9 (FHL)/SCPL-1 (SCP) complex interacts with the C-terminal protein kinase regions of UNC-89 (obscurin) in Caenorhabditis elegans muscle. J Mol Biol 386(4):976–988. https://doi.org/10.1016/j.jmb.2009.01.016
Xu J, Li Z, Ren X, Dong M, Li J, Shi X, Zhang Y, Xie W, Sun Z, Liu X, Dai Q (2015) Investigation of pathogenic genes in Chinese sporadic hypertrophic cardiomyopathy patients by whole exome sequencing. Sci Rep 5:16609. https://doi.org/10.1038/srep16609
Young P, Ehler E, Gautel M (2001) Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol 154(1):123–136. https://doi.org/10.1083/jcb.200102110
Funding
This work was supported by the National Institutes of Health (Training Program in Muscle Biology, T32 AR007592-17 to A.G. and P.T.) and the American Heart Association (Grant In Aid 16GRNT31290010 to A.K.K.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Grogan, A., Tsakiroglou, P. & Kontrogianni-Konstantopoulos, A. Double the trouble: giant proteins with dual kinase activity in the heart. Biophys Rev 12, 1019–1029 (2020). https://doi.org/10.1007/s12551-020-00715-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-020-00715-3