
Abstract
Bacterial proteins belonging to the YczE family are predicted to be membrane proteins of yet unknown function. In many bacterial species, the yczE gene coding for the YczE protein is divergently transcribed with respect to an adjacent transcriptional regulator of the MocR family. According to in silico predictions, proteins named YczR are supposed to regulate the expression of yczE genes. These regulators linked to the yczE genes are predicted to constitute a subfamily within the MocR family. To put forward hypotheses amenable to experimental testing about the possible function of the YczE proteins, a phylogenetic profile strategy was applied. This strategy consists in searching for those genes that, within a set of genomes, co-occur exclusively with a certain gene of interest. Co-occurrence can be suggestive of a functional link. A set of 30 mycobacterial complete proteomes were collected. Of these, only 16 contained YczE proteins. Interestingly, in all cases each yczE gene was divergently transcribed with respect to a yczR gene. Two orthology clustering procedures were applied to find proteins co-occurring exclusively with the YczE proteins. The reported results suggest that YczE may be involved in the membrane translocation and metabolism of sulfur-containing compounds mostly in rapidly growing, low pathogenicity mycobacterial species. These observations may hint at potential targets for therapies to treat the emerging opportunistic infections provoked by the widespread environmental mycobacterial species and may contribute to the delineation of the genomic and physiological differences between the pathogenic and non-pathogenic mycobacterial species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Bacterial proteins belonging to the YczE family [1] are predicted to be membrane proteins of yet unknown function possessing five trans-membrane helices. The Conserved Domain Database (CDD) [2] assigns code COG2364 to the YczE family. These proteins share the presence of one or two so-called DUF161 domains with the YitT family, corresponding to InterPro databank [3] code IPR003740, Pfam [4] code PF02588, and CDD code COG1284. Indeed, YczE and YitT proteins are generically annotated as “membrane proteins containing the DUF161 domain”. Differences between YczE and YitT proteins are not obvious by the annotations reported in the databanks. Possibly, they may be distinguished by the absence of the C-terminal DUF2179 domain in YczE, corresponding to the Protein Data Bank (PDB) structure PDB:3HLU, present in the members of the YitT family. Moreover, in many bacterial species, the yczE gene coding for the YczE protein is divergently transcribed with respect to an adjacent gene coding for a transcriptional regulator of the MocR family [5]. The MocRs linked to the yczE genes, named YczRs, are predicted to constitute a subfamily within the MocR family [6]. Moreover, YczRs are supposed to regulate the expression of the yczE genes as, for example, in Regulog PdxR3—Mycobacteriaceae of the RegPrecise 4.0 database [5].
MocR regulators are a family of proteins belonging to the class of GntR regulators [7] characterized by the presence of two domains. The N-terminal domains, 60 residue-long on average, display the winged-helix–turn–helix architecture (wHTH) and are responsible for DNA recognition and binding [7]. The C-terminal domains [8] are quite large (350 residue on average) and are characterized by a tertiary structure belonging to fold type-I pyridoxal 5′-phosphate (PLP) dependent enzymes [9], of which aspartate aminotransferase (AAT) is the archetypal enzyme. The two domains are linked to each other by a peptide bridge [10, 11]. The three-dimensional structure of GabR [12] from Bacillus subtilis, one of the best characterized MocR [13,14,15,16], confirmed the presence of a C-terminal AAT-like domain and provided fundaments for further investigations. Only a few other MocRs have been experimentally characterized so far: for example, TauR, involved in the regulation of taurine utilization genes in Rhodobacter capsulatus [17]; PdxR, involved in the regulation of the PLP synthesis in several bacteria such as Corynebacterium glutamicum [18], Streptococcus pneumonia [19], Listeria monocytogenes [20], Bacillus clausii [21]; DdlR from Brevibacillus brevis which activates the expression of the gene coding for the enzyme d-alanyl-d-alanine ligase [22]. The entire MocR population can be subdivided into groups characterized by different structural and functional properties [6, 23] such as YczR.
Regarding the possible function of YczE proteins, it has been hypothesized that in Bacillus amyloliquefaciens, they are able to positively regulate the biosynthesis of bacillomycin D although in a yet-unidentified manner [1]. Other authors found that in B. subtilis B3, the genomic region involved in the biosynthesis of surfactin contained the yczEB3 gene coding for a YczE-like protein [24]. Interestingly, the same genomic region hosted the aspB3 gene coding for a putative aminotransferase-like protein. More recently, it has been proposed that YczE may anchor to the membrane the mega-synthases responsible for the biosynthesis of polyketides or lipopeptides in B. amyloliquefaciens FZB42 and subtilis [25]. However, in these two species, yczE is not divergently oriented with a yczR gene. Since, in many cases, yczE genes are presumed to be under the control of transcription factors able to bind PLP, it is reasonable to expect that YczE be involved in transport and/or processing of metabolites related, directly or indirectly, to PLP chemistry.
To put forward hypotheses amenable to experimental testing about the possible function of the YczE proteins, a phylogenetic profile strategy has been applied. The strategy consists in searching for those genes that, within a set of genomes, co-occur exclusively with a certain gene of interest. The co-occurring genes can be considered functionally linked to the target gene [26]. Application of specific in silico methods may also indicate the possible membrane protein type and generic function [27].
