
Abstract
The Spotted sardinella Amblygaster sirm are small pelagic fish that are important protein source to coastal communities in the Indo-West Pacific. In this study, a cytochrome c oxidase subunit 1 (COI) gene of A. sirm from Tanzania was amplified to assess the species’ genetic structure and demographic history. All individuals collected were identified using both morphological examination and genetic barcoding as A. sirm. A total of 19 haplotypes were found in the dataset, with low overall nucleotide (π = 0.13 ± 0.001) and moderate haplotype diversities (h = 0.45 ± 0.07). AMOVA revealed a very low and non-significant genetic differentiation in the dataset (Fst = 0.002, Фst = −0.004, p > 0.05), indicating a lack of population structure. The minimum spanning haplotype network revealed additional evidence for the lack of population structure, which grouped all the sampled haplotypes into one cluster, regardless of their geographical regions. The Tajima’s D, Fu’s Fs tests, and mismatch distribution analyses supported a hypothesis of recent demographic expansion. The lack of population structure identified suggests that the fishery should be treated as a single-stock management unit, consistent with the existing management regime for the species in Mainland Tanzania that currently does not consider genetic structure in managing the fishery. The lack of population structure suggests that populations with low genetic diversity, such as Dar es Salaam, can rebuild by recruiting from other sites if regulations against unsustainable fishing are strictly enforced.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Spotted sardinella (Amblygaster sirm) are small pelagic fishes that are essential protein source for coastal communities in the Indo-West Pacific (IWP) (Isaacs 2016). They play an important role not only as a part of the marine food web but also are used for human consumption, in animal feed manufacturing industries, and as bait in the longline and handline fishery (Pradeep et al. 2014). The species is widely distributed in the IWP (Whitehead 1988), from the Red Sea and Mozambique to the Philippines, Taiwan, Japan, New Guinea, and the Arafura Sea, to the northern coast of Australia and Fiji (Russell and Houston 1989). They exhibit schooling behavior in the coastal shore waters and lagoons (Letourneur et al. 2004; Whitehead 1988) within the relatively shallow waters of up to 75 m deep (Fricke et al. 2009). They live shorter lives, with a reported average life span ranging from 1.2 to 4 years (Hunnam 2021). The fish attains maturity at around 15 cm (Whitehead 1988) and can reach a maximum size of 27.0 cm standard length (Rahimi et al. 2016). The age at first maturity of the species is around 1 year, after which the species experience a high mortality rate (Conand 1991). The juvenile fish feeds on phytoplankton, but adults forage on the nauplii and zoea larvae, larvae of bivalves, gastropods, and adult copepods (Whitehead 1988). The fish are dioecious batch spawners (Milton et al. 1994) and produce up to 96,500 eggs, depending on fish size and environmental conditions (Sululu et al. 2020). The A. sirm spawn in schools (Conand 1991) and breed throughout the year in the IWP, with peaks from May to December (Whitehead 1988). In Tanzania, spawning extends throughout the year but is more prevalent in August and September (Sululu et al. 2020). Because the species exhibit short life spans, their prolonged serial spawning periods increase the likelihood of survival for the species in varying environmental conditions (Fréon et al. 2005; Ganias and Somarakis 2014; Sululu et al. 2020). The fertilization process for the Spotted sardinella is external, and eggs hatch into planktonic larvae, which remain in the water column for a few weeks until they turn into an adult (Conand 1991). During this stage, the larvae can be dispersed by the ocean currents at a varying scale depending on the strength and direction of the currents, size of the former population, landmass, and oceanographic barriers (Cowen and Sponaugle 2009; Mendez et al. 2010).
Connectivity is “the demographic linking local populations through dispersing individuals as larvae, juveniles, or adults” (Cowen et al. 2007). For the same species, organisms can either form a “closed populations” when they are self-recruiting or an “open populations” when there is a significant exchange of individuals among populations either in a planktonic stage (eggs and larvae) or migration in their adulthood (Postaire et al. 2017). It is an essential concept in conservation biology as it determines the recolonization potential of species at some localities when subjected to stressful natural and anthropogenic pressure like overfishing (Sahyoun et al. 2016). According to Sahyoun et al. (2016), management should consider genetic connectivity and stock structure as the essential information when designing a properly functioning marine protected areas (MPAs) network. The lack of considering genetic stock structure may cause fish stocks to experience difficulty recovering (Kerr et al. 2017). One case example is the Atlantic cod fishery which failed to recover because the fishery management ignored the population structure and connectivity of the species (Reiss et al. 2009).
