
Abstract
The discovery of multiple classes of cardiac progenitor cells in the adult mammalian heart has generated hope for their use as a therapeutic in heart failure. However, successful results from animal models have not always yielded similar findings in human studies. Recent Phase I/II trials of c-Kit (SCIPIO) and cardiosphere-based (CADUCEUS) cardiac progenitor cells have demonstrated safety and some therapeutic efficacy. Gaps remain in our understanding of the origins, function and relationships between the different progenitor cell families, many of which are heterogeneous populations with overlapping definitions. Another challenge lies in the limitations of small animal models in replicating the human heart. Cryopreserved human cardiac tissue provides a readily available source of cardiac progenitor cells and may help address these questions. We review important findings and relative unknowns of the main classes of cardiac progenitor cells, highlighting differences between animal and human studies
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Whilst the cardiac regenerative ability of species such as salamanders and zebrafish is now well established, a similar capability of the mammalian heart has only recently been reported (Oberpriller and Oberpriller 1974; Porrello et al. 2011; Poss et al. 2002). The “Holy Grail” is to replicate this in humans. We are not there yet, but we are at a stage where studies using human cardiac tissue have firmly challenged the dogma that the human heart is a terminally differentiated organ.
The adult human heart contains about 3 billion cardiomyocytes, and a large myocardial infarction can eliminate up to 25 % of these cells (Laflamme and Murry 2011). Survival has improved in patients with myocardial infarction and other cardiac diseases. However, despite standard-of-care medical therapy, many patients progress to heart failure, the incidence of which is rising exponentially (Clark et al. 2005). Furthermore, stage IV heart failure carries a grim prognosis with 50 % mortality within 1 year of diagnosis (Cleland 2000). Cardiac regeneration has the potential to permanently improve function in failing hearts. Whilst there are several potential methods by which this could be achieved, these can be broadly grouped into two categories. Firstly, putative stem or progenitor cells can be introduced to generate new cardiomyocytes or other cardiac tissues. Secondly, stimulation of an inherent regenerative capacity (either stem cell mediated or proliferation of de novo cardiomyocytes) may be possible.
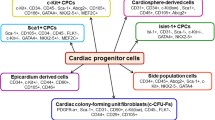
There is an emerging recognition that non-human mammalian hearts have excellent regenerative capacity during development (Porrello et al. 2011). This decreases after birth, but it may be re-activated in post-natal life. Small animal models such as the mouse and rat are popular due to their relative low costs and ease of genetic manipulation (Kooij et al. 2014). Yet, extrapolation to humans is limited by significant differences in proteome, genome, and physiology (Kooij et al. 2014; Siller et al. 2013). Data using murine models of human heart disease are not always applicable (Chong and Murry 2014). Large animal models (including macaque, canine, porcine, and ovine) better approximate human physiology (Dixon and Spinale 2009; Houser et al. 2012) but are expensive to maintain and are more difficult to manipulate genetically. Difficulty in translating promising animal studies into successful therapeutics has led to caution against extrapolation of animal findings to humans (Hackam and Redelmeier 2006; Hartung 2013; Marian 2011). This discrepancy was evidenced when bone marrow mononuclear cells failed to replicate promising animal results in human heart failure (Rehman 2013). More research using large animal models or on human cardiac tissue is needed to bridge this gap. Human cardiac progenitor cells (CPCs) have direct translational significance (Fig. 1), and extension of animal findings to human tissue will aid understanding of their role in human heart failure. However, there are significant hurdles to human research, including difficulty in acquisition of non-diseased specimens, the unpredictability of their availability and rapid degradation (Saha and Hurlbut 2011). In this regard, human heart tissue banks may provide the solution.

Characteristics of various cardiac progenitor cell populations. EPDC epicardium-derived cell, CPC cardiac progenitor cell, CSPC cardiac side population cell, CDC cardiosphere-derived cell, CADUCEUS cardiosphere-derived autologous stem cells to reverse ventricular dysfunction, SCIPIO stem cell infusion in patients with ischemic cardiomyopathy
Examining human cardiac stem cell sources within human cardiac tissue may be the way forward, but first, we need to establish exactly what is a human cardiac stem cell. Here, we review developments in the stem cell field with specific reference to human cardiac stem cells.
c-Kit+ cardiac progenitor cells
The population of cardiac progenitor cells that has been most thoroughly characterised is the c-Kit+ fraction, where c-Kit is the tyrosine kinase receptor for stem cell factor. Beltrami et al. (2003) initially discovered a resident c-Kit+, lineage negativeFootnote 1 population within the murine heart that is self-renewing, clonogenic, and differentiates both in vitro and in vivo to cardiomyocytes, endothelial cells and smooth muscle cells. Although the cells do not contract in vitro, cardiomyocytes formed in vivo couple to resident cardiomyocytes and exhibit similar contractile properties to the host myocardium. However, they are small and did not exhibit striations. Similarly, c-Kit+ CPCs were found in the dog heart and show the greatest cardiomyogenic potential compared to Sca-1+ CPCs or CSPCs. Once again, the generated cardiomyocytes are much smaller than that of the host and cannot directly explain the improvement in cardiac function.
The exact mechanism by which c-Kit stem cells alleviate myocardial dysfunction is contentious. Anversa and co-workers (Rota et al. 2008) found that injection of CPCs into rat myocardium 20 days after infarction reduce infarct size by over 40 % through the formation of new arterioles and small-sized cardiomyocytes in the infarct region. However, intracoronary injection of CPCs in a month-old infarct lead to minimal engraftment with the formation of immature cardiomyocytes. Nevertheless, improvements in cardiac function was noted, with paracrine mechanism suggested (Tang et al. 2010). Although bone marrow-derived c-Kit+ cells can be found in the heart post-MI (Fazel et al. 2006), they do not form cardiomyocytes or vasculature but exert an indirect effect on angiogenesis (Ellison et al. 2013; Jesty et al. 2012). Ellison et al. used an acute diffuse isoproterenol-induced injury model coupled with fate mapping to demonstrate that c-Kit+ cells infused via the tail vein homed to the infarct site differentiate for de novo myocyte formation (Ellison et al. 2013). Although the authors conclude that this proves cardiomyogenesis is the mechanism of cardiac regeneration by c-Kit+ CPCs (Nadal-Ginard et al. 2014), contrasting studies by Hong et al. show lack of engraftment for CPCs with both intramyocardial (Hong et al. 2013) and intracoronary injection (Hong et al. 2014). Recently, van Berlo and co-workers used a similar fate mapping technique to demonstrated that c-Kit+ cells contribute to many cellular fusion events and has little postnatal involvement with cardiomyocyte generation whether in ageing or injury (van Berlo et al. 2014).
The heterogeneity of c-Kit+ CPCs is one reason why it is so difficult to define the exact role of c-Kit cells in the heart (Table 1). Bearzi et al. characterised two separate multipotent c-Kit+ populations in the human heart: those expressing c-Kit and myocyte transcription factors but not vascular endothelial growth factor receptor-2 (VEGFR-2) were classified as myocardial progenitor cells that gave rise to more cardiomyocytes and improved function when injected in a rat MI model (Bearzi et al. 2007). Vascular progenitor cells were c-Kit+/VEGFR-2+ and differentiate in vitro primarily into endothelial and smooth muscle cells. When injected into dogs with stenosed coronary arteries, they formed coronary vessels up to 1.5 mm in diameter (Bearzi et al. 2009). c-Kit+ cells co-expressing CD29 and CD105 differentiate to give cardiac cells but also osteoblasts and adipocytes (Gambini et al. 2011). Indeed, within the same study, He et al. found c-Kit+/VEGFR-2+/CD31+ cells in some patients but not others, and suggested that these differences may be due to discordant medical history (He et al. 2011).
