
Abstract
Hepatocellular carcinoma (HCC) is a common cancer of high mortality, mainly due to the difficulty in diagnosis during its clinical stage. Here we aim to find the gene biomarkers, which are of important significance for diagnosis and treatment. In this work, 3682 differentially expressed genes on HCC were firstly differentiated based on the Cancer Genome Atlas database (TCGA). Co-expression modules of these differentially expressed genes were then constructed based on the weighted correlation network algorithm. The correlation coefficient between the co-expression module and clinical data from the Broad GDAC Firehose was thereafter derived. Finally, the interactive network of genes was then constructed. Then, the hub genes were used to implement enrichment analysis and pathway analysis in the Database for Annotation, Visualization and Integrated Discovery (DAVID) database. Results revealed that the abnormally expressed genes in the module played an important role in the biological process including cell division, sister chromatid cohesion, DNA repair, and G1/S transition of mitotic cell cycle. Meanwhile, these genes also enriched in a few crucial pathways related to Cell cycle, Oocyte meiosis, and p53 signaling. Via investigating the closeness centrality of the interactive network, eight gene biomarkers including the CKAP2, TPX2, CDCA8, KIFC1, MELK, SGO1, RACGAP1, and KIAA1524 gene were discovered, whose functions had been indeed revealed to be correlated with HCC. This study, therefore, suggests that the abnormal expression of those eight genes may be taken as gene biomarkers of HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hepatocellular carcinoma (HCC), together with lung cancer and stomach cancer, is of the most prevalent and fatal malignancies [1,2,3]. The prevalence of HCC primarily arises from the spread of hepatitis virus particularly its subtype B and C. In addition, HCC also originates from Non-alcoholic fatty liver disease and alcoholic liver disease [4]. The pathogenesis at the molecular level is very complex, rendering it hard for diagnosing and treating HCC. Therefore, the 5 year survival rate for HCC patients is less or much less than 10% [5]. However, the survival rate would be substantially increased if surgical resection or liver transplantation was adopted in the early stage.
It is difficult to diagnosis HCC in its early stage by traditional histological examinations. A potentially effective method bypassing this shortage is to exploit the molecular mechanisms of differentially expressed genes. Identifying cancer-specific gene mutations was proved to be fruitful in discovering biological pathways, biomarkers and therapeutic targets [6]. At present, high-throughput transcriptomics analyses uncover the mutation genes in tumor cells and describe the characteristics of genomic rearrangements during tumor oncogenesis [7]. Microarray was applied to study the diversity of transcriptome in many studies. However, whole RNA sequencing (RNA-Seq) data took superiority compared with the microarrays which have many limitations in probe selection, probe hybridization kinetics, and background hybridization [8, 9]. Especially, the integration analysis of RNA-Seq, which have become popular, was utilized to discover the biomarkers and explore the pathogenesis in various cancer comprising breast cancer [10, 11], prostate cancer [12], ovary cancer [13], and lung cancer [14].
In recent years, the explorations on molecular biomarkers not only provided better comprehension for the pathogenesis of disease but a novel method for the diagnosis and targeted therapy [15,16,17]. Chen et al. [18] developed the dynamic network biomarkers based on complex disease, which was applied to acute lung injury, chronic hepatitis B and lymphoma. Shen et al. applied the network biomarker to accurate diagnosis, prediction of the Leukemia, and exploring the pathogenicity mechanisms of prostate cancer [19, 20]. Xavier et al. [21] discovered the potential biomarker for early identification of colon cancer. However, persuasiveness would be enhanced if the biomarker exploration was combined with clinical information.
In this paper, we studied the 424 RNA-Seq data of HCC from the cancer genome atlas database and the 377 clinical data of HCC from Broad GDAC Firehose. The differentially expressed genes were identified. Then, the co-expression module combined with clinical data of differentially expressed genes was then built, followed by the module with a high correlation to clinical information utilized for establishing gene interactive network. The network contains 103 nodes and 1156 edges. Besides, the critical module was used for enrichment and pathway analyses in DAVID database. We identified eight genes as the gene biomarkers of the early stage of HCC, consistent with and supported by the functions of the genes revealed previously.