Although the tropical and subtropical clupeids, including the A. sirm, produced over 2 million tons annually and significantly contributed to global marine capture production in 2016 (FAO 2018), reports on the genetic stock structure, connectivity, and intra-species variations are rare. Despite its commercial importance in Tanzania (Sululu et al. 2020), there is no report on the genetic structure of A. sirm. Most of the studies reported on marine invertebrates (Rumisha et al. 2017, 2018; Silva et al. 2013), Skunk clownfish, and other marine fishes (Huyghe and Kochzius 2018; Johnson et al. 2021; Rumisha et al. 2023). Previous studies on the genetic structure of the A. sirm were conducted in Sri Lanka, the Andaman Sea, and the South China Sea using mitochondrial DNA cytochrome b gene (Jamaludin et al. 2022; Saleh et al. 2020). Since there is no data on the genetic stock structure of the Tanzanian A. sirm, the fishery is managed as one randomly mating fish stock. While separately genetically distinct populations may exist, it is not known whether the current fishery strategy aligns with the genetic stock structure of the fishery. Without incorporating the genetic stock structure of the fishery into management, it can lead to loss of diversity, reduced productivity, and enhanced vulnerability of the fishery stocks to collapsing due to limited recruitment (Kerr et al. 2017). For example, disasters occurred in the USA for Atlantic cod (Gadus morhua), which the fishery failed to rebuild because management did not consider the genetic stock structure of the fishery (Lage et al. 2004); Zemeckis et al. 2014). These resulted in recruitment overfishing and the collapse of the entire Canadian Atlantic cod fishery (Kerr et al. 2017; Reiss et al. 2009).
A diverse array of genetic markers exists to assess the fishery stocks’ genetic structure. However, the cytochrome C oxidase subunit 1 (COI) gene is among the extensively used mitochondrial DNA (mtDNA) to assess genetic diversity and demarcate the genetic population structure of species. The large-scale use of the fragment is due to the relatively low amplification costs, simple isolation, and high abundance within the cell (Zhang and Hewitt 1996). The gene’s fast and slow-evolving regions result in potential variability for studies and an essential universal primer. Consequently, in this study, the COI gene of 78 A. sirm specimens was amplified from four locations in Tanzania to assess the genetic stock structure and demographic history of the A. sirm. The information in this study is important for designing an appropriate management strategy for the sustainability of the fishery.
Material and methods
Study area
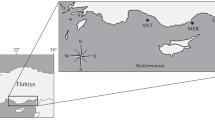
The Dar es Salaam (Ds) is the largest populated city in the country compared to other sampling sites of Kilwa (Kw), Tanga (Ta), and Mtwara (Mt) (Fig. 1). The increasing population in the city exerted pressure on marine resources, accelerated habitat degradation, and unsustainable fishing practices (Muhando and Mohammed 2002). The two monsoons have an impact on the study area, with the strong southeast (SE) monsoon dominating from April to October and the northeast (NE) monsoon dominating from November to March (McClanahan 1988).

Map showing sites where the Spotted sardinella (Amblygaster sirm) were sampled along the Tanzanian coast between 2020 and 2021. Shapefiles from the Database of Global Administrative Areas and the Quantum GIS software version 3.28 were used to create the map. (https://adm.org/download_country.html. Accessed 15 March 2023)
Tissue sample collection
Sampling was conducted between 2020 and 2021. A total of 78 A. sirm were collected from four sites which are Tanga (TA), Dar es Salaam (Ds), Kilwa (Kw), and Mtwara (Mt) in Tanzania (Table 1). Samples were obtained from fishers who used a ring net with a relatively small-mesh size of 8 to 10 mm. The taxonomic identification of the fish was accomplished on-site using the available field guides (Bianchi 1985; Whitehead 1988). The specimens were rinsed with clean distilled water, and fin clips were cut and immediately preserved in 2-ml microcentrifuge tubes containing 95% ethanol. The tissues were then transported to the College of Natural and Applied Sciences (CoNAS) of the Sokoine University of Agriculture (SUA) for laboratory and further analysis.