Alterations in CPC content and function are also seen with ageing. c-Kit+ CPCs can be isolated from the developing mouse embryo (Wu et al. 2006) and decline in numbers postnatally such that only a few are seen in niches of the adult heart (Tallini et al. 2009). These embryonic CPCs differentiate into all three cardiac lineages and, unlike their adult counterparts, exhibit spontaneous contraction (Tallini et al. 2009). Early embryonic c-Kit cells negative for cardiac markers were identified from as early as 6 days post-coitus, and divide both symmetrically and asymmetrically in vitro (Ferreira-Martins et al. 2012). This suggests they are a primitive population contributing to early cardiogenesis. Adult CPCs are quiescent and occupy niches found in greater numbers in the atria and apex and especially lowered in the left ventricle (Saravanakumar and Devaraj 2013; Urbanek et al. 2006). Neonatal c-Kit+ CPCs differentiate to both cardiomyocytes and endothelial cells post-infarct leading to partial regeneration while adult CPCs were only able to adopt a vascular lineage (Jesty et al. 2012).
There is disagreement in the distribution of c-Kit+ CPCs in both healthy and diseased human hearts (Castaldo et al. 2008; Garbade et al. 2010; Matuszczak et al. 2014; Sandstedt et al. 2014). Phase I study of c-Kit+ CPCs in patients with post-infarct ventricular dysfunction was reported to show functional improvement (Bolli et al. 2013), but concerns about data integrity have surfaced (The Lancet 2014). Given the current controversy over efficacy of c-Kit CPCs both in animal and human trials, the complex relationships between the various subpopulations, and unclear mechanisms by which they exert their action, validated, reproducible studies need to be performed to bridge the gap between animal models and human physiology. Such experiments will undoubtedly require both donor and diseased human heart tissue.
Epicardium-derived cells
The embryonic proepicardium is a cluster of extra-cardiac coelomic cells that adhere to the myocardium, and gives rise to the epicardium (visceral pericardium). The epicardium then undergoes an epithelial to mesenchymal transition (EMT) event to give rise to epicardium-derived cells (EPDCs), which migrate into the subepicardium and contribute to vasculature (Carmona et al. 2010; Krenning et al. 2010), and possibly cardiomyocytes (Christoffels et al. 2009; Zhou et al. 2008). Thus, EPDCs in the embryonic heart are highly enriched for stem cell activity (Wessels and Perez-Pomares 2004).
Myocardial infarction (MI) can induce normally quiescent adult mouse EPDCs to proliferate into mesenchymal cells via paracrine mechanisms, promoting subepicardial angiogenesis (Zhou et al. 2011). When treated with thymosin β-4 (Tβ4), explant culture of adult murine epicardium produces outgrowth of cells that differentiate into fibroblasts, smooth muscle cells, and endothelial cells (Smart et al. 2007). A sub-fraction of the resultant primed cells give rise to de novo cardiomyocytes after MI (Smart et al. 2011). Importantly, these cardiomyocytes are seen to structurally integrate with the resident myocardium via gap junctions and exhibit Ca2+ transients synchronous with resident cardiomyocytes, reflecting functional integration. Despite this promising finding, post-MI administration of Tβ4 could not induce EPDCs to adopt cardiac phenotypes or migrate into the subepicardium (Zhou et al. 2012), and its use in MI has not been validated (Smart et al. 2013). Small animal studies are still focused on improving differentiation rates with novel drugs.
Explant culture of human epicardium derived from atrial appendages yields c-Kit−/Islet-1− epithelioid cells with a “cobblestone” appearance before transitioning to a spindle shaped appearance in subculture, mimicking the EMT process. These cells demonstrate ability to differentiate into osteoblasts in vitro, but not endothelial cells nor adipocytes, and therefore appear to have limited plasticity (van Tuyn et al. 2007). When treated with bone morphogenic protein-2 or transforming growth factor-β1, they differentiate to form smooth muscle cells with high efficiency (van Tuyn et al. 2007). In contrast, rat EPDCs attain smooth muscle cell phenotype after stimulation with platelet-derived growth factor with less than 5 % efficiency (Wada et al. 2003). It is unknown whether this is due to differences in intrinsic commitment of these progenitor cells or in the signalling molecule, and whether another molecule such as Tβ4 can induce differentiation into cardiomyocytes. Winter et al. (2009) injected human EPDCs with and without human cardiac progenitor cells marked by Sca-1 into infarcted mouse myocardium and documented improved ejection fraction which is most marked with co-injection. The engrafted cells remained undifferentiated but acted through paracrine channels to increase the vascularity of the infarct area. Di Meglio et al. demonstrated that ischaemia causes mesothelial cells to be lost from the epicardium of adult human hearts while promoting relative accumulation of cells positive for mesenchymal gene products M-cadherein and vimentin in the subepicardium. A subset of EPDCs derived from epicardial mesothelial cells were c-Kit+ and migration and transition of EPDCs into c-Kit+ cardiac progenitor cells (CPCs) is a possible mechanism of endogenous heart regeneration following MI (Di Meglio et al. 2010). A c-Kit+/CD34+/CD45− group of EPDCs has also been isolated in human epicardium, although it is uncertain whether they represent the same population as myocardial derived populations or those investigated in the epicardium of ischaemic hearts (Limana et al. 2007).
Islet-1+ cardiac progenitor cells
Islet-1 (Isl-1) is a transcription factor of the LIM domain family and a marker of the progenitor cells of the second heart field (SHF). The majority of these cells appear to develop from Isl-1+ progenitors, since extensive defects of the atria, right ventricle, and outflow tract occur in mice lacking Isl-1 (Cai et al. 2003). In vivo, Isl-1+ cells give rise to a variety of cardiac lineages including cardiomyocytes, endothelial cells, smooth muscle cells and also pacemaker cells (Moretti et al. 2006; Sun et al. 2007). Ex vivo culture induced differentiation to all three cardiac lineages (Moretti et al. 2006), with cardiomyocytes exhibiting mature phenotype with synchronised [Ca2+] transients (Domian et al. 2009; Laugwitz et al. 2005). After MI, reperfusion induced up-regulation of Isl-1 transcripts points to a possible role in cardiac repair (Genead et al. 2012).
In order for Isl-1+ cells to play a significant role in cardiac regeneration of the adult heart, they must be present at this stage. Expression of Isl-1 declines progressively, and relatively few are present at birth (Bu et al. 2009; Cai et al. 2003; Genead et al. 2010), and this has been attributed to the loss of the Isl-1 marker with commitment to cardiac lineage. However, Genead et al. detected Isl-1+ cells in the outflow tract and its junction with the ventricle, and the inflow tract of the right atrium in young adult rats, some of which actively proliferate into cardiomyocytes (Genead et al. 2010). Subsequently, Khattar et al. characterised in adult mice two different clusters of Isl-1+ cells in mitotically quiescent states: one formed cardiac ganglia, while the other contributed to the cardiomyocyte lineage (Khattar et al. 2011). In order to conclusively characterise these cells in the murine heart, Weinberger et al. examined serial cryosections of adult Isl-1-nLacZ knock-in mice between 10 weeks and 18 months old (Weinberger et al. 2012). Isl-1+ cells were found in four regions and additionally expressed markers for smooth muscle, parasympathetic ganglia, cardiomyocytes and pacemaker cells, respectively. However, no undifferentiated Isl-1+ cells were found. Isl-1+ cells were not found in the ventricular myocardium, and MI did not lead to expansion of Isl-1+ cells in the infarct area, making their role in regeneration questionable (Sussman 2012; Weinberger et al. 2012).