Although YczE is widespread among eubacteria [6], we focused our analysis on a set of mycobacterial species. Mycobacterium genus belongs to the actinobacteria phylum and comprehends many different species several of which are pathogenic to vertebrates. Mycobacterial species are often classified as slow- or fast-grower [28]. Slow-growth Mycobacteria such as M. tuberculosis or leprae are generally highly pathogenic. Moreover, Mycobacteria possess a complex cell envelope consisting of a cytoplasmic membrane and a cell wall which plays a crucial role in the intrinsic drug resistance and in survival under harsh conditions [29]. Mycobacteria represented a very suitable and attractive set of data for our in silico analyses, because: (a) the species are evolutionarily close; (b) YczE and YczR are present only in a subset of the species; (c) in these species, yczE and yczR are divergently transcribed. In general, Mycobacteria are interesting for their increasing relevance to the human health [30]. We collected a set of 30 mycobacterial genomes. Out of the 30 mycobacterial proteomes, only 16 contained YczE proteins. Two orthology clustering procedures were applied to find the proteins co-occurring exclusively with the YczE proteins. Results suggest that YczE may be involved in the membrane translocation and/or metabolism of sulfur-containing compounds such as taurine.
2 Materials and Methods
Mycobacterial complete proteomes have been retrieved from the RefSeq genome databank [31]. RegPrecise version 4.0 was the reference data bank for regulon prediction [5]. Synteny analysis utilized the SyntTax web server [32]. Clusters of orthologs were identified using the program Proteinortho V5.15 [33] and the stand-alone version of OMA [34].
Sequence searches were carried out using Blast, Rps-blast [35], and the HMMER suite [36] implementing the Hidden Markov Models (HMM) profile search. Multiple sequence alignments were obtained with Clustal Omega [37] and displayed with Jalview [38]. Halign Web server [39] was also tested as an alternative multiple sequence alignment procedure. Phylogenetic trees were calculated with the suite MEGA v.7.0 [40]. Fold recognition and structure prediction relied on the Phyre2 [41] and HHpred [42] servers. Phyre2 provides a fold recognition service based on profile alignments. The HHpred suite implements the HMM vs HMM profile comparison to assign a protein to a particular structural fold or, alternatively, to a protein family.
Secondary structure and trans-membrane helix prediction utilized MEMSAT of the PsiPred server [43], TMHMM [44], and CCTOP [45].
Perl, awk, and bash scripts were written to analyze data and parse output text files.
3 Results
3.1 Data Set
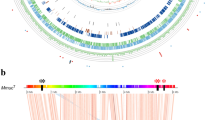
A set of 30 mycobacterial proteomes were retrieved from the RefSeq genome databank (Table 1). The sample was chosen, so that only 16 proteomes contained YczE and YczR proteins (Fig. 1) The corresponding genomes were scanned through the SyntTax Web server [32] using the YczE protein from Mycobacterium smegmatis (RefSeq code YP_886671) as a query, to confirm synteny in correspondence of the genes yczE and yczR. Moreover, scrutiny of the mycobacterial genome maps confirmed that the genes coding for YczE are always divergently transcribed with respect to the genes yczR coding for YczR [6] (Fig. 1). It should also be mentioned that M. smegmatis genome has got two yczE identical copies divergently transcribed to cognate identical yczR genes. Only one pair is reported in Fig. 1. Moreover, in the case of M. smegmatis, goodii and neoaurum short genes coding for non-conserved hypothetical proteins about 50–60 residue-long (WP_023985549, WP_053194597 and YP_886670, respectively) are predicted to occur between the yczR and the yczE genes (Fig. 1).

Scheme showing the gene layout around the genes yczE and yczR (labelled in the scheme) in the mycobacterial genomes considered. Genes coding for homologous proteins are depicted with the same arrow style and their products are listed in the box. Arrow length and distances are not proportional to sequence length except for the “hypothetical” proteins
The presence (or absence) of the YczE proteins in each of the 30 mycobacterial genomes considered was further confirmed by scanning their proteomes with the Hmmsearch program, in the HMMER suite, and Rps-blast. In the first case, an HMM profile calculated with the aligned YczE sequences was employed to scrutinize the mycobacterial proteomes. In the latter, Rps-blast searched for the occurrence of the CDD query profile (code COG2364, PSSM id 225239) representing the YczE domain. It should be reported that HMM searches retrieved also the sequence WP_003885078 from Mycobacterium sp. VKM Ac-1817D originally not included in the YczE set. However, this protein is a distant paralog of WP_003881203 within the same genome, sharing only 29% sequence identity, and it is not divergently transcribed with respect to any MocR regulator.
3.2 MocR Regulators
A census of the MocR regulators occurring in the 30 selected mycobacterial proteomes was obtained with the use of Hmmsearch and Rps-blast. As before, an HMM profile was calculated from the aligned YczR sequences and thereafter used to scan the mycobacterial proteomes. Rps-blast searched the proteomes with the CDD profiles representing the HTH and the AAT domains, respectively (Table 2). A protein was considered a bona-fide MocR only if contained both domains. The number of MocR contained in a single proteome is variable (Table 2). Five proteomes, M. bovis, M. haemophilum, M. leprae, M. tuberculosis, and M. xenopi, apparently lack the regulators.
A cladogram has been generated from the multiple sequence alignments of all the MocRs found in the mycobacterial proteomes. The cladogram substantiates the notion that the regulators divergently transcribed with respect to the yczE genes constitute a subgroup [6] within the MocR family (Fig. 2). Indeed, all the YczRs segregate in the same subtree with the exception of the MocRs from the YczE-free species M. elephantis and thermoresistibile.