DNA extraction
Genomic DNA was extracted individually from each fin clip sample. Each sample was removed from 95% ethanol and suspended in 0.1 TE buffer for 2 h at room temperature. The contents were emptied into a petri dish, and about 2 mm2 of tissue was clipped and transferred to a microcentrifuge tube. Then 95 μl of DNA free water, 95 μl of solid tissue buffer, and 10 μl of proteinase K were added to each sample. Afterwards, the samples were vortexed and incubated at 55 °C for 3 h. DNA extraction was completed using the Zymo gDNA miniprep kit following the manufacturer’s instructions (Zymo Research Corporation, CA, USA). DNA quality was checked on a 1.0% agarose gel, and only samples with visible intact bands were selected for further analysis.
DNA amplification and sequencing
DNA from 78 fish samples were used for PCR and COI sequencing. A partial fragment of COI was amplified by using two sets of universal primers: COIceF (5′-ACTGCCCACGCCCTAGTAATGATATTTTTTATGGTNATGCC-3′) and COIceR (5′-TCGTGTGTCTACGTCCATTCCTACTGTRAACATRTG-3′) according to Hoareau and Boissin (2010). Each reaction (27 μl) contained 2 μl template DNA, 5 mg bovine serum albumin, 0.3 μM of forward and reverse primer, and 1 × OneTaq 2× Master Mix with standard buffer (New England BioLabs Inc., MA, USA). PCR amplification was performed at 94 °C for 3 min, followed by 40 cycles of 94 °C for 45 s, 51 °C for 70 s, and 72 °C for 80 s, and a final step at 72 oC for 15 min. The PCR amplicons were sequenced directly by Macrogen (Rockville, MD, USA) using the primer COIceF.
Data analysis
Genetic diversity
The sequences were edited and inspected using CHROMASPRO (v. 2.1, Technelysium Ltd, Leicester, UK). This was done to ensure that the chromatographic peaks represent the right nucleotides and to correct any sequencing errors. Species identification with DNA barcoding was conducted using BLASTn in the National Center for Biotechnology Information (NCBI) (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 19/05/2022) and the data portal of Barcode of Life Data Systems (BOLD Systems, http://v3.boldsystems.org/, consulted on 19/05/2022). All samples were blasted and identified as A. sirm at 99.9% similarity. Pairwise and multiple alignments of the sample sequences were accomplished using Clustal W (Thompson et al. 1994) in the MEGA ver. 11software. Sequences were trimmed to the least common length, and finally the nucleotide (π) and haplotype (h) diversities were computed in Arlequin version 3.5 (Excoffier and Lischer 2010).
Population genetic structure and phylogeny
The online service FaBox DNA Collapser (Villesen 2007) (https://birc.au.dk/~palle/php/fabox/dnacollapser.php, consulted on 29/05/2022) was used to reduce the sequences into haplotypes. In this process, 19 haplotypes were obtained and used to generate an input file for population structure analyses. Genetic structure was investigated using the analysis of molecular variance (AMOVA) technique (Excoffier et al. 1992) in the Arlequin (Excoffier and Lischer 2010). The software computed the populations’ pairwise differentiation (pairwise Fst) and overall genetic structure (Fst). Using the Bonferroni correction, the pairwise Fst P-values were further corrected (Holm 1979). Finally, a minimum spanning tree was built to show relationships between the haplotypes in the PopART ver. 1.7. software (Bandelt et al. 1999). The evolutionary relationships among the haplotypes sampled from Tanzania and other parts of the world were investigated using the Neighbor-Joining method (Tamura et al. 2021) constructed in the MEGA ver. 11 software. The final alignment used contained 38 nucleotide sequences, the 19 haplotypes from Tanzania, and additional 19 sequences from BOLD Systems. The bootstrap test was used with up to 1000 replicates, and evolutionary distances were computed using the Kimura 2-parameter method according to Russo and Selvatti (2018).
Historical demography
Historical demography analysis was performed in the Arlequin software. In this case, the null hypothesis of the neutral evolution of COI markers was investigated using Tajima’s D (Tajima 1989) and Fu’s FS (Fu 1997) tests. These tests enabled the detection of the signs of a bottleneck or sudden demographic growth in the overall dataset. The demographic expansion was again confirmed using the mismatch distribution analysis (Excoffier and Schneider 1999), computation of the sum of square deviation SSD (Rogers and Harpending 1992), and Harpending’s raggedness index HRI (Harpending 1994; Rogers and Harpending 1992). The program MIGRATE-N ver. 3.6.11 estimated the effective population size (Θ) and pairwise migration rate (m) based on a full migration model and Bayesian inference (Beerli and Palczewski 2010). The Bayesian skyline plot was constructed in BEAST v1.8.2 program (Drummond et al. 2012) to explore and reconstruct the historical population change. The analysis was conducted by setting up a relaxed uncorrelated lognormal molecular clock, with general time reversible (GTR) as the evolutionary model. In this case, the analysis was run for 10 million generations with parameters sampled every 1000 generations. The outputs and summary of the posterior distribution of population size over time were visualized in the Tracer v1.7 (Rambaut et al. 2018).