2 Materials and Methods
2.1 Materials
50 normal cases and 374 tumor cases RNA-Seq data are supplied by TCGA database.Firehose (https://gdac.broadinstitute.org/runs/stddata__2016_01_28/data/LIHC/20160128) offers clinical information composed of 377 liver hepatocellular carcinoma (LIHC) cases. First, we chose the GDC tool to download the RNA-Seq data of 424 LIHC cases from TCGA. The expression matrix of RNA sequencing was extracted with a Perl script. Then, the GTF document contained the human genome and gene annotation downloaded from Ensemble. The expression matrix was annotated with Perl script combine GTF document.
The clinical data included 377 LIHC cases download from firehose database (Table 1). A part of the clinical data was selected, such as pathologic stage (pTNM). The clinical data information was numerically treated, where: For pTNM, the \(i\) corresponds to 1, the \(ii\) is to 2, the \(iii\)a is to 3, the \(iii\)b is to 4, the \(iii\)c is to 5, the \(iv\)a is to 6, the \(iv\)b is to 7.
2.2 Methods
2.2.1 Construction of Co-expression Modules
The co-expression module was constructed by the weighted gene co-expression network algorithm that the typical algorithm in system biology [22]. To set up the non-scale co-expression network to differentiate important modules and genes, the step-by-step network construction and module detection method were taking. The soft threshold power was equivalent to 6 and the minimal value of Module Size was equal to 50. In network analysis, a node represents a gene, it is important to distinguish the dissimilarity among nodes. In this work, the topological overlap matrix (TOM) was used for reflecting the similarity between two nodes.. It is defined as the following by Ravasz et al.[23].
\(A = [a_{ij} ]\) is the adjacency matrix, \(k_{i} = \sum\nolimits_{j = 1}^{n} {a_{ij} }\) equals the number of its direct connection to other nodes, \(l_{ij} = \sum\nolimits_{u} {a_{iu} a_{uj} }\) equals the number of nodes which both \(i\) and \(j\) are connected. Then, dissimilarity on the basis of topological overlap is represented as follow,
Modules were groups of genes with high correlation. In this work, the average linkage hierarchical clustering was used to construct the gene modules. A height cutoff value, cut the tree branches of hierarchical clustering, was chosen based on scale-free topology.
2.2.2 Correlation Between Gene Module and Clinical Data
The first row in right orthogonal matrice extracted from the singular value decomposition of module expression matrix was used to represent the property of module. It is assumed that \(B^{(K)} = (b_{ij}^{K} )\) equals the module expression matrix of \(K\)-th module, where the index \(i = 1,2, \ldots ,n\) indicates the number of the module genes and the index \(j\) indicates the number of the patient samples. The singular value decomposition \(B^{(K)}\) is marked as [24]
\(C_{1}\) and \(C_{2}\) are the orthogonal matrices respectively. The module eigengene \(E^{(K)}\) is represented by the first row of \(C_{2}\). Then, the Pearson correlation coefficient is
adopted to evaluate the relation between module eigengene and clinical data. Simultaneously, The statistical analysis of the student test guarantees the correlation significant in statistics.
2.2.3 Feature Genes and Genes Interactive Network
The Pearson correlation coefficient between 804 genes in the turquoise module and the turquoise module was calculated. The genes that their Pearson correlation coefficients are greater than 0.8 are called feature genes. 112 feature genes were utilized to the constructed gene interaction network. The gene interactive network was aimed to discover the biomarker of module genes. Then, the closeness centrality, one of the evaluation index described the nodes centrality in network analysis, was used for evaluating the gene biomarker. It is represented in the following:
\(d(y,x)\) is the distance between gene \(x\) and \(y\), \(N\) is the number of nodes in the network. Generally, the more central a node is, the closer it is to all other nodes.
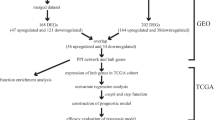
The process for gene biomarkers identification for hepatocellular carcinoma is illustrated in Fig. 1. Here we set up an analysis framework. At first, differential expression analysis screened genes that were differentially expressed in tumor samples. Next, co-expression network analysis was performed, and 13 gene modules were obtained by clustering. Then, the Pearson correlation coefficient between each module and clinical data was calculated, and the module with the largest Pearson correlation coefficient was selected as a representative expression module. After that, the gene interaction network of this module was constructed with a threshold of 0.01. The closeness centrality was used to evaluate the centrality of the network, and the biomarkers of hepatocellular carcinoma were determined. Furthermore, these genes were verified in GEO liver cancer data sets. Afterwards, the eight genes have no aberration in DNA methylation data. Finally, biomarkers were verified through enrichment analysis and pathway analysis and literature research. This workflow can directly determine biomarkers by bypassing the detection of clinical methods, saving a lot of time and effort. In addition, each step of the analysis process has statistical basis and corresponds well to biological significance. The process does not require too complicated steps and can obtain biologically meaningful results.

