
Abstract
Structural changes of heme side-chains of human adult hemoglobin (Hb A) upon ligand (O2 or CO) dissociation have been studied by circular dichroism (CD) and resonance Raman (RR) spectroscopies. We point out the occurrence of appreciable deformation of heme side-chains like vinyl and propionate groups prior to the out-of-plane displacement of heme iron. Referring to the recent fine resolved crystal structure of Hb A, the deformations of heme side-chains take place only in the β subunits. However, these changes are not observed in the isolated β chain (β4 homotetramer) and, therefore, are associated with the α–β inter-subunit interactions. For the communications between α and β subunits in Hb A regarding signals of ligand dissociation, possible routes are proposed on the basis of the time-resolved absorption, CD, MCD (magnetic CD), and RR spectroscopies. Our finding of the movements of heme side-chains would serve as one of the clues to solve the cooperative O2 binding mechanism of Hb A.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemoglobin (Hb) is a well-known O2-transporting allosteric protein. Human adult Hb (Hb A) is a heterotetramer consisting of two α (141 residues) and two β (146 residues) subunits. The active center which binds O2 is a heme (protoporphyrin IX Fe-complex, protoheme) (Fig. 1a). Hb binds O2 in the lung and releases it in tissues. When an O2 molecule is bound to deoxyHb in the lung, its O2 affinity increases and, when a second one binds, its O2 affinity increases further. In this way, O2 affinity increases with increasing number of bound O2 molecules. This phenomenon is called cooperativity and it is a characteristic of Hb. Upon release of O2, the opposite features appear. Structural elucidation of a mechanism of cooperativity is the subject of many studies of Hb.

a The structure of protoheme. b Absorption spectra of oxy (red) and deoxy forms (blue) of Hb A in 0.05 M phosphate buffer, pH 7.0
There are several allosteric effectors of Hb, such as CO2, proton, Cl− (chloride ion), and organic phosphate, similar to other allosteric proteins. However, the most effective regulator for the O2 affinity of Hb is the concentration of O2 itself (partial pressure of O2) (Edelstein 1975; Imai 1982; Bunn and Forget 1986; Dickerson and Geis 1983). When oxyHb, [Hb(O2)4], releases one molecule of O2, the O2 affinity of the remaining O2-bound subunits becomes lower and, thus, the release of one molecule of O2 accelerates the release of the remaining O2. This change of O2 affinity has been interpreted in terms of the quaternary structure transition from relaxed (R) to tense (T), which was first proposed by Monod, Wyman, and Changeux as a two-state model (MWC model) (Monod et al. 1965). Perutz (1970) revealed structures of Hb A with X-ray crystallography and pointed out that oxyHb and deoxyHb correspond to the R and T structures, respectively.
Hb A can also be considered to consist of two αβ dimers. There are two kinds of subunit interfaces, α1β1 (= α2β2) and α1β2 (= α2β1) (Dickerson and Geis 1983). The α1β1 subunit interface is strong and hardly changes, but the α1β2 interface is relatively weak and changes upon O2 binding, resulting in the quaternary structure changes (Perutz 1970). Baldwin and Chothia (1979) have suggested that the rearrangements of the α1β2 subunit interface are essential to the change of O2 affinity of Hb, and, thus, to the cooperativity. In the α1β2 interface, there are two important contacts in the deoxy state; one is an H-bond between Aspα94 and Trpβ37 and the other is an H-bond between Tyrα42 and Aspβ99. The former is called the “flexible joint” or “hinge region” and the latter is called the “switch region” (Dickerson and Geis 1983). These two H-bonds are important for stabilizing the T quaternary structure, but disappear by rotation of one αβ dimer by 15° with regard to the other αβ dimer upon O2 binding, and, thus, by the quaternary structure change from T to R.
Arisaka et al. (2011) examined the tetramer–dimer equilibrium of Hb by analytical ultracentrifugation. The sedimentation coefficient of Hb A and Mb show 4.27S and 2.22S, respectively (Arisaka et al. 2011; Schuck and Demeler 1999). Dimer Hb known as Hb Hirose (Trpβ37 → Ser) gives 3.05S. Using mutant Hbs at the α1β2 interface, they obtained the following s-values and also showed practical effects of an allosteric effector, IHP (inositol hexaphosphate) on the s-value; Hb A 4.27S (+IHP, 4.27S), Hb Hirose 3.05S (+IHP, 4.19S), rHb (Trpβ37 → His) 3.34S (+IHP, 3.82S), and rHb (Tyrα42 → Ser) 3.82S (+IHP, 4.12S) (Arisaka et al. 2011). All these mutants tend to dissociate into two dimers in different ways and the addition of IHP to the dissociated Hbs shifts the equilibrium toward tetramer, and, concomitantly, some mutant Hbs restore cooperativity. These results indicate that the tetramer–dimer equilibrium could be influenced by the substitution of only one amino acid residue. The equilibrium depends on where the substitution occurs and which kind of amino acid is introduced.
Since 1970, the cooperative O2 binding of Hb has been explained in terms of the transition between two quaternary structures (T and R) (Perutz 1970), based on quaternary structure. Recently, Henry et al. (2002) proposed the “tertiary two-state model”, in which tertiary structure is more important rather than quaternary structure, and Yonetani et al. (2002) and Yonetani and Laberge (2008) interpreted the cooperativity by the fluctuation of F- and E-helices as the “global allostery model”, in which quaternary structure is not so important. However, we do not discuss them in this review. Readers who are interested in them are suggested to refer to other reviews (Eaton et al. 1999; Yonetani and Kanaori 2013; Yuan et al. 2015).
The O2-bound heme iron is covalently bound to the proximal (F8) His at the opposite site and stays in the heme plane, but moves out-of-plane (~ 0.5 Å) toward His(F8) upon O2 dissociation (Dickerson and Geis 1983). The movement of iron is conveyed to the other portion of protein via HisF8 and this induces the quaternary structure change. This movement was thought to be skeletal distortion of heme itself (Perutz 1970) and little attention had been paid to the contribution of the individual peripheral group (side-chains) of the heme molecule. We have studied them with circular dichroism (CD) and resonance Raman (RR) spectroscopy and found appreciable changes of heme side-chains occurring prior to the movement of iron upon O2 dissociation (Nagai and Nagai 2011; Nagatomo et al. 2011). Indeed, in the recent 1.25 Å resolution X-ray crystallography of Hb A (Park et al. 2006), we noticed that the changes of heme side-chains occur in the β subunits of Hb A. On the other hand, in an O2 sensor heme protein, HemAT-Bs, an H-bonding of the heme side-chain with a near-by residue seems to be important for selective O2 binding and signal transduction (Yoshimura et al. 2006). These facts may suggest that the participation of heme side-chains in determining the function is common in heme proteins. Therefore, we focused on structural changes of heme side-chains upon ligand dissociation in this review.
Circular dichroism of hemoglobin
Hb A displays absorption spectra in the UV and visible regions (Fig. 1b). The characteristic visible absorption of Hb, called the Soret band, appears at 400–450 nm and derives from an allowed ππ* transition of porphyrin. Soret bands of the isolated α and β chains are alike in both the oxy and deoxy forms, but are different from that of tetrameric Hb. When these isolated chains are mixed, the Soret absorption band profile becomes similar to that of native α2β2 tetramer Hb (Hb A), but the intensity is significantly larger than their sum, especially in the deoxy form (Brunori et al. 1968). This spectral change by reconstitution is derived from some changes in the electronic properties of heme caused by protein conformational change due to interactions between α and β chains. This is the so-called “heme–heme interaction”.