Results
Genetic population structure and connectivity
Successful sequences were uploaded into Genbank and given accession numbers ON631654-106 ON631731. AMOVA revealed very low genetic variations among sites (0.19%) and high variation (99.63%) within populations (Table 2). The analysis showed a small and non-significant index of genetic differentiation (Fst = 0.002, Фst = −0.004, p > 0.05). This indicates a lack of population structure among sites in Tanzania and that the A. sirm fishery in Tanzania constitutes a randomly mating one genetically similar stock. The latter was supported by the non-significant pairwise difference (pairwise Fst) between all the sites (Table 3). The panmictic stock in Tanzania was also illustrated by the haplotype network, which grouped all the sampled haplotypes into one cluster, regardless of their geographical regions. The network contained one highly abundant central haplotype shared by all populations. The majority of haplotypes were closely related, differing by one to three mutation steps (Fig. 2). The migration rate revealed that the populations are connected. Each population exchanged migrants with adjacent and distant populations, supporting the lack of population structure as revealed by AMOVA (Table 4). Further evidence for the lack of population structure was revealed by phylogenetic analysis which clustered together all haplotypes of A. sirm from Tanzania (Fig. 3). It also revealed that A. sirm populations in Tanzania are closely related with populations in Mozambique. However, because A. sirm populations from India, Andaman Sea, and Asia did not cluster together with those from Tanzania and Mozambique, it is possible that there are distinct genetic stocks, but requires further investigation.

The minimum spanning haplotype network of 19 haplotypes of Amblygaster sirm sampled from Tanzania between 2020 and 2021. The size of the circle reflects the number of sequences found in a haplotype (the large central cycle contains 58 samples, smallest surrounding circles have 1 sample). The abbreviations; Ta, Tanga; Ds, Dar es Salaam; Kw, Kilwa; Mt, Mtwara

Evolutionary relationships of Amblygaster sirm haplotypes sampled from Tanzania in relation to the sequences obtained from other parts of the world. The shark Carcharhinus brachyurus was used as the root of the tree
Genetic diversity and effective population size
The study identified 19 haplotypes in the dataset containing sample sequences from Tanzania. The number of haplotypes was highest at Tanga (Ta) and Kilwa (Kw) and lowest in Dar es Salaam (Ds) (Table 5). The analysis revealed moderate overall haplotype (h = 0.45 ± 0.07) and low nucleotide (π = 0.13 ± 0.001) diversities for A. sirm (Table 5). The haplotype diversities were lowest at Dar es Salaam (Ds) and highest at Tanga (Ta). The nucleotide diversities were lowest at Dar es Salaam (Ds) and highest at Tanga and Mtwara (Mt). The effective population size (Θ) was high in Tanga and Kilwa and lowest in Dar es Salaam (Table 5).
Historical demography
The null hypothesis of the neutral evolution of the COI marker was rejected for the overall dataset and individual sites in Tanzania (Table 5). The significant Tajima’s D and Fu’s Fs tests indicate a departure from the population equilibrium or demographic expansion. The latter was confirmed by the model of a sudden demographic expansion which provided non-significant values of SSD and HRI (p-value > 0.05) (Table 5) and the unimodal mismatch distribution (Fig. 4). Bayesian skyline plot on the other hand supported demographic growth by showing a slightly expansion of effective population size overtime (Fig. 5). The sign of demographic growth was supported by the low values of the genetic diversities, implying a need for strengthening protection of the species to prevent further population decline by overfishing. It will ensure a population increase in size or increase genetic diversity and avoid the effect of genetic drift due to overexploitation (Fig 5).