The flow chart of network biomarker identification for hepatocellular carcinoma
The basic assumptions of this analysis process:
-
1
Assuming that the gene expression data obeys normal distribution.
-
2
According to principal component analysis, assuming that the first column of the left-hand orthogonal matrix obtained by singular value decomposition represents module eigengene.
-
3
It is assumed that the gene module represents a highly similar genome.
-
4
It is assumed that Pearson’s correlation coefficient between the clinical data and the module reflects the correlation between each other.
3 Results
Here, the edge R package was selected to differentiate differentially expressed genes. Those genes that an average expression is less than one was removed as the noise. Then, log2 fold change and p value of all genes were calculated. To avoid statistical errors, adjusted p value was adopted with Benjamin-Hochberg method. The log2foldchange values that \(\ge\) 2 and \(\le\) − 2 were selected as obvious differential expression. Meanwhile, the adjusted p value \(\le\) 0.05 was adopted. There are 3697 differential expression genes that 3418 up-regulation and 279 down-regulation genes, and the specific situation is seen in Fig. 2.

Volcano plot of abnormally expressed genes in hepatocellular carcinoma. Red dots refer to high expression genes and green dots mean low expression genes. Black dots show normal expression genes in comparison. Altogether, 3418 up-regulation and 279 down-regulation were identified
3.1 Co-expression Network Module Analysis
Based on results from differential expression analysis, the co-expression module analysis that gene expression data of high similarity is gathered into the same gene module was performed. It aimed to find the similarity of gene expression. 13 gene modules (M1–M13) obtained by co-expression analysis are shown in Table 2. Furthermore, to find the hub gene, the similarity between each module eigengene and clinical data were calculated, the result is presented in Fig. 3. Three modules that M5 (p value = 1e − 04), M10 (p value = 0.04), and M12 (p value = 6e − 05) were marked. In this work, the turquoise module was regarded as the foremost module.

Co-expression module and heat-map with module-clinical relationships, where: a Co-expression module constructed based on abnormally expressed genes; b Heat-map between the module eigengene and the disease progression of HCC stage
3.2 Gene Interaction Network
Set the threshold to 0.01, gene interaction network which contains 103 nodes and 1156 edges was constructed. The visualization of gene interaction network was implemented using Cytoscape software. The network is exhibited in Fig. 4. Subsequently, the interaction network was evaluated with closeness centrality.

The genes interactive network for turquoise module. The bigger the closeness centrality, the bigger the circle and the brighter the color
The closeness centrality range is from 0.6 to 0.85, and the different closeness centrality corresponds to different gene biomarkers, as shown in Table 3. Clustering analysis using different gene biomarkers, situations of classification of diseased samples and normal samples are shown in Fig. 5. The accuracy rate \(R = m/n\) was used to evaluate the effectiveness of the classification. \(m\) is the sum of correctly classified diseased and normal samples, \(n\) is the quantity of all samples. Accuracy rate were 76.42%, 76.89%, 76.89%, 92.45%, 76.41%, and 91.98%, respectively, which corresponded to the closeness centers of 0.85, 0.80, 0.75, 0.70, 0.65, and 0.60, respectively. The gene biomarkers at the closeness centrality corresponding to the classification with the highest accuracy rate were selected as the gene biomarkers of hepatocellular carcinoma in this work. Besides, we used these eight biomarkers in GEO hepatocellular carcinoma datasets to classify normal samples and cancer samples and found that classification by eight gene biomarkers had a high accuracy rate. GSE94660 dataset has 42 samples, of which 21 are normal samples and 21 are cancer samples [25]. The corresponding classification accuracy rate is 100.00%. GSE121248 dataset has 107 samples, of which 37 are normal samples and 70 are cancer samples and its classification accuracy rate is 90.65% [26]. GSE124535 dataset has 70 samples, of which 35 are normal samples and 35 are cancer samples [27]. The classification accuracy rate is 88.57%. GSE138485 dataset has 64 samples, of which 32 are normal samples and 32 are cancer samples. Its classification accuracy rate is 87.50%. Afterwards, we downloaded the methylation data of hepatocellular carcinoma in TCGA. After extracting the methylation expression matrix, we found that all genes except for the KIAA1524 gene have methylation data. After differential expression analysis, it was found that no clear difference occurs in expression of seven genes in normal samples and cancer samples. Ultimately, the eight genes, CKAP2, TPX2, CDCA8, KIFC1, MELK, SGO1, RACGAP1, and KIAA1524, are discovered as the gene biomarkers of hepatocellular carcinoma.