Heme alone cannot exhibit a CD spectrum because the π-orbital of heme has higher symmetry. However, a characteristic CD spectrum appears when the heme is incorporated into the globin. Interaction between the heme and amino acid residues in the globin could induce asymmetry into the π-orbital of heme. Figure 2 shows the CD spectra of the isolated α and isolated β chains of Hb A in the Soret region (Nagai et al. 1969). Although electronic absorption spectra of both isolated chains are alike, different features of CD spectra between the isolated α and isolated β chains probably reflect the different environment around heme in the protein interior. Myoglobin (Mb) exhibits similar CD spectra to those of the isolated α chains (Nagai et al. 1969). In the X-ray crystal structure of Hb A, the orientations of the heme side-chains (vinyl and propionates) are different between the α and β subunits (Park et al. 2006). The different features of CD between the α and β chains partially arise from these differences of orientation of the heme side-chains.

Circular dichroism spectra in the Soret region of the isolated α and β chains at pH 7.0. Dashed line, oxy form; solid line, deoxy form (Nagai et al. 1969)
Figure 3 shows the CD spectra of Hb reconstituted from the isolated α and isolated β chains. The CD spectra of reconstituted oxyHb and deoxyHb are almost the same as those of native Hb A, but are not the same as the arithmetic mean of those of isolated α and isolated β chains (Nagai et al. 1969). Especially, the reconstituted deoxyHb exhibits strong intensity in the Soret region compared with the arithmetic mean. Similar results were also reported by Geraci and Li (1969). The results indicate that the π electronic symmetry of heme changes greatly by tetramerization of α and β chains. In the X-ray crystal structure of Hb A, the heme propionates of the β subunit change the orientation upon ligand dissociation (Park et al. 2006). The intensity enhancement of the Soret CD band in the deoxyHb A might be due to the orientation changes of heme propionates of the β subunit.

Circular dichroism spectra of the reconstituted Hb. Dashed line, reconstituted oxyHb; solid line, reconstituted deoxyHb; dotted line, arithmetic mean of the oxy α and β chains; dot-dashed line, arithmetic mean of the deoxy α and β chains (Nagai et al. 1969)
For the origin of induced heme optical activity in Hb and Mb, Hsu and Woody (1971) proposed a coupled oscillator interaction between heme transition and allowed ππ* transition of near-by aromatic residues, and this idea has been widely accepted. However, this model is contradicted by new observations that Hb with the reversed heme exhibits a negative CD in the Soret region (Nagai et al. 2008). Even if the heme is reversed in the globin pocket, the positions of aromatic residues remain unchanged. Therefore, we proposed a different interpretation. The optical activity of heme is influenced by the orientation of the heme side-chains. Rotation of the heme by 180° about the 5,15-meso axis interchanges the methyl groups at positions 2 and 7 with the vinyl groups at positions 8 and 3, respectively. The 13- and 17-propionates are the same as the 17,13-propionates. However, the orientation of the two propionates are opposite between the normal and reversed hemes, and, therefore, the optical activity will be changed.
In order to examine the effect of heme side-chains on the Soret CD band, we reconstituted Mb with apoMb and unnatural hemes having the substituted side-chains for 3,8-vinyl and 13,17-propionates (Nagai et al. 2015). The upper part of Fig. 4 shows the structures of the artificial hemes used in this experiment: heme 3, 3,8-methyl, 13,17-propionates; heme 5, 3,8-methyl, 13,17-acetates; heme 6, 3,8-methyl, 13,17-carboxylates. The lower part of Fig. 4 shows the CD spectra of the reconstituted Mbs, that is, Mb3 (heme 3), Mb5 (heme 5), and Mb6 (heme 6), respectively.

The Soret circular dichroism spectra of Mbs reconstituted with 3,8-methyl heme modified at 13,17-propionate. Mbs are in the aquomet form at pH 7.0 (Nagai et al. 2015)
The CD spectrum of Mb3 is similar to that of native Mb but the intensity of the Soret band was 70% of that of the native Mb, indicating that 3,8-vinyl side-chains contribute partly (~ 30%) to the chirality of the heme, as suggested by Woody and Pescitelli (2014). The change of the 13,17-propionate to acetate (Mb5) shows little effect, but that to carboxylate (Mb6) considerably diminished the heme chirality. This indicates that the length of 13,17 side-chains is important for the induced CD in the Soret region.
According to X-ray crystallographic analysis of Hb A, two sets of orientations are present for propionates at the 13- and 17-positions; in one, the two side-chains are directed up (proximal side) and down (distal side) against the heme plane (anti-parallel) and in the other, they adopt the same direction (parallel). We performed theoretical analysis on the effect of the propionate orientations on the Soret CD using time-dependent density functional theory (TDDFT) (Nagai et al. 2015). It became clear that up and down orientations of a propionate gave a positive and negative CD, respectively, and their magnitudes are different between the 13- and 17-positions. The great enhancement of the Soret CD band of the deoxyHb A might be caused by an orientational change of the 13,17-propionates upon O2 dissociation, and this will be quantitatively discussed later.
Although Hb and Mb generally exhibit a positive CD band in the Soret region, monomeric Hb from Chironomus thummi thummi (CTTHb) gave a negative Soret CD band (Formanek and Engel 1968). To explain this, Woody and Pescitelli (2014) modified their original idea so that the orientation of the vinyl side-chains of heme is more influential on the induced Soret CD than the interactions between the ππ* transitions of porphyrin and near-by aromatic residues. In the crystal structure of CTTHb (Steigemann and Weber 1979), two vinyl side-chains of heme stand 180° from the heme plane and the 13-propionate extends toward the proximal site, while the 17-propionate extends toward the distal site. This structure is different from those of Mbs (Nagai et al. 2015). These orientations of heme side-chains of CTTHb look similar to that of the Hb A with the reversed heme, suggesting that the orientation of heme side-chains influences the Soret CD.
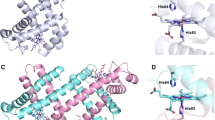
The most recent refinements of the crystallographic structures of oxy, deoxy, and COHb A with 1.25 Å resolution have revealed the movements of heme side-chains upon O2 dissociation in either α or β subunits, which are depicted in Figs. 5 and 6 (Park et al. 2006). In these figures, protein parts are shown by the ribbon model, and the heme moiety is shown by a molecular model. In the heme of the α subunit in the oxyHb A, 3,8-vinyl and 13,17-propionate side-chains seem to be unchanged upon O2 dissociation (Fig. 5). In contrast, these side chains of the β subunit in oxyHb A change greatly upon O2 dissociation; the 3-vinyl side chain changes direction from the proximal side (up) to the distal side (down) against the heme plane, and the 13,17-propionate groups change from the same direction (parallel orientation) to the up and down orientation against the heme (anti-parallel orientation) (Fig. 6). These alterations observed for the β subunit of oxyHb A are the same in COHb A (Park et al. 2006). The isolated β chain forms a homotetramer, β4. The crystal structures of the deoxy and CO forms of the isolated β chains were refined by Borgstahl et al. (1994a, b), who noted that the propionate side-chains of all four β chains take an anti-parallel orientation in both the deoxy and CO forms.