Pairwise mismatch distribution of cytochrome oxidase subunit I haplotype of Amblygaster sirm sampled from the Tanzanian coast. HRI, harpending raggedness index; τ, tau, which corresponds to the time in number of generations since demographic expansion

Bayesian skyline plot of Amblygaster sirm sequences from Tanzania, indicating a slightly expansion of effective population size overtime. Solid line represents median estimates, and shaded areas represent the 95% highest posterior density (HPD) limits
Discussion
Genetic population structure
The AMOVA found higher genetic variation within the overall dataset than between the sites, and no significant structure was detected for the A. sirm in Tanzania. The same was revealed by phylogenetic analysis and nature of the haplotype network with a big central dominant haplotype shared across the populations. These results suggest a lack of population structure and that the fishery in Tanzania is a single randomly mating genetically similar stock. These results match the existing management strategy that does not consider genetic structure in the management. Similarly, the lack of population structure was reported in the Western Indian Ocean (WIO) for the Tuna and tuna-like species (Díaz-Arce et al. 2020; Johnson et al. 2021), prawns (Mwakosya et al. 2018; Rumisha and Kochzius 2022), giant mud crabs (Rumisha et al. 2018), and other species of macroinvertebrates sampled from Tanzania (Obura et al. 2019; Silva et al. 2013).
In contrast, a small but significant genetic differentiation was documented in the WIO region for the Skunk clownfish Amphiprion akallopisos (Huyghe and Kochzius 2017), the East African giant mud crab Scylla serrata (Rumisha et al. 2017), and Octopuses (Van Nieuwenhove et al. 2019). In the South China Sea and the Andaman Sea, studies reported significant genetic differentiation for A. sirm (Jamaludin et al. 2022; Saleh et al. 2020). For Clupeidae species, where the A. sirm belongs, a weak significant stock structure was reported for the Sardinella albella in the Persian Gulf and Sea of Oman (Rahimi et al. 2016). The lack of genetic differentiation among the A. sirm along the coast of Tanzania can be due to the effect of the East African Coastal Current (EACC) that carries fish larvae during their planktonic stage of life from south to north of Tanzania (Rumisha and Kochzius 2022; Semba et al. 2019). The mixing of ocean waters between north and south was reported as a result of the weakening of the EACC during the NE monsoon winds resulting in the slow southward flow of ocean water (Nyandwi 2013). With the NE monsoon ending, the strong SE monsoon takes over and revives the EACC currents’ northward flow with potential fish larvae. Thus, the high gene flow among the A. sirm in Tanzania can result from larvae exchange between sites in Tanzania caused by EACC and under the influence of the prevailing monsoon winds. The A. sirm spawn throughout the year and peak during the SE monsoon (August and September) period (Sululu et al. 2020); this means that more planktonic larvae are also moved north-south direction by the SE monsoon after the weakening of the South-north EACC. The capacity of the species to breed throughout the year and that the species has a relatively short life and larvae of about a month (Conand 1991; Sululu et al. 2020) facilitate an increasing likelihood of availability of fish larvae during the two seasons (NE and SE monsoons). The single stock structure and high connectivity of A. sirm populations along the coast of Tanzania align with the current management regime, which does not consider genetic stocks in the fishery management strategy. Hence, even overfished sites can still rebuild by strengthening the management measures and promoting sustainable fishing practices. Furthermore, the relationships between haplotypes in Tanzania and other parts of the world may suggest the presence of phylogeographic pattern at a global level consisting of Tanzania and Mozambique, India and Andaman Sea, and lastly Taiwan and Australia. However, our results are inconsistent with a previous study on A. sirm that used mitochondrial DNA cytochrome b and identified the genetically diverged stocks supported by a phylogeny distributed in the Andaman and the South China Seas (Jamaludin et al. 2022). But additional samples from Asia, India, and other parts of the world are needed to confirm the phylogeographic pattern observed in the present study.