The heat map of different genes biomarker. The red horizontal axis labels represent the normal samples; the blue horizontal axis labels represent tumor samples. a–f is the clustering by gene biomarkers corresponding to closeness centrality from 0.85 to 0.60 by the step 0.05, respectively
We found a correlation between modules and clinical staging. The eight gene biomarkers were selected from the modules with the largest Pearson correlation coefficient. We further analyzed the link between the classification ability of gene biomarkers and clinical staging. Due to the few quantities of samples in data set classified according to the small clinical stage, we classify cancer patients according to the large clinical stage that stage I, stage II, stage III, and stage IV. The quantity of samples in stage IV is too tiny, so the sample set in stage IV is excluded. We verified the classification ability of gene biomarkers in stages I, II and III, respectively, and the overall classification accuracy rate was above 80%. The accuracy rate for stage I is 90.60%.The accuracy rate for stage II is 81.82%. The accuracy rate for stage III is 81.69%. Accuracy rate of Stage I is largest, so these eight gene markers may play a major role in stage I cancer prediction.
3.3 Enrichment Analysis and Pathway Analysis
To study the function of turquoise module genes, enrichment analysis and pathway analysis was implemented based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). There are 172 significant enrichments in 3 GO categories including biological process (BP, 97), molecular functions (MF, 45), and cellular components (CC, 30). The top ten significant terms of biological process, molecular functions and cellular components are illustrated in Table 4.
There are nine terms pathway based on pathway analysis, as shown in Table 5. In this work, FDR method was adopted to reflect the statistical significance (p value < 0.05).
3.4 Discussion and Analysis
The enrichment analysis and pathway analysis was implemented by GO and KEGG. We find that the biological process of the turquoise module is mostly enriched in cell division, sister chromatid cohesion, DNA repair, cell proliferation, chromosome segregation, DNA replication, regulation of cell cycle, and G1/S transition of mitotic cell cycle. Meanwhile, half the gene biomarkers, KIFC1, CDCA8, TPX2, and SGO1 gene, are enriched in cell division. Genomic instability can raise the rate of tumorigenic mutations by cell division [28, 29]. A few of them, TPX2 and SGO1, are enriched in mitotic nuclear division. The TPX2, an important component of the spindle apparatus, leads to the defects of microtubule organization in mitotic [30]. In sister chromatid cohesion, the SGO1 and CACD8 gene ensure the accuracy of chromosomal separation to prevent chromosomal instability and tumorigenesis [31]. The enrichment analysis reveals that CDK1 exerts a enormous function on in DNA repair and DNA replication. DNA repair and DNA replication are associated with high histopathological grade and HCC oncogenesis [32, 33].
The biomarker of the SGO1 gene is enriched in DNA replication and chromosome segregation respectively. It has been revealed to be early events in the cancer process that DNA replication defect causes Chromosomal segregation abnormality [31, 34]. Furthermore, TPX2 and MELK are regulators of G1/S transition and G2/M transition in the mitotic cell cycle. There is evidence that hepatitis B virus X protein implicated the carcinogenesis of HCC inhibits activation of G1/S and G2/M [35].