Comparison of crystal structures of the α subunits in the oxyHb A (left, PDB: 2DN1) and deoxyHb A forms (right, PDB: 2DN2) (Park et al. 2006). Anti-parallel orientation of the propionate side chains against the heme plane do not change upon O2 dissociation

Comparison of the crystal structures of the β subunits in the oxyHb A (left, PDB: 2DN1) and deoxyHb A forms (right, PDB: 2DN2) (Park et al. 2006). The two propionate side-chains of the heme change from parallel (left) to anti-parallel orientation (right) against the heme plane upon O2 dissociation
Propionates of Mb form the H-bond networks in the upper and lower parts of the heme plane, and these H-bond networks do not change with or without ligand (Hayashi et al. 2002). Therefore, characteristic changes from parallel to anti-parallel orientation of the propionate upon ligand dissociation occur only in the β subunit of the heterotetramer, α2β2 (Hb A).
Gas-sensor proteins such as HemAT-Bs (O2) (Yoshimura et al. 2006), CooA (CO) (Aono et al. 1998), Ec Dos (O2) (Sasakura et al. 2002), and FixL (O2) (Rodgers and Lukat-Rodgers 2005), and P450cam (Hayashi et al. 2009) all have a protoheme (b-type) the same as Hb A. In HemAT-Bs, an H-bond between 13-propionate and His86 is known to be important for O2 sensing and signal transduction, because the mutation of His86 to Ala loses the signal transduction upon O2 binding (Yoshimura et al. 2007). CooA (transcription factor), Ec Dos (phosphodiesterase), and FixL (histidine kinase) are activated by the binding of a specific gas molecule, but there is no report on the contributions of their heme propionates to functions. For the activity of P450cam, 17-propionate is considered to make a salt-bridge with Arg299, regulating the uptake of water molecules to the active site (Hayashi et al. 2009), but the 13-propionate is believed to be essential, because deletion of the 13-propionate changes P450cam to inactive P420 (Hayashi et al. 2009).
Carbon monoxide (CO) is dissociated from HbCO by light irradiation. The change in the absorption spectrum of Hb after photodissociation of CO was pursued using time-resolved absorption spectroscopy (Hofrichter et al. 1983, 1985; Murray et al. 1988; Gelin et al. 1983; Goldbeck et al. 1996), which showed that fast absorption change (0.1 μs) due to conformational change of heme (distortion) was followed by a slow (20–30 μs) quaternary structure change of the protein. Björling et al. (1996) examined the nanosecond time-resolved near-UV CD spectral changes (TRCD) of Hb after CO photodissociation and observed the early time changes of both the heme and the aromatic region of the protein. Perutz et al. (1974) found a negative CD band around 285 nm in the deoxyHb, and called this band “the T-state marker”, because its appearance is correlated to an increase of cooperativity. It is deduced that the 285-nm negative CD band derives from the H-bond formation of Trpβ37-Aspα94 and/or Tyrα42-Aspβ99 at the α1β2 subunit interface (Perutz et al. 1974).
We identified the contribution of aromatic residues at the α1β2 subunit interface contributing to the negative CD band to be Tyrα42 (4%), Trpβ37 (18%), Tyrβ145 (28%), and Tyrα140 (32%), using Hbs, which involves mutation at one of these residues (Aki-Jin et al. 2007; Li et al. 2000a). The negative CD band is not due to the Tyrα42 and Trpβ37, but arises from changes of the penultimate Tyr of both the α and β subunits (Tyrα140 and Tyrβ145). Moreover, the negative CD band also contained an appreciable contribution from the protein tertiary structure changes upon ligand dissociation (Li et al. 2000b). Accordingly, the CD change of the aromatic region observed by Björling et al. (1996) probably reflects the tertiary structure changes after ligand photodissociation. Goldbeck et al. (2002) examined the aromatic region (285–295 nm) of HbCO after CO photodissociation by the time-resolved magnetic CD (TRMCD). They found red-shift of the Trp band at early time (2 μs) and suggested that this red-shift is due to the H-bond formation between Trpβ37 and Aspα94, and that the H-bond formation is the first step of the quaternary structure transition.
As clarified by time-resolved absorption, CD, and MCD studies of Hb mentioned above, fast conformational change of the heme (ns) occurs immediately after CO photodissociation, and the protein tertiary and quaternary structure changes (μs) follow it.
Raman scattering of hemoglobin
Raman scattering is a vibrational spectroscopy like an infra-red absorption spectroscopy. Resonance Raman (RR) is a technique to enhance the Raman scattering from a chromophore of a molecule by approaching the Raman excitation wavelength to the absorption maximum of the target molecule. Accordingly, the fine structure of heme could be examined using the Soret band excitation. If there is a sample of αoxy-βdeoxy Hb, we can obtain the RR spectrum for the α subunit with the 413-nm excitation (absorption peak of oxyHb) and obtain the RR spectrum of the β subunit with the 441.6-nm excitation (near the absorption peak of deoxy Hb).
As cited earlier, Hb with the reversed heme showed a negative Soret CD band (Nagai et al. 2008). In the reversed heme, the methyl groups at positions 2 and 7 interchange for the vinyl groups at positions 3 and 8, but the propionate groups at positions 13 and 17 remain unchanged. Since the spectrum of Mb3 in Fig. 4 suggested that the contribution from vinyl group to Soret CD is approximately 30% (Nagai et al. 2015), it is unlikely to ascribe the Soret negative CD to the exchange of vinyl groups with methyl groups (Nagai et al. 2015). However, it is possible that the orientations of propionates at positions 13 and 17 are opposite to those of native Hb. To examine this possibility, the RR spectrum of Hb with the reversed heme was compared with that of Hb with normal heme, as shown in Fig. 7 (Nagai et al. 2016).

The 441.6-nm excited resonance Raman spectra of native Hb A (lower spectrum) and rHb A with the reversed heme (upper spectrum) in the deoxy form (Nagai et al. 2016)
Many peaks are observed in Fig. 7 and each peak has been assigned to a specific vibration of a heme experimentally and theoretically (Hu et al. 1996; Kitagawa and Teraoka 1979; Li et al. 1990). Assignments of RR bands in Fig. 7 are the ones determined by Hu et al. (1996), who assigned the RR bands on the basis of reconstitution of Mb with isotopically labeled heme. The strong ν4 band at 1357 cm−1 is known as an oxidation state marker and varies with the oxidation of the heme and ligand binding. The ν2 band at 1567 cm−1 and the ν3 band at 1472 cm−1 are called the spin-state and coordination-state markers, respectively. In the higher frequency region (Fig. 7 left panel), RR bands of Hb with the reversed heme (upper spectrum) are the same as those of Hb A with normal heme (lower spectrum), except for the vinyl C = C band around 1620 cm−1. It means that the in-plane skeletal vibrations of the heme are hardly affected by the orientation of heme.