Unrooted consensus tree showing the similarity relationships among the MocRs sequences detected in the selected mycobacterial proteomes. The cladogram has been calculated with the UPGMA method. Pairwise distances between sequences were calculated in units of number of amino acid differences per sequence. The bootstrap tree was inferred from 1000 replicates. Red color of the branches and taxon names denote the YczR subpopulation. Asterisk denote the node of the YczR subtree supported by more than 90% bootstrap frequency
3.3 Mycobacterium YczE
Protein YczE is a conserved membrane protein annotated in RefSeq as a “uncharacterized 5×TM membrane BCR, YitT family”. However, while the TMHMM prediction suggests the presence of only 5 helices, MEMSAT and CCTOP predict six trans-membrane helices (Fig. 3). A multiple sequence alignment of the Mycobacterium proteins contained in the data set and the layout of the predicted trans-membrane helices are displayed in Fig. 3. The proteins do not share any similarity to other structurally characterized proteins. In fact, Phyre2 and HHpred searches were unable to associate YczEs to any known fold.

Multiple sequence alignment of the YczE proteins found in the selected proteomes obtained with Clustal Omega. Sequence codes refer to Table 1. Coloring code follows the ClustalX scheme. Lines labelled with CCTOP, TMHMM and MEMSAT report the results of the trans-membrane helix predictions. Red bars indicate the predicted trans-membrane helices, while grey and blue bars denote “cytoplasmic” and “extracellular” peptide segments, respectively
3.4 Phylogenetic Profile Analysis
To propose hypotheses about possible functions of the conserved YczE protein, a comparative protein phylogenetic profile approach has been applied [26]. The rationale of the strategy consists in looking for proteins uniquely associated to the presence of YczE in every proteome of the considered set. As an example, if proteins A and B are found in a subset of proteomes exclusively in co-occurrence of YczE and viceversa, a functional link between A, B, and YczE may be hypothesized [46].
To identify co-occurring proteins, two ortholog-clustering techniques have been chosen considering their computational efficiency, ease of use, and straightforward result interpretation: Proteinortho V5.15 [33] and stand-alone OMA [34]. Both methods exploit an all-versus-all sequence comparison to quantify the similarity between every pairs of protein sequences followed by a clustering procedure. Differences in the methods used for sequence comparison, for residue exchange scoring, and data clustering can yield partially different results on the same data set. For that reason, we applied a comparative strategy: results delivered by the two methods were compared and combined. The clusters containing homologs exclusively occurring in YczE-positive mycobacterial proteomes were considered functionally associated with yczE gene.
Results derived from the application of the two clustering methods are reported in Table 3. Clusters were denominated according to RefSeq annotation and InterPro assignment [47]. The definitions of the families to which the orthologous clusters are predicted to belong are: (a) S-adenosyl-l-methionine-dependent methyltransferase-like; (b) sulfurtransferase (rhodanese-like domain); (c) monooxygenase (luciferase-like domain); (d) 2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase (OHCU); (e) taurine transporter permease TauC; (f) taurine import ATP-binding protein TauB; (g) ABC-type taurine transport system, periplasmic component, TauA; (h) MarR-like transcriptional regulator. As expected, the two phylogenetic methods were able to correctly cluster the YczE proteins of the 16 mycobacterial proteomes (data not shown).
Composition of each cluster was further validated using HMM profile searches. Sequences of each cluster were aligned with Clustal Omega and a HMM profile was calculated with Hmmbuild from the HMMER suite. Hmmsearch was subsequently applied to compare each query profile against the 30 proteomes considered to detect occurrence of possible orthologs in YczE-free mycobacterial species missed by the clustering programs.
Searches using the profile derived from “S-adenosyl-l-methionine-dependent methyltransferase-like” cluster showed that these proteins are found only in the YczE-containing proteomes. The only exception is a sequence annotated as type-11 methyltransferase in M. gordonae which, however, is assigned a very low E-value (10−6) compared to the average E-value shown by the cluster proteins (about 10−150).
The sulfurtransferase, “rhodanese–like domain” profile, captures the presence of homologous, distant domains also in the YczE-free mycobacterial proteomes. A careful inspection of such sequences shows that they are multi-domain proteins containing a C-terminal rhodanese-like domain and an N-terminal domain of different types. For example: the Ars transcriptional regulators (such as WP_023369982) that contain an N-terminal HTH domain, the molybdopterin biosynthesis-like protein MoeZ (WP_003874621) with an N-terminal domain part of the superfamily of E1-like enzymes, and the sulfurtransferases that possess an N-terminal domain belonging to the MBL-fold metallo-protease superfamily (example WP_003924161).
The search with the monooxygenase “luciferase-like domain” profile captured homologs in all the mycobacterial proteomes; however, those found in the YczE-free species are mostly annotated as “FMN-dependent oxidoreductase” or “urease subunit gamma”. The “luciferase-like” protein displays weak similarities only with the C-terminal portion of these proteins.
The profile corresponding to the putative “2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase” detected distantly related homologs in M. marinum, and ulcerans, while none was present in the other YczE-free mycobacterial species. Interestingly, in most of YczE-containing mycobacterial species, a paralog of OHCU can be observed along with a protein annotated as “uracil–xanthine permease” (for example, YP_001703664 from M. abscessus). This protein contains an N-terminal domain belonging to the family of the xanthine permeases and a C-terminal domain homologous to OHCU.