Early in the discovery of Isl-1 progenitor cells, Laugwitz and co-workers identified these cells as distinct from c-Kit+ and Sca-1+ cells (Laugwitz et al. 2005), but more recently Isl-1+/c-Kit+ cells were reported in foetal and adult human hearts (Fuentes et al. 2013; Serradifalco et al. 2011). Distribution of Isl-1+ cells in the human foetus (Bu et al. 2009) matched that of the mouse (Cai et al. 2003; Moretti et al. 2006) and concentrated in the right atrium, the outflow tract as well as the left atrium and atrial appendage. This reflected their embryological relationship to the SHF. Pandur et al. recently reviewed the role of Isl-1+ progenitors across a number of species and advocated Isl-1+ cells as a common cardiac progenitor for both the SHF and the FHF (Pandur et al. 2013). Studies have found that, in both human embryonic and adult hearts, all progenitor cells expressing Isl-1 also expressed c-Kit, suggesting that Isl-1+ cells may be a subpopulation of the c-Kit+ population (Fuentes et al. 2013; Serradifalco et al. 2011). The relationship between Isl-1 progenitors and Sca-1 is similarly unclear. Interestingly, Sca-1+ cardiospheres also show enrichment in Isl-1+ cells (Ye et al. 2012).
The central role of Isl-1 in heart development was investigated as a cause of congenital heart disease but with conflicting results (Luo et al. 2014; Stevens et al. 2010; Xue et al. 2012). Isl-1+ cardiac progenitor cells are a population worth further investigation, and studies sourcing a similar population from human embryonic stem cells have been performed (Moretti et al. 2010). Fundamental questions about human Isl-1+ CPCs have yet to be answered, including their distribution in both healthy and diseased adult hearts, their relationship with other identified CPCs including c-Kit+ cells, and ultimately their therapeutic value in heart failure.
Sca-1+ stem cells
Stem cell antigen-1 (Sca-1) is a surface protein of the Ly-6 family (Vanderijn et al. 1989) commonly used to purify mouse haematopoietic stem cells (Holmes and Stanford 2007). Similar to c-Kit, Sca-1 is widely expressed by a range of organs (Holmes and Stanford 2007), and therefore Sca-1 selection enriches the “stemness” of a population without being specific for stem cells. A significant proportion of non-myocytes within the murine heart express Sca-1 (Oh et al. 2003; Rosenblatt-Velin et al. 2005), with a maximal density in the atria and ventricular apex that decreases with age (Saravanakumar and Devaraj 2013). While the original study by Oh et al. (2003) found that Sca-1+ progenitor cells were negative for haematopoietic lineage markers CD45, CD34 and c-Kit, different subpopulations were found that were variably positive for CD31, CD38, CD44 (Huang et al. 2011; Meinhardt et al. 2011; Oh et al. 2003; Rosenblatt-Velin et al. 2005), Isl-1 (Takamiya et al. 2011) and even c-Kit (Uchida et al. 2013). Sca-1+ CPCs were shown to differentiate in vitro into endothelial cells and also into functional cardiomyocytes after co-culture with cardiomyocytes or oxytocin treatment (Matsuura et al. 2004; Pfister et al. 2005). In vivo, transplanted Sca-1 stem cells differentiate into both endothelial and cardiomyocytes and improve function post-MI (Takamiya et al. 2011; Wang et al. 2006). Subpopulations have differing lineage potential, and Sca-1+/CD31− cells differentiate primarily into cardiomyocytes in vivo (Pfister et al. 2005; Wang et al. 2006), while Sca-1+/CD31+ cells do not (Pfister et al. 2005).
Although both infused (Oh et al. 2003) and endogenous (Liu et al. 2013) Sca-1+ CPCs homed to the infarct border, in both cases cells show a limited ability to differentiate into cardiomyocytes (Uchida et al. 2013; Wang et al. 2006). Therefore, any improvements in cardiac function conferred by Sca-1+ CPCs may occur mainly via a paracrine route (Huang et al. 2011; Matsuura et al. 2009). Treatment with CPCs or CPC-conditioned culture medium prior to MI acutely improved cardiac function, while treatment with differentiated CPCs failed to show any effect. This early phase effect is attributed to preservation of myocardial tissue by reducing apoptosis, although the reported mechanism differs between groups (Huang et al. 2011; Tateishi et al. 2007). Knockdown of Sca-1 expression reduced colony formation and expansion in vitro, and retarded engraftment and proliferation in vivo (Tateishi et al. 2007). Sca-1+ cells contribute to myocyte turnover during physiological aging (Uchida et al. 2013) and coordinate differentiation of other precursor cells (Rosenblatt-Velin et al. 2011). The importance of Sca-1+ cells in physiological maintenance is underscored by impaired function of c-Kit+ CPCs, development of dilated cardiomyopathy and impairment of heart function in knock-out mice (Bailey et al. 2012; Rosenblatt-Velin et al. 2011).
No human homolog of Sca-1 exists (Holmes and Stanford 2007). Van Vilet and co-workers used anti-Sca-1 antibody to bind an unknown antigen and identified a group of cells with cardiomyogenic potential from atrial biopsies (van Vliet et al. 2008). They further characterised this population in foetal and adult human hearts and demonstrated more mature phenotype in cardiomyocytes derived from adult Sca-1+ cells, which also exhibit less spontaneous contractions than the foetal cells. Adult cells generated more cardiomyocytes and smooth muscle cells, whereas foetal cells preferred endothelial lineage (van Vliet et al. 2010). The method of using Sca-1 to identify human CPCs are still unvalidated. Obvious questions remain as to the exact nature of the epitope target of Sca-1 in humans, and the regenerative potential of these cells.
Side population cells
Side population cells (SPCs) are distinguished by their ability to efflux Hoechst dye due to the presence of membrane ABC transporter proteins (Ding et al. 2010; Zhou et al. 2001). First discovered in the murine haematopoietic system (Goodell et al. 1996), SPCs have since been identified in a number of organs (Ding et al. 2010), including the murine (Hierlihy et al. 2002; Martin et al. 2004; Yoon et al. 2007) and human (Emmert et al. 2013; Meissner et al. 2006; Sandstedt et al. 2012) hearts. SPCs are enriched in Sca-1+ cells while being low in CD34, CD45 and c-Kit (Martin et al. 2004; Pfister et al. 2005).
Cardiac side population cells (CSPCs) constitute a complicated mixture of endothelial and smooth muscle cells of the heart, and also promulgate mesenchymal lineage potential (Yamahara et al. 2008). Under co-culture with cardiomyocytes, mouse CSPCs demonstrated myotube formation (Hierlihy et al. 2002), and α-actinin expression (Martin et al. 2004; Pfister et al. 2005), while expression of Nkx2.5 varied with time after isolation and between subpopulations (Martin et al. 2004; Yamahara et al. 2008). Spontaneous contraction in vitro was achieved by some (Oyama et al. 2007; Pfister et al. 2005), but not others (Yamahara et al. 2008). CD31−/Sca-1+ CSPCs mark a subpopulation with cardiomyogenic potential that is the most well studied. Evidence exists for the homing of this subpopulation to the infarct area post-MI, followed by differentiation into all 3 cardiac lineages with limited efficiency (Liang et al. 2010; Oyama et al. 2007). Liang et al. additionally characterized the CD31+/Sca-1+ cells found in cardiac vasculature, and established their capacity to migrate to the ischaemic myocardium and differentiate into endothelial cells (Liang et al. 2011), confirming earlier in vitro studies (Yoon et al. 2007). It is known that, after an MI, there is an acute depletion of CSPCs followed by gradual replenishment by bone marrow-derived SPCs (Mouquet et al. 2005). Whether the small numbers of differentiated cells generated from CSPCs significantly contribute to function is unknown.