In the low frequency region (Fig. 7 right panel), RR bands at 300 and 340 cm−1 were assigned to the out-of-plane (γ7) and in-plane modes (ν8) of the heme, respectively. The Fe-His mode at 215 cm−1 and the porphyrin ring modes are the same between Hb As with normal and reversed hemes. For side-chain modes, however, the RR band at 364 cm−1 was assigned to the propionate skeletal bending δ(CβCcCd), and the RR bands at 403 and 429 cm−1 were assigned to the 8-vinyl and 3-vinyl bending δ(CβCaCb) modes, respectively. The latter bands merge into a single band at around 418 cm−1 in the spectrum of Hb A with the reversed heme. Moreover, a bending mode δ(CβCcCd) of the 13,17-propionate groups of Hb A with normal heme at 364 cm−1 splits into two bands at 364 cm−1 and 374 cm−1 in the spectrum of Hb A with the reversed heme. The shifts of vinyl Ca = Cb stretching mode at 1619 cm−1 of Hb A with normal heme by 2 cm−1 to higher frequency in Hb A with the reversed heme is probably caused by the change of the orientation of vinyl side-chains. As no change was observed for the γ7 and ν8 bands between Hb A with normal heme and that with the reversed heme, planarity of the heme must be maintained between them. The characteristic negative CD band in the Soret region of Hb A with the reversed heme is probably related to the changes of orientation of the heme side-chains.
In Fig. 8, RR spectra of the isolated α and isolated β chains in the low frequency region are compared with that of native Hb A (α2β2) in the deoxy form (Nagatomo et al. 2011). The νFe-His band of deoxyHb A (T) is observed at 215–216 cm−1, but the corresponding bands of isolated chains appear at 222–223 cm−1, while the frequencies of all other bands are similar between native Hb A and the isolated chains. The frequency of the νFe-His band depends on the O2 affinity: Hb Chesapeake (Argα92 → Leu) with high O2 affinity and low cooperativity (R) gives the νFe-His band at 222 cm−1, although Hb Chesapeake in the presence of IHP with decreased O2 affinity and a significant cooperativity (T) gave the νFe-His at 215 cm−1 (Imai 1974; Matsukawa et al. 1985).

Low frequency resonance Raman spectra of: a Hb A, b the isolated α chains, and c the isolated β chains in the deoxy form (Nagatomo et al. 2011). Excitation wavelength: 441.6 nm
It is a question of whether the νFe-His band of the α subunit is the same as that of the β subunit in the deoxyHb A or not. Nagai and Kitagawa (1980) have determined the νFe-His of each subunit in the T-state Hb using valency hybrid Hbs (ferric heme in either α or β subunit) and metal hybrid Hbs (Ni-heme in either α or β subunit) (Nagai and Kitagawa 1980; Shibayama et al. 1986). They showed that, in the T-state Hb, the νFe-His band appeared at 201–203 cm−1 for the α subunit and at 217–220 cm−1 for the β subunit, respectively. This result indicates that, in the slow relaxation process following CO photolysis, the νFe-His of α subunit shifts to much lower frequencies (222 → 203 cm−1) than that of β subunit (223 → 217 cm−1), and, thus, their relaxation energies would be different.
The relation between Raman intensity and the structure of vinyl side-chain were examined through the selective deuteration of vinyl side-chain by Uchida et al. (1988) and Rwere et al. (2014). They interpreted that the Raman intensity of vinyl side-chain depends on their co-planarity with pyrrole ring, that is, the bands are weaker when vinyl side-chains are out-of-plane. As shown in Fig. 8, the 8- and 3-vinyl δCβCaCb bands are observed at 402 and 429 cm−1, respectively, for Hb A and the α chain. However, the 429 cm−1 band is much weaker for the β chain, suggesting that the 3- and 8-vinyl side-chain of both Hb A and its α chain are in-plane, while 3-vinyl of the β chain is out-of-plane.
Figure 9 shows the RR spectra of equilibrated HbCO (green), deoxyHb (blue), and the time-resolved RR spectra (red) following CO photodissociation (Mizutani and Nagai 2012). The red spectra represent the difference between spectra observed at a specific delay time (Δt) against the spectrum of HbCO. In the spectrum of HbCO (Fig. 9 bottom, green), the RR band at 503 cm−1 is assigned to Fe-CO stretching (νFe-CO) (Tsubaki et al. 1982). It is noted that the spectrum at Δt = 1000 ps is still different from the spectrum of deoxyHb, particularly around the νFe-His band. This means that the structural relaxation of Hb A is extremely slow. With regard to the side-chain modes, 3-vinyl δ(CβCaCb) is present at 429 cm−1 for HbCO, while 8-vinyl δ(CβCaCb) is observed at 404 cm−1 for deoxyHb, suggesting that the co-planarity of vinyl groups with pyrrole ring changes upon CO dissociation. The propionate bending mode, δ(CβCcCd), on the other hand, appears at 365 cm−1 for deoxyHb but splits into two bands at 374 and 353 cm−1 in HbCO, suggesting that the orientation of the propionates changes upon CO dissociation. These changes of RR bands for the heme side-chains upon ligand dissociation are similar to those of Hb A with the reversed heme, as shown in Fig. 7.

Pico-second time-resolved resonance Raman spectra of photodissociated HbCO in the 180–850 cm−1 region. Spectra of the equilibrated states of deoxyHb (blue) and HbCO (green) are depicted at the bottom for comparison (Mizutani and Nagai 2012). The time-resolved spectra (red) are differences of the spectrum observed for specific Δt against the spectrum of HbCO (green)
The propionate bending mode δ(CβCcCd) is known to shift to a higher frequency when the terminal carboxylate of propionate forms an H-bond (Nakashima et al. 1998). As shown in Fig. 6, the 13,17-propionates of β subunit in oxy-Hb A take a parallel orientation and their carboxylates can make an H-bond via a water molecule (Park et al. 2006). Upon O2 dissociation, orientation of the propionates changes from parallel to anti-parallel, and the H-bond disappears. Accordingly, the propionate bending mode, δ(CβCcCd), is shifted to a lower frequency upon ligand dissociation. On the other hand, the 13-propionate of the α subunit of Hb A makes an H-bond with Hisα45, but the H-bond does not change in any of the oxy, CO, and deoxy forms (Park et al. 2006).
In the pico-second time-resolved RR spectra shown in Fig. 9, the νFe-His band appears at 233 cm−1 at Δt = 1 ps and is down-shifted only 2 cm−1 in 1000 ps (Mizutani and Nagai 2012). The 2 cm−1 down-shift of the νFe-His band at 1000 ps was also observed in Mb and the isolated α and β chains, but was not seen in the heme model compound (CO-hemin 2-methyl imidazole) (Mizutani and Kitagawa 2001). Therefore, this 2 cm−1 down-shift is due to protein relaxation. The other RR bands, at 305 cm−1 (out-of-plane mode, γ7) and 406 cm−1 (vinyl bending mode, δCβCaCb), do not change until 1000 ps, but these bands are not the same as the RR bands at 300 cm−1 (γ7) and 403 cm−1 (δCβCaCb) of the deoxyHb. Moreover, the 341 cm−1 band (in-plane mode, ν8) observed in the deoxyHb is not seen even at Δt = 1000 ps. The quaternary structure transition to the T structure takes place at Δt = 20 μs (Yamada et al. 2013).