Pathway analysis shows that genes belonging to the module are mostly enrich in Cell cycle, Oocyte meiosis, Progesterone-mediated oocyte maturation, the Fanconi anemia pathway, HTLV-I infection, MicroRNAs in cancer, Homologous recombination, p53 signaling pathway and FoxO signaling pathway. The evidence is revealed that CDKs family is one of participator in the cell cycle [36, 37]. The oocyte meiosis pathway regulates stem cell transformation, cancer development, and stem cell differentiation [38]. In this study, it is confirmed that the progesterone-mediated oocyte maturation pathway has a strong correlation with HCC [39]. Fanconi anemia mutations change homologous recombination in DNA repair, which can cause cell death. The defects of fanconi anemia increase cancer susceptibility to various cancer. Human T-lymphotropic virus is correlated with adult T-cell leukemia especially in Japan [40, 41]. HTLV-I infection is highly associated with the oncogenesis in HCC infected from chronic hepatitis virus [42]. Homologous recombination can promote the DNA double-strand breaks, explaining the genetic instability of cancer development. Besides, homologous recombination deficiency is regarded as potential therapeutic strategy in ovarian cancer and breast cancer [43,44,45]. p53 signaling pathway, which regulates tumor suppression and growth inhibition, pushes forward a immense influence on HCC carcinogenesis [46]. FOXO proteins promoted cell cycle arrest, stress resistance, and apoptosis by regulating many of the target genes. It inhibits tumor by translating environmental stimuli into the gene expression [47].
The gene cytoskeleton-associated protein 2 (CKAP2), which is relevant to the early stage and the extensive recurrence of HCC, takes significant functions in cell proliferation of mitosis. Besides, it shows up-regulation in various human malignancies including gastric carcinoma, diffuse large B-cell lymphomas, and breast cancer [48, 49]. The TPX2 gene, which contributes to tumor cell invasion by inactivation of AKT signaling, has an essential influence on mitotic nuclear division and cell proliferation [30]. The CDCA8 gene, which mainly functions to inhibit HCC cell proliferation, influence cell cycle progression, and induce apoptosis in HCC cells, is treated as a fresh therapeutic target [50]. The KIFC1 gene reduces HCC cell viability, promoting apoptosis and cell death. Due to high correlation with HCC, the KIFC1 gene is a therapeutic target and gene biomarker [51]. The mainly biological processes participated by MELK are apoptosis and mitosis. Low expression of MELK inhibits cell growth, invasion, stemness, and tumorigenicity. The MELK gene, which is a target gene of miR-214-3p that leads to cell proliferation reduction, cell cycle arrest, and cell apoptosis enhancement, is in close connection with the early stage of HCC [52, 53]. The SGO1 gene, which is correlated with the onset of HCC, takes key roles in mitosis. It guarantees the fidelity of chromosome separation by safeguarding sister chromatid cohesion [31]. The RACGAP1 gene participates in the processes of microtubule-based movement and mitotic cytokinesis. It is found that the RACGAP1 has correlation with pathological grading including T, N, and M stage and lymph node metastasis in HCC. It can be used as a biomarker in the T, N, and M stage and poor prognosis of HCC [54]. The KIAA1524 gene produces cancerous inhibitor of phosphatase 2A (CIP2A), which inhibits the activity of protein phosphatase 2A regulated phosphor-Akt and apoptosis in HCC [55, 56]. Besides, CIP2A facilitates the malignant growth in human breast cancer [57] and prevents cell growth, senescence, and differentiation in gastric cancer [58, 59].
4 Conclusions
In conclusion, the work integrated and exploited the RNA-Seq data and the clinical data of HCC. A total of eight genes were identified as the biomarkers of HCC. The functions of these genes were indeed revealed to be closely related to HCC. Of the eight genes, the CKAP2L, MELK, and SGO1 gene were confirmed to have a high correlation with the HCC. The RACGAP1 gene participates in the T, N, and M stage of HCC. The TPX2, CDCA8, KIFC1, and KIAA1524 gene play significant roles in the mitotic division, cell proliferation, cell cycle, apoptosis, and malignant growth, which begin in the early stage of HCC oncogenesis. Therefore, the abnormal expression of those eight genes may be taken as a mark of HCC. The work of searching key stage-specific biomarker will be carried out in the further.