Genetic diversity and historical demography
Similar to our findings, the combination of high haplotype diversity and low nucleotide diversity is common in pelagic marine fishes (Chanthran et al. 2020). The low genetic diversity indicates a sign of a sudden demographic expansion from a few founders (Grant and Bowen 1998; Ivanova et al. 2021). The overall haplotype diversity estimates in this study are comparable to the previous reports (h = 0.53 ± 0.100) for the Skunk clownfish Amphiprion akallopisos (Huyghe and Kochzius 2017) in the WIO but low than the overall value (h = 0.98 ± 0.050) in the East Indian Ocean (EIO) region documented by the same study. The haplotype diversities were within the range of the reports for the Mud crabs Scylla serrata in the WIO (Fratini et al. 2016; Rumisha et al. 2017), the scalloped hammerhead shark Sphyrna lewini (Hadi et al. 2020), but low than the reports on the narrow-barred Spanish mackerel Scomberomorus commerson (h = 0.934 ± 0.002) from Tanzania using mtDNA control region (Johnson et al. 2021). The nucleotide diversities, on the other hand, are far low than the records in the WIO for the invertebrates like the East African Perisesarma guttatum (π = 0.42 ± 0.25%), the Indo pacific mangrove crabs Uca hesperiae (θπ = 0.25 ± 0.16%) and Neosarmatium africanum (π = 0.46 ± 0.26%) (Fratini et al. 2016). Our study’s relatively moderate haplotype and low nucleotide diversity suggest demographic expansion from a bottleneck event (Alves et al. 2001). The recent population growth in Tanzania was well supported by the negative and significant Tajima’s D, Fu’s FS tests, the Bayesian skyline plot, and non-significant parameters of the mismatch distribution analyses (Fig. 4 and Table 2). Since Dar es Salaam (Ds) showed the lowest Θ, the lowest genetic diversities at the site could be due to the impact of low effective size on genetic diversity. Low Θ at the site may result from overfishing due to high fishing pressure and degraded marine habitats (Rumisha et al. 2018). The latter was supported by the reef surveys that reported low fish diversity and abundance in Dar es Salaam compared to other sites in Tanzania (Muhando and Mohammed 2002). Because there is high genetic connectivity across sites in Tanzania, strengthening the management through enforcement can help rebuild the population’s diversities, especially at sites with the lowest genetic diversity, such as Dar es Salaam (Ds). Therefore, this study recommends increasing control and management measures to reduce overfishing, prevent further decline in populations, and avoid the effect of genetic drift.
Conclusion and recommendations
This study found a single genetic structure and high gene flow of the A. sirm along the coast of Tanzania, indicating that the fishery is a single genetically similar stock. These findings match with the current fishery management regime that does not consider the genetic structure of the A. sirm fishery. Therefore, future management approaches should consider other biological, ecological, and social-economic factors. The demographic history indicated a recent expansion of the A. sirm population in Tanzania after a bottleneck event. Because the population is genetically homogenous, the lowest genetic diversity at Dar es Salaam can be boosted by promoting sustainable fishing practices. The low overall genetic diversities in the overall dataset imply a need to strengthen enforcement and management to reduce overfishing and ensure the population increase in size by preventing overfishing and avoiding further effects of genetic drift. Nonetheless, because the COI marker used is based on a single locus and is maternally inherited (Bazin and Glémin 2006), the findings of this study should be validated using other markers, particularly those that assess genetic divergence across multiple loci like microsatellites (Zink and Barrowclough 2008). Additionally, future studies should collect samples from neighboring countries in the Western Indian Ocean (WIO) to confirm the panmictic stock and demographic growth documented by this study.
References
Alves MJ, Coelho H, Collares-Pereira MJ, Coelho MM (2001) Mitochondrial DNA variation in the highly endangered cyprinid fish Anaecypris hispanica: importance for conservation. Heredity 87:463–473. https://doi.org/10.1046/j.1365-2540.2001.00929.x
Bandelt H, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Bazin E, Glémin S (2006) Population size does not influence mitochondrial genetic diversity in animals. Science 312:570–572. https://doi.org/10.1126/science.1122033