Recently, Jones et al. (2014) have shown an interesting result using the proto-meso heme hybrid Hb. An RR spectrum of the subunit having proto-heme could be obtained upon excitation at 441.6-nm, because the Soret absorption peak of the meso-heme is shifted to a shorter wavelength than that of the proto-heme by 10 nm. From the RR spectra after photodissociation and theoretical calculations with quantum mechanics/molecular mechanics (QM/MM), using the PELE program, it became clear that the shift of the νFe-His band to the frequency of the T structure is much faster (3 μs) in the β subunit than that in the α subunit (20 μs).
The ultra-violet RR (UVRR) spectroscopy allows us to monitor the conformational changes of proteins (Miura et al. 1988; Chi and Asher 1998). Upon excitation at 235 nm, the vibrations of tyrosine (Tyr) and tryptophan (Trp) are selectively detected. Accordingly, we examined the UVRR spectral changes of these residues due to the quaternary structure transition upon ligand dissociation, and observed high-frequency shifts for Tyr residues and intensity enhancement for Trp residues (Nagai et al. 1995, 1996). We clarified that the frequency shifts of Tyr are due to Tyrα42 and Tyrα140 and that the intensity enhancement of Trp RR bands is due to Trpβ37 (Nagai et al. 1995, 1996, 2012). All these aromatic residues are located at the α1β2 subunit interface and form an intra- or inter-subunit H-bond in deoxyHb. Thus the frequency shifts and intensity enhancements of RR bands upon ligand dissociation seem to be caused by H-bond formations or change of hydrophobic circumstances around these aromatic residues in proteins. Specific H-bond formations of Tyrα42 and Trpβ37 in the deoxyHb were clarified by the 1H NMR spectra. The characteristic 1H NMR signals of these residues are used as “T-state marker bands” (Ho 1992).
Rodgers and Spiro (1994) have examined the conformational changes of protein after photodissociation by time-resolved UVRR and revealed that tertiary structure changes occur at 0.3 μs and quaternary structure changes occur at 20 μs. The tertiary H-bond changes arise from Trpα14-Thrα67 and Trpβ15-Serβ72 (Balakrishnan et al. 2004b), although these authors improved their data (Balakrishnan et al. 2004a), which indicated that the T-state quaternary contacts are formed in two steps, with time constants at 2.9 μs and 21 μs, instead of a single 20-μs process. The first step involves the hinge region by the formation of an H-bond between Trpβ37 and Aspα94 and the second step involves the switch region by an H-bond formation between Tyrα42 and Aspβ99 at the α1β2 subunit interface. Kavanaugh et al. (2005) have investigated how the Tyrα42 contributes to the cooperativity using a mutant with Tyrα42 replaced by alanine, rHb (Tyrα42 → Ala). This mutant Hb showed increased O2 affinity but exhibited a significant cooperativity (Hill’s n = 1.9). Intensity enhancement of Trp RR bands upon O2 dissociation was almost the same as those in native Hb. From these results, they concluded that Tyrα42 significantly stabilizes the quaternary T structure but is not a major player in yielding quaternary constraints in the deoxyHb.
Conformational changes of the heme upon ligand dissociation and its relation to cooperativity
Cooperativity in O2 binding of Hb has been attributed to the T–R quaternary structure transition. It was known from the crystal structure of the porphyrin model compounds that a five-coordinated high-spin Fe(II) sits roughly 0.45 Å out-of-heme plane, whereas a six-coordinated low-spin Fe(II) sits on the heme plane (Perutz 1979). Fermi et al. (1984) pointed out that, in the deoxyHb with T structure, the iron atom stays out-of-plane of the porphyrin ring toward the proximal HisF8 by ∼ 0.4 Å. RR measurements indicated that the Fe-Nε(HisF8) stretching mode (νFe-His) of deoxyHb appeared at 215–218 cm−1 for the T state and at 220–221 cm−1 for the R state (Nagai et al. 1980; Nagai and Kitagawa 1980). This suggested that the Fe-Nε(HisF8) bond was weakened in the T state due to a strain exerted by globin. The binding of O2 to Hb would force the iron atom to move into the heme plane, pulling the HisF8 and, thus, the F-helix toward the heme against the strain. This motion could trigger an eventual T-to-R (deoxy-to-oxy) structure transition. The Perutz group (Perutz et al. 1974; Fermi et al. 1984) proposed this trigger mechanism, in which the displacement of iron is the first event for the quaternary structure change.
However, our finding suggests that the changes of heme side-chains upon O2 dissociation precede the quaternary structure transition. The shift of the νFe-His band to 215 cm−1 after CO photodissociation (233 cm−1) takes 20 μs (Yamada et al. 2013), but the changes of heme side-chains start immediately after CO photodissociation (1 ps), as shown in Fig. 9. A possible transient structure of heme in the β subunit of Hb A upon O2 dissociation is illustrated in Fig. 10.

A possible transient structure of heme in the β subunits of Hb A upon O2 dissociation
As shown in Fig. 10, O2 dissociation from the β subunit in Hb A changes the orientation of the propionate side-chains from parallel to anti-parallel, and also changes the orientation of the 3-vinyl side-chain from the proximal side to the distal side, in addition to the shift of heme iron from in-plane to out-of-heme plane. However, just after O2 dissociation, heme planarity is supposed to be unstable because the RR band at 305 cm−1, an out-of-plane mode (γ7), is different from that of the deoxyHb (at 300 cm−1), and the RR band at 340 cm−1, in-plane stretching mode of the iron and pyrrole nitrogens (ν8), does not yet appear in the transient state, though it appears in the deoxyHb (Mizutani and Nagai 2012).
The great intensity enhancement of CD and the changes of low frequency RR spectra suggest that structural changes of heme moiety take place immediately after O2 dissociation and that they might derive from the alteration of the orientation of heme-side chains. According to X-ray crystallographic analysis of Hb A, the alterations of the vinyl and propionate side-chains of the heme upon ligand dissociation occur only in the β subunit of the α2β2 tetramer but do not occur in the isolated β chains (β4 homotetramer). The presumed changes of the β subunit would be transmitted to the α subunit via the α1β2 subunit interface and then the α subunit would start to change. Thus, the constraints for the T structure of Hb would be completed.
We have shown the CD spectra of isolated α and β subunits, which are different from each other, and their arithmetic mean is not the same as that of α2β2 tetramer, especially the two-fold intensity increase in the deoxy form (Nagai et al. 1969). We demonstrated with Fig. 4 that ca. 70% of the induced Soret CD of Hb arises from the 13,17-propionates (Nagai et al. 2015). As shown in Figs. 5 and 6, the propionates of α subunit always take anti-parallel orientation, but those of β subunit take parallel and anti-parallel orientations in the oxy and deoxy forms, respectively (Park et al. 2006). On the other hand, TDDFT calculations (Nagai et al. 2015) is compatible with the assumption that the contributions from 17- and 13- propionates to Soret CD are +ε and −3ε when both are oriented along the proximal side of heme.
Since the orientation of 17-propionate is along the proximal side in the deoxy and oxy forms, its contribution should be +ε. On the other hand, since the orientation of 13-propionate is along the proximal or distal sides, its contribution should be −3ε or +3ε, respectively. Then, the parallel and ant-parallel orientations of 13- and 17-propionates are expected to yield −2ε (= (+ε) + (−3ε)) and +4ε (= (+ε) + (+3ε)), respectively. This is the case of the β subunit. Since the propionates in both the deoxy and oxy forms of the α subunit adopt the anti-parallel orientation, their contributions are +4ε. This is illustrated in Fig. 11, where the CD band is assumed to be Gaussian, with the band center at the wavelength of Fig. 2. When the peak wavelengths of α and β subunits are close, the apparent CD intensities of α2β2 tetramer would be +8ε and +2ε for the deoxy and oxy forms, respectively. This explains the strong CD intensity for deoxyHb. If we assume 1.8 × 10−4 for ε, we obtain ellipticity (θ) values of 14.4 × 10−4 and 3.6 × 10−4 for deoxy and oxyHb A, respectively. These values are very close to the observed values of Fig. 3.