There is a gap in the literature with regards to large animal and human studies of CSPCs, and current studies have been limited in their scope. Immunohistochemistry of the human endomyocardial biopsy samples reveal highest levels of CSPCs in the left atria and elevation with ischaemia (Emmert et al. 2013; Sandstedt et al. 2012). Unlike murine CSPCs, these cells did not differentiate into functional cardiomyocytes in vitro. Further research will reveal whether human CSPCs can attain cardiomyogenic potential and improve function in heart failure.
Cardiospheres
Cardiospheres (CSs) can be derived from murine and human hearts as an explant culture of myocardial tissue on poly-D-lysine-coated multiwell plates (Messina et al. 2004). They refer to clusters of small, round, phase bright cells that emerge from the explant and are clonogenic, self-renewing and differentiable into cardiomyocytes, endothelial cells, and smooth muscle cells, both in vitro and in vivo (Davis et al. 2009; Messina et al. 2004). CSs are composed of a mixture of stem cells in their core while progenitor cells, and more differentiated lineage cells aggregate at the periphery to form a niche-like environment (Li et al. 2010). Markers for cardiospheres include those for progenitor cells (c-Kit, Sca-1), endothelial cells (CD31, CD34, CD105, CD133), mesenchymal cells (CD90, CD105) and cardiomyocyte-related proteins (Cx43, Nkx2.5, αMHC) (Davis et al. 2009; Messina et al. 2004; Ye et al. 2012). While murine CSs beat spontaneously soon after formation, human CSs require co-culture with rat cardiomyocyte before they can contract. In ascertaining the origins of CSs, it was posited that contractions may be due to contaminating cardiomyocytes and that CSs form by aggregation rather than expansion (Andersen et al. 2009). These propositions were later refuted by Davis and co-workers, who also demonstrated that small variations in culture conditions can greatly impact on the phenotype and yield of cardiospheres (Davis et al. 2009).
Smith et al. (2007) used percutaneous biopsies to obtain human myocardial samples and generated cardiosphere derived cells (CDCs) by plating CSs onto fibronectin-coated plates. CDCs are now commonly used due to the relative ease by which they are produced and expanded (Aminzadeh et al. 2014; Chan et al. 2012; Hsiao et al. 2014; Makkar et al. 2012). CDCs come from a more homogeneous population of cells, mainly of a stromal phenotype with high expression of CD105 along with reduced expression of CD45 (Makkar et al. 2012; Marban and Cingolani 2012). However, after prolonged culture, CDCs also adopt a heterogeneous phenotype (Davis et al. 2009). CDCs can be delivered via an intracoronary route without infarction, whereas CSs require intramyocardial injection (Johnston et al. 2009). They also do not exhibit major histocompatibility class II (MHCII) antigen nor B7 costimulatory molecules, and it is hoped that allogenic CDCs can one day be used as “off the shelf” products in heart failure (Malliaras et al. 2012), a premise investigated in the ongoing ALLSTAR clinical trial (NCT01458405).
Ageing and non-ischaemic cardiac disease impact on the quantity and regenerative potential of CDCs, while transplantation of CDCs may also alleviate the functional decline associated with these conditions. Allogenic transplantation of CDCs in a murine model of dilated cardiomyopathy showed promising results and again hint at the potential of CDCs as an allogenic therapeutic (Aminzadeh et al. 2014). Heart samples from neonatal (<30 days) congenital heart disease patients express three times the level of c-Kit seen in those >2 years of age, with the right atrium showing the greatest proliferative capacity (Mishra et al. 2012). This finding agrees with murine models of ageing, which also show decreased c-Kit, Sca-1, and reduced CDC proliferation, migration and differentiation (Hsiao et al. 2014). Interestingly, Mdx mice, which exhibit some of the phenotype of Duchenne muscular dystrophy, do not experience decline in either quantity or capacity of derived CDCs (Hsiao et al. 2014). In contrast, ventricle-derived CDCs from golden retriever muscular dystrophy (GRMD) dogs have reduced self-renewal and myogenic potential compared to healthy controls (Cassano et al. 2012). Better understanding of the disease process in humans would help resolve these conflicting findings and help transform current treatment.
CSs and CDCs have proven beneficial in ischaemic cardiomyopathy in many murine and porcine studies by increasing cardiac function while minimising adverse remodelling, MI mass and scar size (Johnston et al. 2009; Lee et al. 2011; Shen et al. 2012; Tseliou et al. 2013, 2014; Ye et al. 2012). However, direct differentiation of the stem cells into cardiomyocytes appears to be a minor contributor and generated cardiomyocytes lacked calcium transients (Chimenti et al. 2010; Malliaras et al. 2013; Shenje et al. 2008; Ye et al. 2012). Instead, paracrine mediators such as VEGF, HGF and IGF1 appear to preserve cardiac function by enhancing cardiomyocyte proliferation, increasing angiogenesis, and decreasing apoptosis (Chimenti et al. 2010; Malliaras et al. 2013; Tseliou et al. 2014). This hypothesis is supported by observations that retention of injected stem cells is less than 1 % at 3 weeks (Aminzadeh et al. 2014; Tseliou et al. 2014). Improving engraftment has been shown to increase functional benefit of stem cells (Cheng et al. 2010). Although CDCs may be more easily prepared, their efficacy compared to CSs have been questioned. A porcine infarct model demonstrated the superiority of SCs over CDCs in improving regional function and ventricular remodelling (Lee et al. 2011). Murine CS culture enhances the expression of c-Kit (taken as a marker of “stemness”), and shows greater improvement in left ventricular ejection fraction (LVEF) than CDCs (Li et al. 2010). The significance of enhanced c-Kit expression in CSs was recently questioned when c-Kit+-purified CDCs exhibited reduced potency both in terms of functional benefit and paracrine secretion compared to the unrefined mixture (Li et al. 2012). The authors suggest that mesenchymal and stromal cells present within the heterogeneous cells present within CSs and CDCs act synergistically with cardiac progenitors cells after exogenous delivery to the injured heart. Therefore, removal of these cells during c-Kit purification leads to decreased paracrine effects of transplanted cells. Nevertheless, CDCs have superior myogenic potency, angiogenesis, and paracrine effects in vitro and in vivo compared to marrow and adipose mesenchymal stem cells, as well as bone marrow-derived mononuclear cells (Li et al. 2012). CDCs from heart failure patients have improved engraftment, and induce greater endogenous repair than equivalent cells from healthy donors (Cheng et al. 2014). Beyond the application of CSs and CDCs, secondary cardiospheres have been cultured from CDCs in 3-dimensional culture and may have an event greater therapeutic effect than CSs (Cho et al. 2012).
The CADUCEUS phase I clinical trial found that intracoronary CDCs transplanted into patients with post-MI ventricular dysfunction significantly reduced scar size but did not improve LVEF or heart failure symptoms. Better understanding of the relative contributions of each subgroup of cells in CS and CDCs will enlighten with regards to their comparative efficacy, and improved outcomes in future studies may result.
PDGFRα+ progenitor cells
Bone marrow colony-forming unit-fibroblasts (CFU-Fs) are a group of perivascular cells that grow in vitro to form mesenchymal stromal cells (Friedenstein et al. 1970). Perivascular cells resembling CFU-Fs reside in a number of solid organs (Crisan et al. 2008) and exhibit multipotent differentiation potential, including myogenicity in long-term culture. This population isolated from both murine (Chong et al. 2011) and human (Chong et al. 2013) hearts was enabled by expression of the tyrosine kinase, platelet-derived growth factor receptor α (PDGFRα). Comprehensive lineage tracing and bone marrow transplantation studies have shown these cells to be distinct from their bone marrow-derived counterpart (Slukvin 2011). Interestingly, there is significant overlap in gene expression, differentiation potential and immunophenotype between CFU-Fs from different organs, with important differences related to tissue-specific roles (Pelekanos et al. 2012). It is also possible that cardiac CFU-Fs are a subpopulation of the cardiac fibroblastic pool (Furtado et al. 2014). The distinction between fibroblasts and mesenchymal stromal cells are not well characterised and in vitro they are indistinguishable (Bianco et al. 2008; Hematti 2012). Both cardiac fibroblasts and cardiac CFU-Fs originate from the proepicardium, and undergo EMT before populating the subepicardium and myocardial interstitium where they adopt a perivascular location (Chong et al. 2011; Krenning et al. 2010).