References
Han LL, Lv Y, Guo H et al (2014) Implications of biomarkers in human hepatocellular carcinoma pathogenesis and therapy. World J Gastroenterol 20(30):10249–10261. https://doi.org/10.3748/wjg.v20.i30.10249
Kim JU, Cox IJ, Taylor-Robinson SD (2017) The quest for relevant hepatocellular carcinoma biomarkers. Cell Mol Gastroenterol Hepatol 4(2):283–284. https://doi.org/10.1016/j.jcmgh.2017.06.003
Miura T, Ban D, Tanaka S et al (2015) Distinct clinicopathological phenotype of hepatocellular carcinoma with ethoxybenzyl-magnetic resonance imaging hyperintensity: association with gene expression signature. Am J Surg 210(3):561–569. https://doi.org/10.1016/j.amjsurg.2015.03.027
Zhou C, Zhang W, Chen W et al (2017) Integrated analysis of copy number variations and gene expression profiling in hepatocellular carcinoma. Sci Rep 7(1):10570. https://doi.org/10.1038/s41598-017-11029-y
Wei L, Lian B, Zhang Y et al (2014) Application of microRNA and mRNA expression profiling on prognostic biomarker discovery for hepatocellular carcinoma. BMC Genomics 15(1):S13. https://doi.org/10.1186/1471-2164-15-S1-S13
Maher CA, Kumar-Sinha C, Cao XH et al (2009) Transcriptome sequencing to detect gene fusions in cancer. Nature 458:97–101. https://doi.org/10.1038/nature07638
Griffith M, Griffith OL, Mwenifumbo J et al (2010) Alternative expression analysis by RNA sequencing. Nat Methods 7:843–847. https://doi.org/10.1038/nmeth.1503
Maher CA, Palanisamy N, Brenner JC et al (2009) Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci 106(30):12353–12358. https://doi.org/10.1073/pnas.0904720106
McCarthy DJ, Chen Y, Smyth GK et al (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40(10):4288–4297. https://doi.org/10.1093/nar/gks042
Zhao Q, Caballero OL, Levy S et al (2009) Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc Natl Acad Sci 106(6):1886–1891. https://doi.org/10.1073/pnas.0812945106
Sinicropi D, Qu K, Collin F et al (2012) Whole transcriptome RNA-Seq analysis of breast cancer recurrence risk using formalin-fixed paraffin-embedded tumor tissue. PLoS ONE 7(7):e40092. https://doi.org/10.1371/journal.pone.0040092
Ren S, Peng Z, Mao JH et al (2012) RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res 22:806–821. https://doi.org/10.1038/cr.2012.30
Shah SP, Köbel M, Senz J et al (2009) Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med 360(26):2719–2729. https://doi.org/10.1056/NEJMoa0902542
Beane J, Vick J, Schembri F et al (2011) Characterizing the impact of smoking and lung cancer on the airway transcriptome using RNA-Seq. Cancer Prev Res 4(6):803–817. https://doi.org/10.1158/1940-6207.CAPR-11-0212
Liu J, Yu Z, Sun M et al (2019) Identification of cancer/testis antigen 2 gene as a potential hepatocellular carcinoma therapeutic target by hub gene screening with topological analysis. Oncol Lett 18(5):4778–4788. https://doi.org/10.3892/ol.2019.10811
Guo Y, Bao Y, Ma M et al (2017) Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int J Mol Sci 18(4):722. https://doi.org/10.3390/ijms18040722
Agarwal R, Narayan J, Bhattacharyya A et al (2017) Gene expression profiling, pathway analysis and subtype classification reveal molecular heterogeneity in hepatocellular carcinoma and suggest subtype specific therapeutic targets. Cancer Genetics 216:37–51. https://doi.org/10.1016/j.cancergen.2017.06.002
Chen L, Liu R, Liu ZP et al (2012) Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci Rep 2:342. https://doi.org/10.1038/srep00342
Li Y, Vongsangnak W, Chen L et al (2014) Integrative analysis reveals disease-associated genes and biomarkers for prostate cancer progression. BMC Med Genomics 7(1):S3. https://doi.org/10.1186/1755-8794-7-S1-S3
Yuan X, Chen J, Lin Y et al (2017) Network biomarkers constructed from gene expression and protein-protein interaction data for accurate prediction of leukemia. J Cancer 8(2):278–286. https://doi.org/10.7150/jca.17302
Solé X, Crous-Bou M, Cordero D et al (2014) Discovery and validation of new potential biomarkers for early detection of colon cancer. PLoS ONE 9(9):e106748. https://doi.org/10.1371/journal.pone.0106748
Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. https://doi.org/10.2202/1544-6115.1128
Ravasz E, Somera AL, Mongru DA et al (2002) Hierarchical organization of modularity in metabolic networks. Science 297(5586):1551–1555. https://doi.org/10.1126/science.1073374
Langfelder P, Horvath S (2007) Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol 1(1):54. https://doi.org/10.1186/1752-0509-1-54
Yoo S, Wang W, Wang Q et al (2017) A pilot systematic genomic comparison of recurrence risks of hepatitis B virus-associated hepatocellular carcinoma with low- and high-degree liver fibrosis. BMC Med 15:214. https://doi.org/10.1186/s12916-017-0973-7
Wang SM, Ooi LL, Hui KM (2007) Identification and validation of a novel gene signature associated with the recurrence of human hepatocellular carcinoma. Clin Cancer Res 13(21):6275–6283. https://doi.org/10.1158/1078-0432.CCR-06-2236
Jiang Y, Sun A, Zhao Y et al (2019) Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567:257–261. https://doi.org/10.1038/s41586-019-0987-8
Marte B (2004) Cell division and cancer. Nature 432:293. https://doi.org/10.1038/432293a
Liu C, Liu L, Chen X et al (2016) Sox9 regulates self-renewal and tumorigenicity by promoting symmetrical cell division of cancer stem cells in hepatocellular carcinoma. Hepatology 64:117–129. https://doi.org/10.1002/hep.28509
Liu Q, Yang P, Tu K et al (2014) TPX2 knockdown suppressed hepatocellular carcinoma cell invasion via inactivating AKT signaling and inhibiting MMP2 and MMP9 expression. Chin J Cancer Res 26(4):410–417. https://doi.org/10.3978/j.issn.1000-9604.2014.08.01
Wang LH, Yen CJ, Li TN et al (2015) Sgo1 is a potential therapeutic target for hepatocellular carcinoma. Oncotarget 6(4):2023–2033. https://doi.org/10.18632/oncotarget.2764
Gramantieri L, Trerè D, Chieco P et al (2003) In human hepatocellular carcinoma in cirrhosis proliferating cell nuclear antigen (PCNA) is involved in cell proliferation and cooperates with P21 in DNA repair. J Hepatol 39(6):997–1003. https://doi.org/10.1016/S0168-8278(03)00458-6
Lukish JR, Muro K, DeNobile J et al (1998) Prognostic significance of DNA replication errors in young patients with colorectal cancer. Ann Surg 227(1):51–56. https://doi.org/10.1097/00000658-199801000-00008
Martin-Lluesma S, Schaeffer C, Robert EI et al (2008) Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 48:1467–1476. https://doi.org/10.1002/hep.22542
Wu BK, Li CC, Chen HJ et al (2006) Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem Biophys Res Commun 340(3):916–928. https://doi.org/10.1016/j.bbrc.2005.12.089
Zhang L, Guo Y, Li B et al (2013) Identification of biomarkers for hepatocellular carcinoma using network-based bioinformatics methods. Eur J Med Res 18(1):35. https://doi.org/10.1186/2047-783X-18-35
Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9:153–166. https://doi.org/10.1038/nrc2602
ElHefnawi M, Soliman B, Abu-Shahba N et al (2013) An integrative meta-analysis of microRNAs in hepatocellular carcinoma. Genom Proteom Bioinf 11(6):354–367. https://doi.org/10.1016/j.gpb.2013.05.007
Wong YH, Wu CC, Lin CL et al (2015) Applying NGS data to find evolutionary network biomarkers from the early and late stages of hepatocellular carcinoma. Biomed Res Int 2015:391475. https://doi.org/10.1155/2015/391475
Hucl T (2011) Gallmeier E (2011) DNA repair: exploiting the Fanconi anemia pathway as a potential therapeutic target. Physiol Res 60(3):453–465. https://doi.org/10.33549/physiolres.932115
Palagyi A, Neveling K, Plinninger U et al (2010) Genetic inactivation of the Fanconi anemia gene FANCC identified in the hepatocellular carcinoma cell line HuH-7 confers sensitivity towards DNA-interstrand crosslinking agents. Mol Cancer 9:127. https://doi.org/10.1186/1476-4598-9-127
Okayama A, Maruyama T, Tachibana N et al (1995) Increased prevalence of HTLV-I infection in patients with hepatocellular carcinoma associated with Hepatitis C virus. Cancer Sci 86:1–4. https://doi.org/10.1111/j.1349-7006.1995.tb02979.x