Calculated circular dichroism spectra of oxyHb A (left panel) and deoxyHb A (right panel). For each band of propionate, Gauss functions are assumed. The thin lines are the spectra of 17-propionate and 13-propionate of the heme. The bold lines are the spectra of each subunit in α2β2 tetramer. Red, α subunit; blue, β subunit; black, the sum of α and β subunits. Parameters used for the Gauss function, θ = Aexp(−(λ – λ0)/width), are as follows: width = 10 nm; A = 1.8 for α subunit, 5.4 for β subunit (distal), and −5.4 for β subunit (proximal); λ0 = 418 nm for α subunit (oxy), 430 nm for α subunit (deoxy), 417 nm for β subunit of 13-propionate (oxy), 422 nm for β subunit of 17-propionate (oxy), 431 nm for β subunit of 13-propionate (deoxy), and 436 nm for α subunit of 17-propionate (deoxy)
Time-resolved RR spectra of HbCO indicate that the orientation change of heme side-chains occurs at early times (ps) after CO photodissociation, but that it takes a long time (μs) to stabilize the heme structure (Yamada et al. 2013). Stabilization of the heme structure and the shift of νFe-His to low frequency (215 cm−1) take place in almost the same time regime (20 μs) (Yamada et al. 2013; Jones et al. 2014). Presumably, the changes of heme side-chains initiate the tertiary structure changes of protein, which is followed by the quaternary structure transition.
A possible route of signal transmission between α and β subunits upon O2 dissociation is illustrated in Fig. 12. Changes of heme side-chains of the β subunit occur at 1 ps following CO photodissociation. The change of the νFe-His band in the β subunit (2.9 μs) is much faster than that in the α subunit (20 μs) (Balakrishnan et al. 2004a; Jones et al. 2014). The H-bond formation at the hinge region (Trpβ37-Aspα94) (3 μs) is also faster than that of the switch region (Tyrα42-Aspβ99) (21 μs) (Jones et al. 2014). Accordingly, the first event in the sequence is orientation changes of the heme side-chains in the β subunit (1 ps) and the subsequent changes involve stabilization of the unstable heme (transient form) by tertiary structure changes, and then the shift of the νFe-His band to lower frequency and an H-bond formation at the hinge region (3 μs) at the α1β2 subunit interface take place. After these structure changes of the β subunit, the signal would be transmitted to the α subunit through the α1β2 subunit interface. The changes of the α subunit begin with a shift of the νFe-His band from 222 cm−1 (R) to 203 cm−1 (T). When the ligand dissociation occurs at the α subunit, the νFe-His band must shift to a lower frequency and then forms an H-bond in the switch region, and the signal would be transmitted to the β subunit via the α1β2 subunit interface.

A possible route of signal transmission between the α and β subunits of Hb A upon O2 dissociation
Concluding remarks
Heme alone cannot exhibit a circular dichroism (CD) spectrum, but gives a distinct CD when it is incorporated into the globin. We have found experimentally that the deformation of propionate side-chains makes a considerable change in the Soret CD spectrum of hemoglobin (Hb), and it is supported by theoretical calculations. CD spectra of the isolated α and β chains are different from each other, and the CD spectrum of recombined α2β2 tetramer (Hb A) is not the same as the arithmetic mean of α- and β-isolated chains, especially about the two-fold increased intensity in deoxyHb A. We could demonstrate theoretically that the enhanced positive CD in the deoxyHb A is due to the changed orientation of propionate in the β subunit. Changes of propionate side-chain were also detected by resonance Raman (RR) spectra. Time-resolved RR spectra obtained after the photodissociation of HbCO revealed that the deformation of propionates side-chains starts immediately after the ligand dissociation. Therefore, deformation of propionates would be associated with the cooperative O2 binding of Hb as a trigger for initiating protein conformational transition prior to the movement of heme iron.
References
Aki-Jin Y, Nagai Y, Imai K, Nagai M (2007) Changes of near-UV circular dichroism spectra of human hemoglobin upon the R → T quaternary structure transition. In: Kneipp K, Aroca R, Kneipp H, Wentrup-Byrne E (eds) New approaches in biomedical spectroscopy, ACS symposium series, vol. 963, chapter 19. American Chemical Society, Washington, DC, pp 297–311
Aono S, Ohkubo K, Matsuo T, Nakajima H (1998) Redox-controlled ligand exchange of the heme in the CO-sensing transcriptional activator CooA. J Biol Chem 273:25757–25764
Arisaka F, Nagai Y, Nagai M (2011) Dimer–tetramer association equilibria of human adult hemoglobin and its mutants as observed by analytical ultracentrifugation. Methods 54:175–180
Balakrishnan G, Case MA, Pevsner A, Zhao X, Tengroth C, McLendon GL, Spiro TG (2004a) Time-resolved absorption and UV resonance Raman spectra reveal stepwise formation of T quaternary contacts in the allosteric pathway of hemoglobin. J Mol Biol 340:843–856
Balakrishnan G, Tsai C-H, Wu Q, Case MA, Pevsner A, McLendon GL, Ho C, Spiro TG (2004b) Hemoglobin site-mutants reveal dynamical role of interhelical H-bonds in the allosteric pathway: time-resolved UV resonance Raman evidence for intra-dimer coupling. J Mol Biol 340:857–668
Baldwin J, Chothia C (1979) Haemoglobin: the structural changes related to ligand binding and its allosteric mechanism. J Mol Biol 129:175–220
Björling SC, Goldbeck RA, Paquette SJ, Milder SJ, Kliger DS (1996) Allosteric intermediates in hemoglobin. 1. Nanosecond time-resolved circular dichroism spectroscopy. Biochemistry 35:8619–8627
Borgstahl GE, Rogers PH, Arnone A (1994a) The 1.8 Å structure of carbonmonoxy-β4 hemoglobin: analysis of a homotetramer with the R quaternary structure of liganded α2β2 hemoglobin. J Mol Biol 236:817–830
Borgstahl GE, Rogers PH, Arnone A (1994b) The 1.9 Å structure of deoxyβ4 hemoglobin: analysis of the partitioning of quaternary-associated and ligand-induced changes in tertiary structure. J Mol Biol 236:831–843
Brunori M, Antonini E, Wyman J, Anderson SR (1968) Spectral differences between haemoglobin and isolated haemoglobin chains in the deoxygenated state. J Mol Biol 34:357–359
Bunn HF, Forget BG (1986) Hemoglobin: molecular, genetic and clinical aspects. W. B. Saunders Company, Philadelphia
Chi Z, Asher SA (1998) UV Raman determination of the environment and solvent exposure of Tyr and Trp residues. J Phys Chem B 102:9595–9602