Although murine PDGFRα+ progenitor cells can differentiate into cardiomyocytes when co-cultured with neonatal rat cardiomyocytes or after transplantation into the infarcted heart, it is unclear whether this cardiomyogenic role is dominant during normal cardiac homeostasis (Chong et al. 2009, 2011). In human hearts, PDGFRα+ progenitors were predominantly the smooth muscle cells of coronary vessels and interstitial cells throughout the ventricles. Co-expression with troponin-T was rare, suggesting minimal myogenesis (Chong et al. 2013). Interestingly, in hearts from patients with heart failure, PDGFRα+ cardiomyocytes were more frequent than in hearts from patients without overt heart failure (unpublished data, not shown). Following our previous work, we have recently isolated cardiovascular cells from cryopreserved donor left ventricles from an established tissue bank (see below). Explant culture of this tissue in media containing high serum yields spindle-shaped PDGFRα+ cells (data not shown). Detailed characterisation of their biological properties and their relationship to other cardiac progenitor cell types is currently ongoing. From a translational perspective, the regenerative potential of these PDGFRα+ cardiac progenitor cells is currently being tested in small and large animal models of heart failure.
Pluripotent Stem Cells
Another category of stem cells requires mentioning, although detailed discussion is beyond the scope of this review. Human pluripotent stem cells have the ability to form bona fide cardiomyocytes that robustly form spontaneously contracting syncytia after in vitro differentiation. Resulting cells can be used as a potentially unlimited source of cardiomyocytes for replacement therapy to treat heart failure. Transplantation studies of human embryonic stem cell-derived cardiomyocytes (hESC-CMs) in rodents and non-human primates have shown encouraging results in terms of both regeneration of new myocardium and improvement of cardiac function in infarcted hearts [Shiba et al. 2012; Chong and Murry 2014; Chong et al. 2014]. hESC-CM, however, require further preclinical optimisation and are currently still some time from clinical trials.
Cardiac stem cells and tissue banking
The clinical application of human heart tissue-derived multipotent CPCs requires expansion, cryopreservation, and transportation from the laboratory to the site of cell implantation. Thus, the preferred setting for tissue engineering and regenerative medicine applications is the availability of human samples and cell products. Current studies have relied on the availability of fresh heart samples, often obtained during interventional procedures on diseased hearts. Donor heart tissue is not always available for use and cryopreserved tissue represents an alternative approach. However, there are few published data characterising CPCs isolated from cryopreserved heart tissue.
The approach to isolation and preparation of stem cells with cryopreserved tissue has been successfully demonstrated in various tissues including human periodontal ligament, cord and adipose tissue (Choudhery et al. 2013, 2014; Seo et al. 2005). In these studies, mesenchymal stem cells (MSCs) isolated from fresh and frozen tissue were capable of differentiating into adipogenic, chondrogenic, osteogenic and neurogenic lineages, and no significant functional differences were observed. Furthermore, the cryopreservation and thawing process of MSCs did not alter the fundamental characteristics (growth properties, phenotypes and gene expression patterns, as well as similar differentiation potential in vitro) of these cells (Mamidi et al. 2012). In contrast to fresh tissue sources, harvesting CPCs from frozen tissue is a non-invasive procedure and poses no risk to the donor.
In order to facilitate utilisation of cryopreserved heart tissue, human bio-repositories containing a sufficient range of failing hearts (dilated, hypertrophic and ischaemic cardiomyopathies) and non-failing hearts is required. These are from hearts procured but not used for orthotopic heart transplantation. The broad spectrum of pre-phenotyped tissue will expedite pre-clinical biological studies. Non-diseased cryopreserved cardiac tissue will provide a readily available source of cardiac progenitor cells for allogeneic cell transplantation albeit with necessary implications of immune suppression. Success using this human cardiac tissue for regenerative studies relies on there being no significant degradation between the last heartbeat and the start of an experiment. All tissues should be frozen within minutes and maintained at liquid nitrogen temperature (−196 °C) to preserve tissue quality (intact mRNA, no proteolysis). Under these conditions, our experience (Fig. 2) is that the quality of the samples does not deteriorate even after 20 years of storage. In contrast, tissue stored at −80 °C appears to undergo slow degradation over a period of 6–12 months. Given the merits of such an approach and the large number of successful collaborative projects sourcing from the Sydney Heart Bank (Li et al. 2013), we believe that cryopreserved heart tissue will prove an invaluable resource for future stem cell-mediated therapies.

The Sydney Heart Bank currently contains about 20,000 tissue samples from approximately 600 human hearts. Of these, 190 are from healthy organ donors and the remainder are from failing hearts. The upper cluster of the figures shows the donor hearts (blue) and the major subsets of failing hearts (red). The lower cluster provides more detail about the types of tissue (myocardium, vessels) obtained from these hearts. IDCM idiopathic dilated cardiomyopathy, FDCM familial dilated cardiomyopathy, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, IHD ischaemic heart disease, RV right ventricle, RA right atrium, LV left ventricle, LA left atrium, RCA right coronary artery, LCA left coronary artery
Conclusion
Although the existence of resident cardiac stem cells has now been validated in a large number of animal and human studies, many questions remain as to their origin, developmental relationships, and influence in heart regeneration. Variation with both age and disease and inter-individual diversity poses particular challenge to the resolution of these issues. It is essential that these be addressed as they have direct therapeutic implications.
Over the last decade, attempts to treat patients with various cardiac stem cells from peripheral blood, bone marrow (Fisher et al. 2014) and skeletal myoblasts (Menasche et al. 2008) have produced varied results with no conclusive evidence of clinically significant regeneration. More recently, encouraging results from clinical trials using human cardiac progenitors such as c-Kit+ cells (Bolli et al. 2013) and CDCs (Makkar et al. 2012; Takehara et al. 2012) have emerged. An alternative approach to exogenous cell is to stimulate intrinsic mechanisms of regeneration. Biological insights into the varied cardiac progenitor populations will be required for meaningful progress in either of these approaches.