The profile calculated with the proteins annotated as “taurine transporter permease TauC” collected several, more distant, homologs in all the other mycobacterial species except M. leprae. This result reflects the presence, within a species, of a variety of homologous membrane transporters responsible for the translocation of different classes of metabolites. However, the membrane proteins from the YczE-positive species constitute a distinct subpopulation which suggests that they may have different function and/or possess a different substrate specificity.
A similar pattern can be observed for the protein cluster annotated as “taurine import ATP-binding protein TauB”, namely the ATP-binding subunit of the ABC transporter. In this case, only OMA detected the cluster (Table 3). For that reason, a more careful analysis of the distribution of homologs among the mycobacterial species was carried out. Several distant homologs could be observed in all species. However, the TauB protein from the YczE-positive species constitutes a distinct subgroup within the set as shown by the cladogram reported in Supplementary material Fig. 1.
ABC importers have cognate periplasmic-binding proteins that capture the solute and feed it to the periplasmic side of the transporter [48, 49]. Both orthology programs were able to detect a cluster containing a periplasmic-binding protein, namely “ABC-type taurine transport system, periplasmic component TauA” (Table 3). The results of the HMM search revealed the presence of several homologs within the same proteomes, whereas the TauA-like proteins were missing in the M. bovis, haemophilum, leprae, marinum, parascrofulaceum, tuberculosis and ulcerans species. Sequence comparison confirms that the TauA-like periplasmic proteins represent a conserved subgroup within the considered proteomes. All these considerations substantiate the notion that a complete ABC system is specifically co-occurring with the gene yczE.
MarR (multiple antibiotic resistance) transcriptional regulators are a family of small proteins (about 140 residue-long) containing a winged-helix DNA-binding domain [50]. Proteins of the MarR family are involved in a variety of biological functions, such as resistance to multiple antibiotics, organic solvents, and oxidative stress agents. These proteins also regulate the synthesis of pathogenic factors in bacteria able to infect humans and plants [51]. The HMM profile of the 16 MarR orthologs found by the clustering programs retrieved distant homologs in all the considered mycobacterial species. Once more, the MarRs from the YczE-containing mycobacterial species represent a subfamily clearly denoted within the set of homologous regulators (Supplementary material Fig. 2). Interestingly, in all the YczE-positive species, MarR genes are adjacent to a Major Facilitator Superfamily (MFS) transporter gene [52]. On the contrary, MarR is often adjacent to a peptide ABC transporter ATP-binding protein in the other mycobacterial species.
3.5 Putative Structural and Functional Features of the Cluster Proteins
3.5.1 S-Adenosylmethionine-Dependent Methyltransferase-Like Proteins
Phyre2 fold recognition using one of the proteins of the S-adenosylmethionine-dependent methyltransferase-like cluster as a query (WP_005085951 from Mycobacterium abscessus) suggests that these proteins are structurally compatible with the N-terminal portion of several proteins annotated as methyltransferases. Among the most compatible are: tfu_2867 from Thermobifida fusca (PDB:2QE6) that shares with the query about 29% sequence identity over a 130 residue-long alignment; the S-adenosylmethionine-dependent methyltransferase from C. glutamicum (PDB:3CGG), 23% sequence identity; the trans-aconitate 2-methyltransferase from Agrobacterium tumefaciens (PDB:2P35), 20% identity; the methyltransferase from antibiotic biosynthesis pathway from Anabaena variabilis (PDB:3EGE), 19% identity. A corresponding multiple sequence alignment is reported in Supplementary material Fig. 3. Moreover, HHpred assigns the portion encompassed by the positions 55–173 of the multiple sequence alignment of the mycobacterial methyltransferase-like proteins, to several CDD profiles pertaining to different methylases such as COG2230, representing the “Cyclopropane fatty-acyl-phospholipid synthase and related methyltransferases” (Table 3).
3.5.2 Sulfurtransferase Rhodanese-Like Domains
The sulfurtransferase “rhodanese-like domains” are structurally affine to the rhodanese domains contained in several multi-domain proteins. For example, the sequence from M. smegmatis (WP_003883953) shares 36% sequence identity with the homologous domain of the Alicyclobacillus acidocaldarius protein that contains also a N-terminal β-lactamase domain (PDB:3TP9). Likewise, the sequence is structurally compatible to many other sulfurtransferase enzymes; among the most similar are the uncharacterized rhodanese-related protein from Thermoplasma volcanium (PDB:3GK5) that shares 26% sequence identity with the query over about 100 aligned residue, and the thiosulfate sulfurtransferase GLPE from E. coli (PDB:1GMX), 26% identity. A corresponding alignment is reported in Supplementary material Fig. 4. Accordingly, HHpred assigns the sequences to CDD profiles related to sulfurtransferases such as: CD01524 (pyridine nucleotide-disulphide oxidoreductase), or CD01534 representing “Rhodanese-related sulfurtransferase” (Table 3).