Dickerson RE, Geis I (1983) Hemoglobin: structure, function, evolution, and pathology. Benjamin/Cummings, Menlo Park
Eaton WA, Henry ER, Hofrichter J, Mozzarelli A (1999) Is cooperative oxygen binding by hemoglobin really understood? Nat Struct Biol 6:351–358
Edelstein SJ (1975) Cooperative interactions of hemoglobin. Annu Rev Biochem 44:209–232
Fermi G, Perutz MF, Shaanan B, Fourme R (1984) The crystal structure of human deoxyhaemoglobin at 1.74 Å resolution. J Mol Biol 175:159–174
Formanek H, Engel J (1968) Optical rotatory dispersion of a respiratory hemeprotein of Chrinomus thummi. Biochim Biophys Acta 160:151–158
Gelin BR, Lee AW-M, Karplus M (1983) Hemoglobin tertiary structural change on ligand binding its role in the co-operative mechanism. J Mol Biol 171:489–559
Geraci G, Li T-K (1969) Circular dichroism of isolated and recombined hemoglobin chains. Biochemistry 8:1848–1854
Goldbeck RA, Paquette SJ, Björling SC, Kliger DS (1996) Allosteric intermediates in hemoglobin. 2. Kinetic modeling of HbCO photolysis. Biochemistry 35:8628–8639
Goldbeck RA, Esquerra RM, Kliger DS (2002) Hydrogen bonding to Trp β37 is the first step in a compound pathway for hemoglobin allostery. J Am Chem Soc 124:7646–7647
Hayashi T, Matsuo T, Hitomi Y, Okawa K, Suzuki A, Shiro Y, Iizuka T, Hisaeda Y, Ogoshi H (2002) Contribution of heme-propionate side chains to structure and function of myoglobin: chemical approach by artificially created prosthetic groups. J Inorg Biochem 91:94–100
Hayashi T, Harada K, Sakurai K, Shimada H, Hirota S (2009) A role of the heme-7-propionate side chain in cytochrome P450cam as a gate for regulating the access of water molecules to the substrate-binding site. J Am Chem Soc 131:1398–1400
Henry ER, Bettati S, Hofrichter J, Eaton WA (2002) A tertiary two-state allosteric model for hemoglobin. Biophys Chem 98:149–164
Ho C (1992) Proton nuclear magnetic resonance studies on hemoglobin: cooperative interactions and partially ligated intermediates. Adv Protein Chem 43:153–312
Hofrichter J, Sommer JH, Henry ER, Eaton WA (1983) Nanosecond absorption spectroscopy of hemoglobin: elementary processes in kinetic cooperativity. Proc Natl Acad Sci U S A 80:2235–2239
Hofrichter J, Henry ER, Sommer JH, Deutsch R, Ikeda-Saito M, Yonetani T, Eaton WA (1985) Nanosecond optical spectra of iron–cobalt hybrid hemoglobins: geminate recombination, conformational changes, and intersubunit communication. Biochemistry 24:2667–2679
Hsu M-C, Woody RW (1971) The origin of the heme Cotton effects in myoglobin and hemoglobin. J Am Chem Soc 93:3515–3525
Hu S, Smith KM, Spiro TG (1996) Assignment of protoheme resonance Raman spectrum by heme labeling in myoglobin. J Am Chem Soc 118:12638–12646
Imai K (1974) Hemoglobin Chesapeake (92α, arginine → leucine). Precise measurements and analyses of oxygen equilibrium. J Biol Chem 249:7607–7612
Imai K (1982) Allosteric effects in haemoglobin. Cambridge University Press, Cambridge
Jones EM, Monza E, Balakrishnan G, Blouin GC, Mak PJ, Zhu Q, Kincaid JR, Guallar V, Spiro TG (2014) Differential control of heme reactivity in alpha and beta subunits of hemoglobin: a combined Raman spectroscopic and computational study. J Am Chem Soc 136:10325–10339
Kavanaugh JS, Rogers PH, Arnone A, Hui HL, Wierzba A, DeYoung A, Kwiatkowski LD, Noble RW, Juszczak LJ, Peterson ES, Friedman JM (2005) Intersubunit interactions associated with Tyr42α stabilize the quaternary-T tetramer but are not major quaternary constraints in deoxyhemoglobin. Biochemistry 44:3806–3820
Kitagawa T, Teraoka J (1979) The resonance Raman spectra of intermediate-spin ferrous porphyrin. Chem Phys Lett 63:443–446
Li X-Y, Czernuszewicz RS, Kincaid JR, Stein P, Spiro TG (1990) Consistent porphyrin force field. 2. Nickel octaethylporphyrin skeletal and substituent mode assignments from 15N, meso-d 4, and methylene-d 16 Raman and infrared isotope shifts. J Phys Chem 94:47–61
Li R, Nagai Y, Nagai M (2000a) Contribution of α140Tyr and β37Trp to the near-UV CD spectra on quaternary structure transition of human hemoglobin A. Chirality 12:216–220
Li R, Nagai Y, Nagai M (2000b) Changes of tyrosine and tryptophan residues in human hemoglobin by oxygen binding: near- and far-UV circular dichroism of isolated chains and recombined hemoglobin. J Inorg Biochem 82:93–101
Matsukawa S, Mawatari K, Yoneyama Y, Kitagawa T (1985) Correlation between the iron-histidine stretching frequencies and oxygen affinity of hemoglobins. A continuous strain model. J Am Chem Soc 107:1108–1113
Miura T, Takeuchi H, Harada I (1988) Characterization of individual tryptophan side chains in proteins using Raman spectroscopy and hydrogen–deuterium exchange kinetics. Biochemistry 27:88–94
Mizutani Y, Kitagawa T (2001) Ultrafast structural relaxation of myoglobin following photodissociation of carbon monoxide probed by time-resolved resonance Raman spectroscopy. J Phys Chem B 105:10992–10999
Mizutani Y, Nagai M (2012) Ultrafast protein dynamics of hemoglobin as studied by picosecond time-resolved resonance Raman spectroscopy. Chem Phys 396:45–52
Monod J, Wyman J, Changeux J-P (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118
Murray LP, Hofrichter J, Henry ER, Eaton WA (1988) Time-resolved optical spectroscopy and structural dynamics following photodissociation of carbonmonoxyhemoglobin. Biophys Chem 29:63–76
Nagai K, Kitagawa T (1980) Differences in Fe(II)-Nε(His-F8) stretching frequencies between deoxyhemoglobins in the two alternative quaternary structures. Proc Natl Acad Sci U S A 77:2033–2037
Nagai M, Nagai Y (2011) Studies on hemoglobin quaternary structure transition and heme orientation by circular dichroism. In: Nagai M (ed) Hemoglobin: recent developments and topics. Research Signpost, Kerala, pp 63–77
Nagai M, Sugita Y, Yoneyama Y (1969) Circular dichroism of hemoglobin and its subunits in the Soret region. J Biol Chem 244:1651–1653
Nagai K, Kitagawa T, Morimoto H (1980) Quaternary structures and low frequency molecular vibrations of haems of deoxy and oxyhaemoglobin studied by resonance Raman scattering. J Mol Biol 136:271–289
Nagai M, Kaminaka S, Ohba Y, Nagai Y, Mizutani Y, Kitagawa T (1995) Ultraviolet resonance Raman studies of quaternary structure of hemoglobin using a tryptophan β37 mutant. J Biol Chem 270:1636–1642
Nagai M, Imai K, Kaminaka S, Mizutani Y, Kitagawa T (1996) Ultraviolet resonance Raman studies of hemoglobin quaternary structure using a tyrosine-α42 mutant: changes in the α1β2 subunit interface upon the T → R transition. J Mol Struct 379:65–75