Notes
Lineage markers are found on mature haematopoietic cells and lineage depletion selects for a subpopulation enriched in early progenitor cells not yet expressing lineage markers
Reference
Aminzadeh MA, Tseliou E, Sun B, Cheng K, Malliaras K, Makkar RR, Marbán E (2014) Therapeutic efficacy of cardiosphere-derived cells in a transgenic mouse model of non-ischaemic dilated cardiomyopathy. Eur Heart J. doi:10.1093/eurheartj/ehu196. Published Online First: 27 May 2014
Andersen DC, Andersen P, Schneider M, Jensen HB, Sheikh SP (2009) Murine “cardiospheres” are not a source of stem cells with cardiomyogenic potential. Stem Cells 27:1571–1581
Bailey B, Fransioli J, Gude NA, Alvarez R, Zhan XX, Gustafsson AB, Sussman MA (2012) Sca-1 knockout impairs myocardial and cardiac progenitor cell function. Circ Res 111:750–760
Bax NA et al (2011) Epithelial-to-mesenchymal transformation alters electrical conductivity of human epicardial cells. J Cell Mol Med 15:2675–2683
Bearzi C et al (2007) Human cardiac stem cells. Proc Natl Acad Sci U S A 104:14068–14073
Bearzi C et al (2009) Identification of a coronary vascular progenitor cell in the human heart. Proc Natl Acad Sci U S A 106:15885–15890
Beltrami AP et al (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114:763–776
Bianco P, Robey PG, Simmons PJ (2008) Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2:313–319
Bolli R et al (2013) Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378:1847–1857
Bu L et al (2009) Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 460:113–U130
Cai CL, Liang XQ, Shi YQ, Chu PH, Pfaff SL, Chen J, Evans S (2003) Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5:877–889
Carmona R, Guadix JA, Cano E, Ruiz-Villalba A, Portillo-Sanchez V, Perez-Pomares JM, Munoz-Chapuli R (2010) The embryonic epicardium: an essential element of cardiac development. J Cell Mol Med 14:2066–2072
Cassano M et al (2012) Alteration of cardiac progenitor cell potency in GRMD dogs. Cell Transplant 21:1945–1967
Castaldo C et al (2008) CD117-positive cells in adult human heart are localized in the subepicardium, and their activation is associated with laminin-1 and alpha6 integrin expression. Stem Cells 26:1723–1731
Chan HHL et al (2012) Human cardiosphere-derived cells from patients with chronic ischaemic heart disease can be routinely expanded from atrial but not epicardial ventricular biopsies. J Cardiovasc Transl Res 5:678–687
Cheng K, Li TS, Malliaras K, Davis DR, Zhang YQ, Marban E (2010) Magnetic targeting enhances engraftment and functional benefit of iron-labeled cardiosphere-derived cells in myocardial infarction. Circ Res 106:1570–U1554
Cheng K et al (2014) Human cardiosphere-derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. JACC Heart Fail 2:49–61
Chimenti I, Smith RR, Li T-S, Gerstenblith G, Messina E, Giacomello A, Marban E (2010) Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ Res 106:971–U304
Cho HJ et al (2012) Secondary sphere formation enhances the functionality of cardiac progenitor cells. Mol Ther 20:1750–1766
Chong JJH, Murry CE (2014) Cardiac regeneration using pluripotent stem cells - progression to large animal models. Stem Cell Res. doi:10.1016/j.scr.2014.06.005. Published Online First: 6 July 2014
Chong JJH et al (2009) Sca1+/CD31−/PDGFRα + cardiac stem cells are from an epicardial/mesodermal but not neural-crest, cardiomyocyte or bone-marrow origin. Heart Lung Circ 18(3):S3
Chong JJH et al (2011) Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 9:527–540
Chong JJH, Reinecke H, Iwata M, Torok-Storb B, Stempien-Otero A, Murry CE (2013) Progenitor cells Identified by PDGFR-alpha expression in the developing and diseased human heart. Stem Cells Dev 22:1932–1943
Chong JJH et al (2014) Human embryonic-stem-cell-derived cardiomyocytesregenerate non-humanprimate hearts. Nature 510:273
Choudhery MS, Badowski M, Muise A, Harris DT (2013) Utility of cryopreserved umbilical cord tissue for regenerative medicine. Curr Stem Cell Res Ther 8:370–380
Choudhery MS, Badowski M, Muise A, Pierce J, Harris DT (2014) Cryopreservation of whole adipose tissue for future use in regenerative medicine. J Surg Res 187:24–35
Christoffels VM, Grieskamp T, Norden J, Mommersteeg MTM, Rudat C, Kispert A (2009) Tbx18 and the fate of epicardial progenitors. Nature 458:E8–E9
Clark RA, McLennan S, Eckert K, Dawson A, Wilkinson D, Stewart S (2005) Chronic heart failure beyond city limits. Rural Remote Health 5:443
Cleland JGF (2000) Improving patient outcomes in heart failure: evidence and barriers. Heart 84:i8–i10
Crisan M et al (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313
Davis DR et al. (2009) Validation of the cardiosphere method to culture cardiac progenitor cells from myocardial tissue. Plos ONE 4
Di Meglio F et al (2010) Epithelial–mesenchymal transition of epicardial mesothelium is a source of cardiac CD117-positive stem cells in adult human heart. J Mol Cell Cardiol 49:719–727
Ding XW, Wu JH, Jiang CP (2010) ABCG2: A potential marker of stem cells and novel target in stern cell and cancer therapy. Life Sci 86:631–637
Dixon JA, Spinale FG (2009) Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail 2:262–271
Domian IJ et al (2009) Generation of functional ventricular heart muscle from mouse ventricular progenitor cells. Science 326:426–429
Ellison GM et al (2013) Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 154:827–842
Emmert MY et al (2013) Higher frequencies of BCRP + cardiac resident cells in ischaemic human myocardium. Eur Heart J 34:2830–2838
Fazel S et al (2006) Cardioprotective c-kit(+) cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Investig 116:1865–1877
Ferreira-Martins J et al (2012) Cardiomyogenesis in the developing heart is regulated by c-kit-positive cardiac stem cells. Circ Res 110:701–U171
Fisher SA, Brunskill SJ, Doree C, Mathur A, Taggart David P, Martin-Rendon E (2014) Stem cell therapy for chronic ischaemic heart disease and congestive heart failure. Cochrane Database Syst Rev. doi:10.1002/14651858.CD007888.pub2. Issue 4. Art. No.: CD007888. Published Online First: 29 April 2014
Friedenstein AJ, Chailakhjan RK, Lalykina KS (1970) The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet 3:393–403
Fuentes TI, Appleby N, Tsay E, Martinez JJ, Bailey L, Hasaniya N, Kearns-Jonker M (2013) Human neonatal cardiovascular progenitors: unlocking the secret to regenerative ability. PLoS ONE 8:e77464
Furtado MB et al (2014) Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ Res 114:1422–1434
Gambini E, Pompilio G, Biondi A, Alamanni F, Capogrossi MC, Agrifoglio M, Pesce M (2011) C-kit(+) cardiac progenitors exhibit mesenchymal markers and preferential cardiovascular commitment. Cardiovasc Res 89:362–373
Garbade J et al (2010) There is a clear distribution pattern of viable resident c-kit positive cardiac stem cells in the human heart in patients suffering from ischemic cardiomyopathy. Thorac Cardiovasc Surg 58:MP55
Genead R, Danielsson C, Andersson AB, Corbascio M, Franco-Cereceda A, Sylven C, Grinnemo KH (2010) Islet-1 cells are cardiac progenitors present during the entire lifespan: from the embryonic stage to adulthood. Stem Cells Dev 19:1601–1615
Genead R et al (2012) Ischemia-reperfusion injury and pregnancy initiate time-dependent and robust signs of up-regulation of cardiac progenitor cells. Plos ONE 7:e36804
Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC (1996) Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 183:1797–1806
Hackam DG, Redelmeier DA (2006) Translation of research evidence from animals to humans. JAMA 296:1727–1732. This systematic review graded animal studies published in leading journals on their quality, and followed them up to subsequent human trials. Only a third of these highly cited animal studies were met with successes in human translation. There is therefore an imperative to better assess preclinical research, and human tissues are one way of approximating this gap
Hartung T (2013) Look back in anger - what clinical studies tell us about preclinical work. ALTEX 30:275–291