Lips EH, Mulder L, Hannemann J et al (2010) Indicators of homologous recombination deficiency in breast cancer and association with response to neoadjuvant chemotherapy. Ann Oncol 22(4):870–876. https://doi.org/10.1093/annonc/mdq468
Abkevich V, Timms KM, Hennessy BT et al (2012) Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 107:1776–1782. https://doi.org/10.1038/bjc.2012.451
Helleday T (2010) Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 31(6):955–960. https://doi.org/10.1093/carcin/bgq064
Kiss A, Wang NJ, Xie JP et al (1997) Analysis of transforming growth factor (TGF)-alpha/epidermal growth factor receptor, hepatocyte growth Factor/c-met, TGF-beta receptor type II, and p53 expression in human hepatocellular carcinomas. Clin Cancer Res 3(7):1059–1066
Greer EL, Brunet A (2005) FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24(50):7410–7425. https://doi.org/10.1038/sj.onc.1209086
Guo Q, Song Y, Hua K et al (2017) Involvement of FAK-ERK2 signaling pathway in CKAP2-induced proliferation and motility in cervical carcinoma cell lines. Sci Rep 7(1):2117. https://doi.org/10.1038/s41598-017-01832-y
Hayashi T, Ohtsuka M, Okamura D et al (2014) Cytoskeleton-associated protein 2 is a potential predictive marker for risk of early and extensive recurrence of hepatocellular carcinoma after operative resection. Surgery 155(1):114–123. https://doi.org/10.1016/j.surg.2013.06.009
Jeon TW, Min JK, Seo YR et al (2017) Abstract 3113: Knockdown of cell division cycle-associated 8 (CDCA8) suppresses hepatocellular carcinoma growth via the upregulation of tumor suppressor ATF3. Can Res 77(13):3113–3113. https://doi.org/10.1158/1538-7445.AM2017-3113
Fu X, Zhu Y, Zheng B et al (2018) KIFC1, a novel potential prognostic factor and therapeutic target in hepatocellular carcinoma. Int J Oncol 52(6):1912–1922. https://doi.org/10.3892/ijo.2018.4348
Xia H, Kong SN, Chen J et al (2016) MELK is an oncogenic kinase essential for early hepatocellular carcinoma recurrence. Cancer Lett 383(1):85–93. https://doi.org/10.1016/j.canlet.2016.09.017
Li Y, Li Y, Chen Y et al (2017) MicroRNA-214-3p inhibits proliferation and cell cycle progression by targeting MELK in hepatocellular carcinoma and correlates cancer prognosis. Cancer Cell Int 17(1):102. https://doi.org/10.1186/s12935-017-0471-1
Li Y, Zheng J, Yao J et al (2017) Aberrant expression and prognostic value of RacGAP1 in hepatocellular carcinoma. Int J Clin Exp Pathol 10:1747–1755
Junttila MR, Puustinen P, Niemelä M et al (2007) CIP2A inhibits PP2A in human malignancies. Cell 130(1):51–62. https://doi.org/10.1016/j.cell.2007.04.044
Chen KF, Liu CY, Lin YC et al (2010) CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene 29:6257–6266. https://doi.org/10.1038/onc.2010.357
Côme C, Laine A, Chanrion M et al (2009) CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res 15(16):5092–5100. https://doi.org/10.1158/1078-0432.CCR-08-3283
Li W, Ge Z, Liu C et al (2008) CIP2A is overexpressed in gastric cancer and its depletion leads to impaired clonogenicity, senescence, or differentiation of tumor cells. Clin Cancer Res 14(12):3722–3728. https://doi.org/10.1158/1078-0432.CCR-07-4137
Khanna A, Böckelman C, Hemmes A et al (2009) MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst 101(11):793–805. https://doi.org/10.1093/jnci/djp103
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grand Nos. 11371174 and 11804123) and Postgraduate Research and Practice Innovation Program of Jiangsu Province (No. KYCX18_1866). This manuscript had been accepted for presentation in CBC2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflicts of interest in this paper.
Rights and permissions
About this article

Cite this article
Qi, J., Zhou, J., Tang, XQ. et al. Gene Biomarkers Derived from Clinical Data of Hepatocellular Carcinoma. Interdiscip Sci Comput Life Sci 12, 226–236 (2020). https://doi.org/10.1007/s12539-020-00366-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-020-00366-8