Nagai M, Nagai Y, Aki Y, Imai K, Wada Y, Nagatomo S, Yamamoto Y (2008) Effect of reversed heme orientation on circular dichroism and cooperative oxygen binding of human adult hemoglobin. Biochemistry 47:517–525
Nagai M, Nagatomo S, Nagai Y, Ohkubo K, Imai K, Kitagawa T (2012) Near-UV circular dichroism and UV resonance Raman spectra of individual tryptophan residues in human hemoglobin and their changes upon the quaternary structure transition. Biochemistry 51:5932–5941
Nagai M, Kobayashi C, Nagai Y, Imai K, Mizusawa N, Sakurai H, Neya S, Kayanuma M, Shoji M, Nagatomo S (2015) Involvement of propionate side chains of the heme in circular dichroism of myoglobin: experimental and theoretical analyses. J Phys Chem B 119:1275–1287
Nagai M, Nagai Y, Aki Y, Sakurai H, Mizusawa N, Ogura T, Kitagawa T, Yamamoto Y, Nagatomo S (2016) Heme orientation of cavity mutant hemoglobins (His F8 → Gly) in either α or β subunits: circular dichroism, 1H NMR, and resonance Raman studies. Chirality 28:585–592
Nagatomo S, Nagai M, Kitagawa T (2011) Resonance Raman investigation of quaternary structure change in hemoglobin upon ligand binding. In: Nagai M (ed) Hemoglobin: recent developments and topics. Research Signpost, Kerala, pp 37–61
Nakashima S, Kitagawa T, Olson JS (1998) Time-resolved resonance Raman study of intermediates generated after photodissociation of wild-type and mutant CO-myoglobins. Chem Phys 228:323–336
Park S-Y, Yokoyama T, Shibayama N, Shiro Y, Tame JRH (2006) 1.25 Å resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J Mol Biol 360:690–701
Perutz MF (1970) Stereochemistry of cooperative effects in haemoglobin: haem–haem interaction and the problem of allostery. Nature 228:726–734
Perutz MF (1979) Regulation of oxygen affinity of hemoglobin: influence of structure of the globin on the heme iron. Annu Rev Biochem 48:327–386
Perutz MF, Ladner JE, Simon SR, Ho C (1974) Influence of globin structure on the state of the heme. I. Human deoxyhemoglobin. Biochemistry 13:2163–2173
Rodgers KR, Lukat-Rodgers GS (2005) Insights into heme-based O2 sensing from structure–function relationships in the FixL proteins. J Inorg Biochem 99:963–977
Rodgers KR, Spiro TG (1994) Nanosecond dynamics of the R → T transition in hemoglobin: ultraviolet Raman studies. Science 265:1697–1699
Rwere F, Mak PJ, Kincaid JR (2014) Resonance Raman determination of vinyl group disposition in different derivatives of native myoglobin and its heme-disoriented form. J Raman Spectrosc 45:97–104
Sasakura Y, Hirata S, Sugiyama S, Suzuki S, Taguchi S, Watanabe M, Matsui T, Sagami I, Shimizu T (2002) Characterization of a direct oxygen sensor heme protein from Escherichia coli. J Biol Chem 277:23821–23827
Shibayama N, Morimoto H, Kitagawa T (1986) Properties of chemically modified Ni(II)–Fe(II) hybrid hemoglobins: Ni(II) protoporphyrin IX as a model for a permanent deoxy-heme. J Mol Biol 192:331–336
Schuck P, Demeler B (1999) Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys J 76:2288–2296
Steigemann W, Weber E (1979) Structure of erythrocruorin in different ligand states refined at 1.4 Å resolution. J Mol Biol 127:309–338
Tsubaki M, Srivastava RB, Yu N-T (1982) Resonance Raman investigation of carbon monoxide bonding in (carbon monoxy)hemoglobin and -myoglobin: detection of Fe-CO stretching and Fe-C-O bending vibrations and influence of the quaternary structure change. Biochemistry 21:1132–1140
Uchida K, Susai Y, Hirotani E, Kimura T, Yoneya T, Takeuchi H, Harada I (1988) 4-Vinyl and 2,4-divinyl deuteration effects on the low frequency resonance Raman bands of myoglobin: correlation with the structure of vinyl group. J Biochem 103:979–985
Woody RW, Pescitelli G (2014) The role of heme chirality in the circular dichroism of heme proteins. Z Naturforsch A 69:313–325
Yamada K, Ishikawa H, Mizuno M, Shibayama N, Mizutani Y (2013) Intersubunit communication via changes in hemoglobin quaternary structures revealed by time-resolved resonance Raman spectroscopy: direct observation of the Perutz mechanism. J Phys Chem B 117:12461–12468
Yonetani T, Kanaori K (2013) How does hemoglobin generate such diverse functionality of physiological relevance? Biochim Biophys Acta 1834:1873–1884
Yonetani T, Laberge M (2008) Protein dynamics explain the allosteric behaviors of hemoglobin. Biochim Biophys Acta 1784:1146–1158
Yonetani T, Park SI, Tsuneshige A, Imai K, Kanaori K (2002) Global allostery model of hemoglobin. Modulation of O2 affinity, cooperativity, and Bohr effect by heterotropic allosteric effectors. J Biol Chem 277:34508–34520
Yoshimura H, Yoshioka S, Kobayashi K, Ohta T, Uchida T, Kubo M, Kitagawa T, Aono S (2006) Specific hydrogen-bonding networks responsible for selective O2 sensing of the oxygen sensor protein HemAT from Bacillus subtilis. Biochemistry 45:8301–8307
Yoshimura H, Yoshioka S, Mizutani Y, Aono S (2007) The formation of hydrogen bond in the proximal heme pocket of HemAT-Bs upon ligand binding. Biochem Biophys Res Commun 357:1053–1057
Yuan Y, Tam MF, Simplaceanu V, Ho C (2015) New look at hemoglobin allostery. Chem Rev 115:1702–1724
Acknowledgements
We are grateful to the Japanese Red Cross Kanto-Koshinetsu Block Blood Center for the gift of concentrated red cells to advance this human hemoglobin study. This study was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology for Scientific Research (C) to S.N. (17K05606) and Scientific Research (B) to T.K. (24350086), and also by a research grant from the Research Center for Micro-Nano Technology, Hosei University to M.N. and N.M.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Masako Nagai declares that she has no conflict of interest. Naoki Mizusawa declares that he has no conflict of interest. Teizo Kitagawa declares that he has no conflict of interest. Shigenori Nagatomo declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This article is part of a Special Issue on ‘Biomolecules to Bio-nanomachines - Fumio Arisaka 70th Birthday’ edited by Damien Hall, Junichi Takagi and Haruki Nakamura.
Rights and permissions
About this article

Cite this article
Nagai, M., Mizusawa, N., Kitagawa, T. et al. A role of heme side-chains of human hemoglobin in its function revealed by circular dichroism and resonance Raman spectroscopy. Biophys Rev 10, 271–284 (2018). https://doi.org/10.1007/s12551-017-0364-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-017-0364-5