He JQ, Vu DM, Hunt G, Chugh A, Bhatnagar A, Bolli R (2011) Human cardiac stem cells isolated from atrial appendages stably express c-kit. PLoS ONE 6:e27719
Hematti P (2012) Mesenchymal stromal cells and fibroblasts: a case of mistaken identity? Cytotherapy 14:516–521
Hierlihy AM, Seale P, Lobe CG, Rudnicki MA, Megeney LA (2002) The post-natal heart contains a myocardial stem cell population. FEBS Lett 530:239–243
Holmes C, Stanford WL (2007) Concise review: Stem cell antigen-1: Expression, function, and enigma. Stem Cells 25:1339–1347
Hong KU, Li QH, Guo YR, Patton NS, Moktar A, Bhatnagar A, Bolli R (2013) A highly sensitive and accurate method to quantify absolute numbers of c-kit plus cardiac stem cells following transplantation in mice. Basic Res Cardiol 108:346
Hong KU et al (2014) c-kit + cardiac stem cells alleviate post-myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PLoS ONE 9:e96725
Houser SR et al (2012) Animal models of heart failure: A scientific statement from the american heart association. Circ Res 111:131–150
Hsiao LC et al (2014) Murine cardiosphere-derived cells are impaired by age but not by cardiac dystrophic dysfunction. Stem Cells Dev 23:1027–1036
Huang C, Gu H, Yu Q, Manukyan MC, Poynter JA, Wang M (2011) Sca-1+ cardiac stem cells mediate acute cardioprotection via paracrine factor SDF-1 following myocardial ischemia/reperfusion. PLoS ONE 6:e29246
Jesty SA et al (2012) c-kit(+) precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci U S A 109:13380–13385
Johnston PV et al (2009) Engraftment, differentiation, and functional benefits of autologous cardiosphere-derived cells in porcine ischemic cardiomyopathy. Circulation 120:1075–U1095
Khattar P et al (2011) Distinction between two populations of islet-1-positive cells in hearts of different murine strains. Stem Cells Dev 20:1043–1052
Kooij V et al (2014) Sizing up models of heart failure: Proteomics from flies to humans. Proteomics Clin Appl 8:653–664
Krenning G, Zeisberg EM, Kalluri R (2010) The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 225:631–637
Laflamme MA, Murry CE (2011) Heart regeneration. Nature 473:326–335
Laugwitz KL et al (2005) Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433:647–653
Lee S-T et al (2011) Intramyocardial injection of autologous cardiospheres or cardiosphere-derived cells preserves function and minimizes adverse ventricular remodeling in pigs with heart failure post-myocardial infarction. J Am Coll Cardiol 57:455–465
Li TS et al (2010) Cardiospheres recapitulate a niche-like microenvironment rich in stemness and cell-matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells 28:2088–2098
Li TS et al (2012) Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere-derived cells. J Am Coll Cardiol 59:942–953
Li A et al (2013) Heart research advances using database search engines, Human Protein Atlas and the Sydney Heart Bank. Heart Lung Circ 22:819–826. The Sydney Heart Bank provides a wide range of both donor and diseased hearts and is the source of tissue for ongoing cardiac stem cell research. The paper explores the basic characteristics of the preserved hearts and summarises the successful collaborations to date
Liang SX, Tan TYL, Gaudry L, Chong B (2010) Differentiation and migration of Sca1+/CD31-cardiac side population cells in a murine myocardial ischemic model. Int J Cardiol 138:40–49
Liang SX, Khachigian LM, Ahmadi Z, Yang M, Liu S, Chong BH (2011) In vitro and in vivo proliferation, differentiation and migration of cardiac endothelial progenitor cells (SCA1(+)/CD31(+) side-population cells). J Thromb Haemost 9:1628–1637
Limana F et al (2007) Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ Res 101:1255–1265
Liu J, Wang Y, Du W, Yu B (2013) Sca-1-positive cardiac stem cell migration in a cardiac infarction model. Inflammation 36:738–749
Luo ZL et al (2014) Genetic variations of ISL1 associated with human congenital heart disease in Chinese Han people. Genet Mol Res 13:1329–1338
Makkar RR et al (2012) Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379:895–904
Malliaras K et al (2012) Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 125:100–U500
Malliaras K et al (2013) Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol Med 5:191–209
Mamidi MK et al (2012) Comparative cellular and molecular analyses of pooled bone marrow multipotent mesenchymal stromal cells during continuous passaging and after successive cryopreservation. J Cell Biochem 113:3153–3164
Marban E, Cingolani E (2012) Heart to heart: Cardiospheres for myocardial regeneration. Heart Rhythm 9:1727–1731
Marian AJ (2011) Modeling human disease phenotype in model organisms: "It's only a model!". Circ Res 109:356–359
Martin CM et al (2004) Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev Biol 265:262–275
Matsuura K et al (2004) Adult cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. J Biol Chem 279:11384–11391
Matsuura K et al (2009) Transplantation of cardiac progenitor cells ameliorates cardiac dysfunction after myocardial infarction in mice. J Clin Investig 119:2204–2217
Matuszczak S et al (2014) Characteristic of c-Kit + progenitor cells in explanted human hearts. Clin Res Cardiol 103:711–718
Meinhardt A, Spicher A, Roehrich ME, Glauche I, Vogt P, Vassalli G (2011) Immunohistochemical and flow cytometric analysis of long-term label-retaining cells in the adult heart. Stem Cells Dev 20:211–222
Meissner K et al (2006) The ATP-binding cassette transporter ABCG2 (BCRP), a marker for side population stem cells, is expressed in human heart. J Histochem Cytochem 54:215–221
Menasche P et al (2008) The myoblast autologous grafting in ischemic cardiomyopathy (MAGIC) trial - First randomized placebo-controlled study of myoblast transplantation. Circulation 117:1189–1200
Messina E et al (2004) Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res 95:911–921
Mishra R et al (2012) Characterization and functionality of cardiac progenitor cells in congenital heart patients. Circulation 123:364–373
Moretti A et al (2006) Multipotent embryonic Isl1(+) progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 127:1151–1165
Moretti A et al (2010) Mouse and human induced pluripotent stem cells as a source for multipotent Isl1(+) cardiovascular progenitors. FASEB J 24:700–711
Mouquet F et al (2005) Restoration of cardiac progenitor cells after myocardial infarction by self-proliferation and selective homing of bone marrow-derived stem cells. Circ Res 97:1090–1092
Nadal-Ginard B, Ellison GM, Torella D (2014) The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res. doi:10.1016/j.scr.2014.04.008. Published Online First: 29 April 2014
Oberpriller JO, Oberpriller JC (1974) Response of the adult newt ventricle to injury. J Exp Zool 187:249–253
Oh H et al (2003) Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A 100:12313–12318
Oyama T et al (2007) Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J Cell Biol 176:329–341
Pandur P, Sirbu IO, Kuhl SJ, Philipp M, Kuhl M (2013) Islet1-expressing cardiac progenitor cells: a comparison across species. Dev Genes Evol 223:117–129
Pelekanos RA et al (2012) Comprehensive transcriptome and immunophenotype analysis of renal and cardiac MSC-like populations supports strong congruence with bone marrow MSC despite maintenance of distinct identities. Stem Cell Res 8:58–73
Pfister O et al (2005) CD31(-) but not CD31(+) cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ Res 97:52–61
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA (2011) Transient regenerative potential of the neonatal mouse heart. Science 331:1078–1080
Poss KD, Wilson LG, Keating MT (2002) Heart regeneration in zebrafish. Science 298:2188–2190
Rehman J (2013) Bone marrow tinctures for cardiovascular disease lost in translation. Circulation 127:1935–1937
Rosenblatt-Velin N, Lepore MG, Cartoni C, Beermann F, Pedrazzini T (2005) FGF-2 controls the differentiation of resident cardiac precursors into functional cardiomyocytes. J Clin Investig 115:1724–1733