3.5.3 Monooxygenase (Luciferase-Like) Domains
Fold recognition for the “luciferase-like domains” detects structural compatibility with several monooxygenases. For example, the protein from M. smegmatis (WP_003882562) shares a significant structural compatibility with the C-terminal portion (sequence interval 225–406) of alkane monooxygenase from Geobacillus thermodenitrificans (PDB:3B9N) although with only 16% sequence identity. Likewise, HHpred indicates structural compatibility with portions of several monooxygenases. The most similar appear: nitrilotriacetate monooxygenase from Burkholderia pseudomallei (PDB:3SDO; sequence interval 226–424, 17% identity); alkane monooxygenase from G. thermodenitrificans (PDB:3B90; sequence interval: 234–405, 16% identity); alkanesulfonate monooxygenase from E. coli (PDB:1NQK; sequence interval 184–357, 12% identity). A sequence alignment is reported in Supplementary material Fig. 5. According to these findings, HHpred detects the presence of the CDD domains pertaining mainly FMN-dependent monooxygenases: for example, TIGR03612 corresponding to “pyrimidine utilization protein A” that is a FMN-dependent monooxygenase; TIGR03565 (alkanesulfonate monooxygenase, FMNH2-dependent); cd01095 (nitrilotriacetate monooxygenase); COG2141 (flavin-dependent oxidoreductase, luciferase family, including alkanesulfonate monooxygenase SsuD and methylene tetrahydromethanopterin reductase) (Table 3). Moreover, HHpred also identify the presence of a signature corresponding to the TIGR03854 profile corresponding to “probable F420-dependent oxidoreductase”. Interestingly, it has been found that coenzyme F420 is particularly abundant in mycobacterial species [53].
3.5.4 2-Oxo-4-Hydroxy-4-Carboxy-5-Ureidoimidazoline Decarboxylases (OHCU)
Phyre2 searches confirm the identity of the protein cluster denominated “2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase”. The protein from M. smegmatis (WP_005064897) shares 33% sequence identity with PDB ID:3O7K, the 2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline decarboxylase from Klebsiella pneumoniae.
3.5.5 Taurine Transporter Permease TauC
Proteins within this cluster are predicted to possess six trans-membrane helices. Blast searches through the UniProt/Swissprot databank retrieve several proteins of the Taurine permease transporter of type-I such as UniProt:Q47539 from E. coli (37% identity to the query sequence from Mycobacterium abscessus). Fold recognition confirms structural compatibility with several membrane transporters such as: molybdate/tungstate ABC transporter from Archaeoglobus fulgidus (PDB:2ONK), 14% sequence identity, and sulfate/molybdate ABC transporter from Methanosarcina acetivorans (PDB:3D31), 11% identity (Supplementary material Fig. 6). HHpred search of the CDD databank detects many domain signatures representing membrane transporters. Among the most significant are: COG0600 corresponding to “ABC-type nitrate/sulfonate/bicarbonate transport system, permease component”, TIGR01183 (“nitrate ABC transporter, permease protein”), and COG1174 (“ABC-type proline/glycine betaine transport system, permease component”) (Table 3).
3.5.6 Taurine Import ATP-Binding Protein TauB
Although this cluster is not detected by ProteinOrtho, its proteins should be considered as integral components of the Taurine-like ABC transporter in the YczE mycobacterial genomes. A Blast search in the Uniprot/Swissprot databank retrieves many ATP-binding components of ABC membrane transporters such as, for example, the “uncharacterized ABC transporter ATP-binding protein MJ0412” from Methanocaldococcus jannaschii (UniProt:Q57855) at 40% sequence identity and the “Taurine import ATP-binding protein TauB” from Paracoccus pantotrophus (UniProt:Q6RH47) at 43% identity. Fold recognition confirms that the cluster proteins are compatible with the structure of the ATP-binding components of membrane transporters, for example the “ABC transporter ATP-binding protein” from Thermotoga maritima (PDB:4YER) with which it shares 28% sequence identity, “CYSA, putative ABC transporter ATP-binding protein” from Alicyclobacillus acidocaldarius (PDB:1Z47), 42% identity, or the “maltose/maltodextrin transport ATP-binding protein” PDB:1Q1B from E. coli, 40% identity (Supplementary material Fig. 7). Likewise, HHpred CDD search assigns the cluster proteins to families of ATP-binding of ABC transporters, such as: COG1116 (“ABC-type nitrate/sulfonate/bicarbonate transport system, ATPase component”) or COG1126 (“ABC-type polar amino acid transport system, ATPase component”).
3.5.7 TauA Periplasmic-Binding Protein
Fold recognition procedures assigns this cluster to the structures of periplasmic proteins such as “alkanesulfonate binding protein2” from Xanthomonas axonopodis pv. citri (PDB:3E4R) with 25% sequence identity, and “periplasmic aliphatic sulphonates-binding protein” from E. coli (PDB:2X26), 18% identity (Supplementary material Fig. 8). Accordingly, HHpred assigns the binding protein to the family TauA (CDD code COG4521) denominated “ABC-type taurine transport system, periplasmic component”, or “ABC-type nitrate/sulfonate/bicarbonate transport system, periplasmic component” (COG0715) (Table 3).
3.5.8 MarR-Like Transcriptional Regulator
Fold recognition links the proteins of this cluster to structures of MarR family transcriptional regulators such as the regulator from Bacillus stearothermophilus (PDB: 2RDP) at about 17% sequence identity; from Salicibacter pomeroyi (3E6M) at 22% identity; from Streptomyces coelicolor (PDB:3ZPL), 24% identity; the regulator BldR from Sulfolobus solfataricus (PDB:3F3X) with 25% sequence identity. As expected, HHPred relates the ortholog cluster to the domain COG1846 corresponding to the MarR family (Supplementary material Fig. 9).