Beerli P, Palczewski M (2010) Unified Framework to evaluate panmixia and migration direction among Multiple Sampling Locations. Genetics 185:313–326. https://doi.org/10.1534/GENETICS.109.112532
Bianchi G (1985) Field guide to the commercial marine and brackish-water species of Tanzania. FAO species identification sheets for fishery purposes. TCP/URT/4406 and FAO (FIRM) Regular Programme FAO, Rome
Chanthran SS, Lim PE, Li Y, Liao TY, Poong SW, Du J, Hussein MA, Sade A, Rumpet R, Loh KH (2020) Genetic diversity and population structure of Terapon jarbua (Forskål, 1775) (Teleostei, Terapontidae) in Malaysian waters. Zookeys 911:139–160. https://doi.org/10.3897/ZOOKEYS.911.39222
Conand F (1991) Biology and phenology of Amblygaster sirm (Clupeidae) in New Caledonia, a sardine of the coral environment. Bull Mar Sci 48:137–149
Cowen R, Gawarkiewicz G, Pineda J (2007) Population connectivity in marine systems an overview. Oceanography 20:14–21
Cowen R, Sponaugle S (2009) Larval dispersal and marine population connectivity. Annu Rev Mar 2009:443–466. https://doi.org/10.1146/annurev.marine.010908.163757
Díaz-Arce N, Grewe P, Krug I, Artetxe I, Ruiz J, Nikolic N, Medieu A, Pernak M, Lansdell M, Aulich J, Clear N (2020) Evidence of connectivity of bigeye tuna (Thunnus obesus) throughout the Indian Ocean inferred from genome-wide genetic markers. IOTC-2020-WPTT22 (AS) https://doi.org/www.hal.archivesouvertes. fr/hal-03206834
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/J.1755-0998.2010.02847.X
Excoffier L, Schneider S (1999) Why hunter-gatherer populations do not show signs of Pleistocene demographic expansions. Proc Natl Acad Sci U S A 96:10597–10602. https://doi.org/10.1073/PNAS.96.19.10597
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1093/GENETICS/131.2.479
FAO (2018) The state of world fisheries and aquaculture - meeting the sustainable goals. FAO, Rome
Fratini S, Ragionieri L, Cannicci S (2016) Demographic history and reproductive output correlates with intraspecific genetic variation in seven species of Indo-Pacific mangrove crabs. PLoS One 11:e0158582. https://doi.org/10.1371/JOURNAL.PONE.0158582
Fréon P, Cury P, Shannon L, Roy C (2005) Sustainable exploitation of small pelagic fish stocks challenged by environmental and ecosystem changes: a review. Bull Mar Sci 76:385–462
Fricke R, Mulochau T, Chabanet P et al (2009) Annotated checklist of the fish species (Pisces) of La Réunion, including a red list of threatened and declining species. Stuttg Beitr Naturk A, N Ser 2:1–168
Fu XY (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Ganias K, Somarakis S, Nunes (2014) Reproductive potential. In: Ganias K (ed) Biology and Ecology of Sardines and Anchovies. CRC Press: Boca Raton, FL, USA, pp 79–121
Grant WS, Bowen BW (1998) Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J Hered 89:415–426. https://doi.org/10.1093/JHERED/89.5.415
Hadi S, Andayani N, Muttaqin E et al (2020) Genetic connectivity of the scalloped hammerhead shark Sphyrna lewini across Indonesia and the Western Indian Ocean. PLoS One 15:e0230763. https://doi.org/10.1371/JOURNAL.PONE.0230763
Harpending H (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch Distribution. Hum Biol 66:591–600
Hoareau TB, Boissin E (2010) Design of phylum-specific hybrid primers for DNA barcoding: addressing the need for efficient COI amplification in the Echinodermata. Mol Ecol Resour 10:960–967. https://doi.org/10.1111/j.1755-0998.2010.02848.x
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6:65–70 https://doi.org/http://www.jstor.org/stable/4615733
Hunnam K (2021) The biology and ecology of tropical marine sardines and herrings in Indo-West Pacific fisheries: a review. Rev Fish Biol Fish 31:449–484. https://doi.org/10.1007/S11160-021-09649-9
Huyghe F, Kochzius M (2017) Highly restricted gene flow between disjunct populations of the skunk clownfish (Amphiprion akallopisos) in the Indian Ocean. Mar Ecol 38:e12357. https://doi.org/10.1111/MAEC.12357
Huyghe F, Kochzius M (2018) Sea surface currents and geographic isolation shape the genetic population structure of a coral reef fish in the Indian Ocean. PLoS One 13:e0193825. https://doi.org/10.1371/JOURNAL.PONE.0193825
Isaacs M (2016) The humble sardine (small pelagics): fish as food or fodder. Agric Food Secur 5:1–14. https://doi.org/10.1186/s40066-016-0073-5
Ivanova P, Dzhembekova N, Atanassov I, Rusanov K, Raykov V, Zlateva I, Yankova M, Raev Y, Nikolov G (2021) Genetic diversity and morphological characterisation of three turbot (Scophthalmus maximus L., 1758) populations along the Bulgarian Black Sea coast. Nat Conserv 43:123–146. https://doi.org/10.3897/natureconservation.43.64195