Rosenblatt-Velin N, Ogay S, Felley A, Stanford WL, Pedrazzini T (2011) Cardiac dysfunction and impaired compensatory response to pressure overload in mice deficient in stem cell antigen-1. FASEB J 26:229–239
Rota M et al (2008) Local activation or implantation of cardiac progenitor cells rescues scarred infarcted myocardium improving cardiac function. Circ Res 103:107–116
Saha K, Hurlbut JB (2011) Research ethics: Treat donors as partners in biobank research. Nature 478:312–313
Sandstedt J et al (2012) Left atrium of the human adult heart contains a population of side population cells. Basic Res Cardiol 107:255
Sandstedt J, Jonsson M, Dellgren G, Lindahl A, Jeppsson A, Asp J (2014) Human C-kit + CD45-cardiac stem cells are heterogeneous and display both cardiac and endothelial commitment by single-cell qPCR analysis. Biochem Biophys Res Commun 443:234–238
Saravanakumar M, Devaraj H (2013) Distribution and homing pattern of c-kit(+) Sca-1(+) CXCR4(+) resident cardiac stem cells in neonatal, postnatal, and adult mouse heart. Cardiovasc Pathol 22:257–263
Seo BM, Miura M, Sonoyama W, Coppe C, Stanyon R, Shi S (2005) Recovery of stem cells from cryopreserved periodontal ligament. J Dent Res 84:907–912. Periodontal ligament stem cells were retrieved from cryopreserved human tissue validating the approach of utilising frozen tissue as a source of stem cells. The authors also note preserved differentiation and surface expression of stem cell markers
Serradifalco C et al (2011) Embryonic and foetal Islet-1 positive cells in human hearts are also positive to c-Kit. Eur J Histochem 55:229–234
Shen DL, Cheng K, Marban E (2012) Dose-dependent functional benefit of human cardiosphere transplantation in mice with acute myocardial infarction. J Cell Mol Med 16:2112–2116
Shenje LT et al (2008) Lineage tracing of cardiac explant derived cells. Plos ONE 3:e1929
Shiba Y et al (2012) Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 489:322
Siller R, Greenhough S, Park IH, Sullivan GJ (2013) Modelling human disease with pluripotent stem cells. Curr Gene Ther 13:99–110
Slukvin I (2011) Epicardial origin of cardiac CFU-Fs. Cell Stem Cell 9:492–493
Smart N, Risebro CA, Melville AAD, Moses K, Schwartz RJ, Chien KR, Riley PR (2007) Thymosin beta 4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445:177–182
Smart N et al (2011) De novo cardiomyocytes from within the activated adult heart after injury. Nature 474:640–U117
Smart N, Dube KN, Riley PR (2013) Epicardial progenitor cells in cardiac regeneration and neovascularisation. Vasc Pharmacol 58:164–173
Smith RR et al (2007) Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 115:896–908
Stevens KN et al (2010) Common variation in ISL1 confers genetic susceptibility for human congenital heart disease. Plos ONE 5:e10855
Sun YF et al (2007) Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev Biol 304:286–296
Sussman MA (2012) Myocardial Isl(+) land a place with lots of rhythm, but no beat. Circ Res 110:1267–1269
Takamiya M, Haider KH, Ashraf M (2011) Identification and characterization of a novel multipotent sub-population of Sca-1(+) cardiac progenitor cells for myocardial regeneration. Plos ONE 6:e25265
Takehara N, Nagata M, Ogata T, Nakamura T, Matoba S, Gojo S, Sawada T, Yaku H, Matsubara H. (2012) The ALCADIA (Autologous Human Cardiac-derived Stem Cell To Treat Ischemic Cardiomyopathy) trial. AHA 2012
Tallini YN et al (2009) c-kit expression identifies cardiovascular precursors in the neonatal heart. Proc Natl Acad Sci U S A 106:1808–1813
Tang XL et al (2010) Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 121:293–U235
Tateishi K et al (2007) Clonally amplified cardiac stem cells are regulated by Sca-1 signaling for efficient cardiovascular regeneration. J Cell Sci 120:1791–1800
The Lancet E (2014) Expression of concern: the SCIPIO trial. Lancet 383:1279
Tseliou E et al (2013) Allogeneic cardiospheres safely boost cardiac function and attenuate adverse remodeling after myocardial infarction in immunologically mismatched rat strains. J Am Coll Cardiol 61:1108–1119
Tseliou E, de Couto G, Terrovitis J, Sun BM, Liu WX, Marban L, Marban E (2014) Angiogenesis, cardiomyocyte proliferation and anti-fibrotic effects underlie structural preservation post-Infarction by intramyocardially-injected cardiospheres. Plos ONE 9:e88590
Uchida S et al (2013) Sca1-derived cells are a source of myocardial renewal in the murine adult heart. Stem Cell Reports 1:397–410
Urbanek K et al (2006) Stem cell niches in the adult mouse heart. Proc Natl Acad Sci U S A 103:9226–9231
van Berlo JH et al (2014) c-kit(+) cells minimally contribute cardiomyocytes to the heart. Nature 509:337–341
van Tuyn J et al (2007) Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells 25:271–278
van Vliet P et al (2008) Progenitor cells isolated from the human heart: a potential cell source for regenerative therapy. Neth Heart J 16:163–169
van Vliet P et al (2010) Foetal and adult cardiomyocyte progenitor cells have different developmental potential. J Cell Mol Med 14:861–870
Vanderijn M, Heimfeld S, Spangrude GJ, Weissman IL (1989) Mouse hematopoietic stem-cell antigen Sca-1 is a member of the LY-6 antigen family. Proc Natl Acad Sci U S A 86:4634–4638
Wada AM, Smith TK, Osler ME, Reese DE, Bader DM (2003) Epicardial/mesothelial cell line retains vasculogenic potential of embryonic epicardium. Circ Res 92:525–531
Wang XH, Hu QS, Nakamura Y, Lee J, Zhang G, From AHL, Zhang JY (2006) The role of the Sca-1(+)/CD31(-) cardiac progenitor cell population in postinfarction left ventricular remodeling. Stem Cells 24:1779–1788
Weinberger F et al (2012) Localization of Islet-1-positive cells in the healthy and infarcted adult murine heart. Circ Res 110:1303–U1395
Wessels A, Perez-Pomares JM (2004) The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat Rec A 276A:43–57
Winter EM et al (2009) A new direction for cardiac regeneration therapy: application of synergistically acting epicardium-derived cells and cardiomyocyte progenitor cells. Circ Heart Fail 2:643–653
Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien CL, Schultheiss TM, Orkin SH (2006) Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell 127:1137–1150
Xue L et al (2012) ISL1 common variant rs1017 is not associated with susceptibility to congenital heart disease in a Chinese population. Genet Test Mol Biomarkers 16:679–683
Yamahara K et al (2008) Heterogeneic nature of adult cardiac side population cells. Biochem Biophys Res Commun 371:615–620
Ye JQ et al (2012) Sca-1(+) cardiosphere-derived cells are enriched for Isl1-expressing cardiac precursors and improve cardiac function after myocardial injury. Plos ONE 7:e30329
Yoon J, Choi SC, Park CY, Shim WJ, Lim DS (2007) Cardiac side population cells exhibit endothelial differentiation potential. Exp Mol Med 39:653–662
Zhou S et al (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7:1028–1034
Zhou B et al (2008) Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454:109–U105
Zhou B et al (2011) Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Investig 121:1894–1904
Zhou B et al (2012) Thymosin beta 4 treatment after myocardial infarction does not reprogram epicardial cells into cardiomyocytes. J Mol Cell Cardiol 52:43–47
Acknowledgments
The authors are grateful for funding from the Bosch Institute.
Conflict of interest
Zijun Ge, Sean Lal, Thi YL Le, Cris dos Remedios, James JH Chong declare that they do not have any conflict of interest regarding the present manuscript.
Human and animal studies
This article does not contain any original studies with either human participants or with animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: Biophysics of Human Heart Failure
Rights and permissions
About this article

Cite this article
Ge, Z., Lal, S., Le, T.Y.L. et al. Cardiac stem cells: translation to human studies. Biophys Rev 7, 127–139 (2015). https://doi.org/10.1007/s12551-014-0148-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-014-0148-0