4 Discussion
In an attempt to delineate possible functions of the putative membrane YczE protein, phylogenetic profile analysis has been applied to a set of 30 mycobacterial species to look for co-occurring genes. Since phylogenetic profiling can be affected by serious artifacts mainly caused by the difficulty of discrimination between orthologs and paralogs [54], we applied two clustering programs the results of which have been combined by a consensus approach to minimize inaccuracies.
Our results suggest that YczE is consistently associated with the presence of at least eight other proteins (Table 3) in the YczE-positive mycobacterial species. Although uncharacterized, in silico analyses of these proteins have provided useful hypotheses about their potential role. The common trait of most of the co-occurring proteins is the apparent structural affinity with enzymes or transporters involved in the metabolism of sulfur-containing compounds. Indeed, among pathogenic bacteria only, Mycobacteria have been reported to produce sulfated metabolites [55]. To this respect, the most interesting result is the co-occurrence of YczE with an ABC importer (the trans-membrane, the ATP-binding components, and the periplasmic-binding proteins) that shares some similarity to other bacterial transport systems such as the E. coli TauABC complex involved in the uptake and subsequent processing of taurine in conditions of sulfate or cysteine starvation [56]. It is, therefore, conceivable that the homologous mycobacterial complex is functionally linked to membrane translocation of taurine and/or related sulfur compounds.
All these considerations point to a potential role of YczE in the context of a metabolic pathways involving transport and processing of sulfur-containing compounds, at least in the Mycobacterium genus. YczE is a membrane protein: it can be speculated that it interacts with the ABC transporter possibly triggering transport or coupling the transport to metabolite processing. Expression of yczE is under the putative control of YczR: therefore, it can be hypothesized that the sulfur compound uptaken by the membrane transporter or one of its metabolites may be the effector of YczR. The binding of this molecule to the regulator should be able to influence the expression of the yczE gene.
Distribution of YczE in the mycobacterial proteomes overlaps very well with the rate of growth: rapidly growing bacteria possess YczE, whereas the slow-growing ones, mostly highly pathogenic, do not. Only exception are Mycobacterium elephantis and thermoresistibile, which tolerate [57] higher temperature compared to other mycobacterial species. It is indeed well known that slow-growing mycobacterial species possess fewer membrane transporters [58, 59] that may limit uptake of external nutrients. This fact strengthens the notion that YczE may be involved in uptake processes able to contribute to sustain the activity of the metabolic machinery of fast-growing Mycobacteria. As a consequence, YczE cannot be considered per se a target for therapies for the most severe infections by the slow-growing pathogenic mycobacterial species but can be a potential target for the emerging opportunistic infections provoked by the widespread environmental Mycobacteria. Moreover, this comparative work contributes to the delineation of genomic and physiological differences between the pathogenic and non-pathogenic mycobacterial species.
In conclusion, this report proposes a putative functional role for the mycobacterial protein YczE and provides a conceptual framework for the design of rational experiments aimed at validating the theoretical observations. It may also contribute to a deeper understanding of the genomic differences between slow- and fast-growing mycobacterial species relevant to human health.
References
Koumoutsi A, Chen XH, Vater J, Borriss R (2007) DegU and YczE positively regulate the synthesis of bacillomycin D by Bacillus amyloliquefaciens strain FZB42. Appl Environ Microbiol 73:6953–6964. https://doi.org/10.1128/AEM.00565-07
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI’s conserved domain database. Nucleic Acids Res 43:D222–D226. https://doi.org/10.1093/nar/gku1221
Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, Yong SY, Bateman A, Punta M, Attwood TK, Sigrist CJ, Redaschi N, Rivoire C, Xenarios I, Kahn D, Guyot D, Bork P, Letunic I, Gough J, Oates M, Haft D, Huang H, Natale DA, Wu CH, Orengo C, Sillitoe I, Mi H, Thomas PD, Finn RD (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43:D213–D221. https://doi.org/10.1093/nar/gku1243
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. https://doi.org/10.1093/nar/gkv1344
Novichkov PS, Kazakov AE, Ravcheev DA, Leyn SA, Kovaleva GY, Sutormin RA, Kazanov MD, Riehl W, Arkin AP, Dubchak I, Rodionov DA (2013) RegPrecise 3.0—a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genom 14:745. https://doi.org/10.1186/1471-2164-14-745
Milano T, Angelaccio S, Tramonti A, Di Salvo ML, Contestabile R, Pascarella S (2016) A bioinformatics analysis reveals a group of MocR bacterial transcriptional regulators linked to a family of genes coding for membrane proteins. Biochem Res Int 2016:4360285. https://doi.org/10.1155/2016/4360285
Hoskisson PA, Rigali S (2009) Chapter 1: variation in form and function the helix-turn-helix regulators of the GntR superfamily. Adv Appl Microbiol 69:1–22. https://doi.org/10.1016/S0065-2164(09)69001-8
Bramucci E, Milano T, Pascarella S (2011) Genomic distribution and heterogeneity of MocR-like transcriptional factors containing a domain belonging to the superfamily of the pyridoxal-5′-phosphate dependent enzymes of fold type I. Biochem Biophys Res Commun 415:88–93. https://doi.org/10.1016/j.bbrc.2011.10.017
Schneider G, Kack H, Lindqvist Y (2000) The manifold of vitamin B6 dependent enzymes. Structure 8:R1–R6
Milano T, Angelaccio S, Tramonti A, di Salvo ML, Contestabile R, Pascarella S (2016) Structural properties of the linkers connecting the N- and C-terminal domains in the MocR bacterial transcriptional regulators. Biochimie Open 3:8–18. https://doi.org/10.1016/j.biopen.2016.07.002
Angelaccio S, Milano T, Tramonti A, Di Salvo ML, Contestabile R, Pascarella S (2016) Data from computational analysis of the peptide linkers in the MocR bacterial transcriptional regulators. Data Brief 9:292–313. https://doi.org/10.1016/j.dib.2016.08.064
Belitsky BR (2004) Bacillus subtilis GabR, a protein with DNA-binding and aminotransferase domains, is a PLP-dependent transcriptional regulator. J Mol Biol 340:655–664. https://doi.org/10.1016/j.jmb.2004.05.020
Edayathumangalam R, Wu R, Garcia R, Wang Y, Wang W, Kreinbring CA, Bach A, Liao J, Stone TA, Terwilliger TC, Hoang QQ, Belitsky BR, Petsko GA, Ringe D, Liu D (2013) Crystal structure of Bacillus subtilis GabR, an autorepressor and transcriptional activator of gabT. Proc Natl Acad Sci USA 110:17820–17825. https://doi.org/10.1073/pnas.1315887110
Okuda K, Ito T, Goto M, Takenaka T, Hemmi H, Yoshimura T (2015) Domain characterization of Bacillus subtilis GabR, a pyridoxal 5′-phosphate-dependent transcriptional regulator. J Biochem 158:225–234. https://doi.org/10.1093/jb/mvv040
Al-Zyoud WA, Hynson RM, Ganuelas LA, Coster AC, Duff AP, Baker MA, Stewart AG, Giannoulatou E, Ho JW, Gaus K, Liu D, Lee LK, Bocking T (2016) Binding of transcription factor GabR to DNA requires recognition of DNA shape at a location distinct from its cognate binding site. Nucleic Acids Res 44:1411–1420. https://doi.org/10.1093/nar/gkv1466
Amidani D, Tramonti A, Canosa AV, Campanini B, Maggi S, Milano T, di Salvo ML, Pascarella S, Contestabile R, Bettati S, Rivetti C (2016) Study of DNA binding and bending by Bacillus subtilis GabR, a PLP-dependent transcription factor. Biochem Biophys Acta 1861:3474–3489. https://doi.org/10.1016/j.bbagen.2016.09.013
Wiethaus J, Schubert B, Pfander Y, Narberhaus F, Masepohl B (2008) The GntR-like regulator TauR activates expression of taurine utilization genes in Rhodobacter capsulatus. J Bacteriol 190:487–493. https://doi.org/10.1128/JB.01510-07
Jochmann N, Gotker S, Tauch A (2011) Positive transcriptional control of the pyridoxal phosphate biosynthesis genes pdxST by the MocR-type regulator PdxR of Corynebacterium glutamicum ATCC 13032. Microbiology 157:77–88. https://doi.org/10.1099/mic.0.044818-0
El Qaidi S, Yang J, Zhang JR, Metzger DW, Bai G (2013) The vitamin B6 biosynthesis pathway in Streptococcus pneumoniae is controlled by pyridoxal 5′-phosphate and the transcription factor PdxR and has an impact on ear infection. J Bacteriol 195:2187–2196. https://doi.org/10.1128/JB.00041-13
Belitsky BR (2014) Role of PdxR in the activation of vitamin B6 biosynthesis in Listeria monocytogenes. Mol Microbiol 92:1113–1128. https://doi.org/10.1111/mmi.12618
Tramonti A, Fiascarelli A, Milano T, di Salvo ML, Nogues I, Pascarella S, Contestabile R (2015) Molecular mechanism of PdxR—a transcriptional activator involved in the regulation of vitamin B6 biosynthesis in the probiotic bacterium Bacillus clausii. FEBS J 282:2966–2984. https://doi.org/10.1111/febs.13338
Takenaka T, Ito T, Miyahara I, Hemmi H, Yoshimura T (2015) A new member of MocR/GabR-type PLP-binding regulator of d-alanyl-d-alanine ligase in Brevibacillus brevis. FEBS J 282:4201–4217. https://doi.org/10.1111/febs.13415
Milano T, Contestabile R, Lo Presti A, Ciccozzi M, Pascarella S (2015) The aspartate aminotransferase-like domain of Firmicutes MocR transcriptional regulators. Comput Biol Chem 58:55–61. https://doi.org/10.1016/j.compbiolchem.2015.05.003
Yao S, Gao X, Fuchsbauer N, Hillen W, Vater J, Wang J (2003) Cloning, sequencing, and characterization of the genetic region relevant to biosynthesis of the lipopeptides iturin A and surfactin in Bacillus subtilis. Curr Microbiol 47:272–277
Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess WR, Reva O, Junge H, Voigt B, Jungblut PR, Vater J, Sussmuth R, Liesegang H, Strittmatter A, Gottschalk G, Borriss R (2007) Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol 25:1007–1014. https://doi.org/10.1038/nbt1325
Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO (1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA 96:4285–4288
Zou Q, Li XB, Jiang Y, Zhao YM, Wang GH (2013) BinMemPredict: a web server and software for predicting membrane protein types. Curr Proteom 10:2–9
Cook GM, Berney M, Gebhard S, Heinemann M, Cox RA, Danilchanka O, Niederweis M (2009) Physiology of mycobacteria. Adv Microb Physiol 55:81–182. https://doi.org/10.1016/S0065-2911(09)05502-7
Niederweis M, Danilchanka O, Huff J, Hoffmann C, Engelhardt H (2010) Mycobacterial outer membranes: in search of proteins. Trends Microbiol 18:109–116. https://doi.org/10.1016/j.tim.2009.12.005
Prevots DR, Marras TK (2015) Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin Chest Med 36:13–34. https://doi.org/10.1016/j.ccm.2014.10.002
Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I (2014) RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res 42:D553–D559. https://doi.org/10.1093/nar/gkt1274
Oberto J (2013) SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinform 14:4. https://doi.org/10.1186/1471-2105-14-4
Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ (2011) Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinform 12:124. https://doi.org/10.1186/1471-2105-12-124
Altenhoff AM, Gil M, Gonnet GH, Dessimoz C (2013) Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS One 8:e53786. https://doi.org/10.1371/journal.pone.0053786
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. https://doi.org/10.1093/nar/gkr367
Sievers F, Higgins DG (2014) Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079:105–116. https://doi.org/10.1007/978-1-62703-646-7_6
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Zou Q, Hu Q, Guo M, Wang G (2015) HAlign: fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 31:2475–2481. https://doi.org/10.1093/bioinformatics/btv177
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. https://doi.org/10.1038/nprot.2009.2
Hildebrand A, Remmert M, Biegert A, Soding J (2009) Fast and accurate automatic structure prediction with HHpred. Proteins 77:128–132. https://doi.org/10.1002/prot.22499
Buchan DW, Minneci F, Nugent TC, Bryson K, Jones DT (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res 41:W349–W357. https://doi.org/10.1093/nar/gkt381
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. https://doi.org/10.1006/jmbi.2000.4315
Dobson L, Remenyi I, Tusnady GE (2015) CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res 43:W408–W412. https://doi.org/10.1093/nar/gkv451
Cheng Y, Perocchi F (2015) ProtPhylo: identification of protein–phenotype and protein–protein functional associations via phylogenetic profiling. Nucleic Acids Res 43:W160–W168. https://doi.org/10.1093/nar/gkv455
Mulder NJ, Kersey P, Pruess M, Apweiler R (2008) In silico characterization of proteins: UniProt, InterPro and Integr8. Mol Biotechnol 38:165–177. https://doi.org/10.1007/s12033-007-9003-x
Hvorup RN, Goetz BA, Niederer M, Hollenstein K, Perozo E, Locher KP (2007) Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD–BtuF. Science 317:1387–1390. https://doi.org/10.1126/science.1145950
Locher KP (2016) Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol 23:487–493. https://doi.org/10.1038/nsmb.3216
Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF (2001) The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution. Nat Struct Biol 8:710–714. https://doi.org/10.1038/90429
Wilkinson SP, Grove A (2006) Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr Issues Mol Biol 8:51–62
Ranaweera I, Shrestha U, Ranjana KC, Kakarla P, Willmon TM, Hernandez AJ, Mukherjee MM, Barr SR, Varela MF (2015) Structural comparison of bacterial multidrug efflux pumps of the major facilitator superfamily. Trends Cell Mol Biol 10:131–140
Selengut JD, Haft DH (2010) Unexpected abundance of coenzyme F(420)-dependent enzymes in Mycobacterium tuberculosis and other actinobacteria. J Bacteriol 192:5788–5798. https://doi.org/10.1128/JB.00425-10
Studer RA, Robinson-Rechavi M (2009) How confident can we be that orthologs are similar, but paralogs differ? Trends Genet 25:210–216. https://doi.org/10.1016/j.tig.2009.03.004
Paritala H, Carroll KS (2013) New targets and inhibitors of mycobacterial sulfur metabolism. Infect Disord Drug Targets 13:85–115
Eichhorn E, van der Ploeg JR, Leisinger T (2000) Deletion analysis of the Escherichia coli taurine and alkanesulfonate transport systems. J Bacteriol 182:2687–2695
Edwards TE, Liao R, Phan I, Myler PJ, Grundner C (2012) Mycobacterium thermoresistibile as a source of thermostable orthologs of Mycobacterium tuberculosis proteins. Protein Sci 21:1093–1096. https://doi.org/10.1002/pro.2084
Song H, Niederweis M (2012) Uptake of sulfate but not phosphate by Mycobacterium tuberculosis is slower than that for Mycobacterium smegmatis. J Bacteriol 194:956–964. https://doi.org/10.1128/JB.06132-11
Prasanna AN, Mehra S (2013) Comparative phylogenomics of pathogenic and non-pathogenic mycobacterium. PLoS One 8:e71248. https://doi.org/10.1371/journal.pone.0071248
Acknowledgements
This work was supported by Regione Lazio [Grant code FILAS-RU-2014-1020 A/15/2015 to TM], by University of Rome “La Sapienza” and by the Italian Education, University and Research Ministry (MIUR) [Grant numbers C26N158EP9; C26A14SY4E to MLDS and C26N158EP9 to TM]. Authors thank Fabio Francescato for the help in the synteny analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Milano, T., Angelaccio, S., Tramonti, A. et al. A Comprehensive Computational Analysis of Mycobacterium Genomes Pinpoints the Genes Co-occurring with YczE, a Membrane Protein Coding Gene Under the Putative Control of a MocR, and Predicts its Function. Interdiscip Sci Comput Life Sci 10, 111–125 (2018). https://doi.org/10.1007/s12539-017-0266-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-017-0266-z