Jamaludin NA, Jamaluddin JA, Rahim MA, Akib NA, Ratmuangkhwang S, Arshaad WM, Nor SA (2022) Mitochondrial marker implies fishery separate management units for spotted sardinella, Amblygaster sirm (Walbaum, 1792) populations in the South China Sea and the Andaman Sea. PeerJ 10:e13706. https://doi.org/10.7717/peerj.13706
Johnson MG, Mgaya YD, Shaghude YW (2021) Analysis of the genetic stock structure and phylogenetic relationship of narrow-barred Spanish mackerel Scomberomorus commerson (Lacépède, 1800) along the northern Tanzanian coastal waters using mitochondrial DNA. Reg Stud Mar Sci 46:101862. https://doi.org/10.1016/J.RSMA.2021.101862
Kerr LA, Hintzen NT, Cadrin SX, Clausen LW, Dickey-Collas M, Goethel DR, Hatfield EM, Kritzer JP, Nash RD (2017) Lessons learned from practical approaches to reconcile mismatches between biological population structure and stock units of marine fish. ICES J Mar Sci 74:1708–1722. https://doi.org/10.1093/icesjms/fsw188
Lage C, Kuhn K, Kornfield I (2004) Genetic differentiation among Atlantic cod (Gadus morhua) from Browns Bank, Georges Bank, and Nantucket Shoals. Fish Bull 102:289–298
Letourneur Y, Chabanet P, Durville P et al (2004) An updated checklist of the marine fish fauna of Reunion Island, south-western Indian Ocean. Cybium 28:199–216. https://doi.org/10.26028/cybium/2004-283-002
McClanahan T (1988) Seasonality in East Africa’s coastal waters. Mar Ecol Prog Ser 44:191–199. https://doi.org/10.3354/meps044191
Mendez M, Rosenbaum HC, Subramaniam A, Yackulic C, Bordino P (2010) Isolation by environmental distance in mobile marine species: molecular ecology of franciscana dolphins at their southern range. Wiley Online Libr 19:2212–2228. https://doi.org/10.1111/j.1365-294X.2010.04647.x
Milton D, Blaber S, Rawlinson N (1994) Reproductive biology and egg production of three species of Clupeidae from Kiribati, tropical central Pacific. Fish Bull 92:102–121
Muhando CA, Mohammed MS (2002) Coral reef benthos and fisheries in Tanzania before and after the 1998 bleaching and mortality event. West Indian Ocean J Mar Sci 1:43–52
Mwakosya CA, Mgaya YD, Jiddawi NS (2018) Genetic connectivity of Fenneropenaeus indicus (H. Milne Edwards 1837) among three prawn fishing grounds of Tanzanian coastal waters. Reg Stud Mar Sci 24:107–112. https://doi.org/10.1016/j.rsma.2018.08.001
Nyandwi N (2013) The effects of monsoons on the East African Coastal Current through the Zanzibar Channel, Tanzania. J Ocean Technol 8:63–74
Obura D, Bandeira S, Bodin N, Braulik G (2019) Chapter 4 - the northern Mozambique channel. In: Sheppard C (ed) World seas: an environmental evaluation, 2nd edn. Academic Press, pp 75–99
Postaire B, Gélin P, Bruggemann JH, Magalon H (2017) One species for one island? Unexpected diversity and weak connectivity in a widely distributed tropical hydrozoan. Heredity 118:385–394. https://doi.org/10.1038/hdy.2016.126
Pradeep HD, Shirke SS, Kar AB (2014) Age, growth and mortality of Amblygaster sirm (Walbaum, 1792) from Andaman waters. J Andaman Sci Assoc 19:201–208
Rahimi P, Rezvani Gilkolaie S, Ghavam Mostafavi P, Jamili S, Rahnema M (2016) Population genetic structure of the white sardine, Sardinella albella, in the Persian Gulf and Sea of Oman by analysis of mitochondrial control region. Iran J Fish Sci 15:995–1008
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Reiss H, Hoarau G, Dickey-Collas M, Wolff WJ (2009) Genetic population structure of marine fish: mismatch between biological and fisheries management units. Fish Fish 10:361–395. https://doi.org/10.1111/j.1467-2979.2008.00324.x
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569. https://doi.org/10.1093/OXFORDJOURNALS.MOLBEV.A040727
Rumisha C, Bugota VJ, Mtonga CJ (2023) Evidence of extensive genetic connectivity and recent demographic expansion of the queen mackerel Scomberomorus plurilineatus in Tanzanian coastal waters. Mar Biodivers 53:9. https://doi.org/10.1007/s12526-022-01315-y
Rumisha C, Huyghe F, Rapanoel D, Mascaux N, Kochzius M (2017) Genetic diversity and connectivity in the East African giant mud crab Scylla serrata: Implications for fisheries management. PLoS One 12:e0186817
Rumisha C, Kochzius M (2022) Genetic evidence for a single stock of giant tiger prawns Penaeus monodon in demarcated prawn fishing zones of Tanzania. Fish Manag Ecol 30:36–43. https://doi.org/10.1111/FME.12598
Rumisha C, Mdegela RH, Gwakisa PS, Kochzius M (2018) Genetic diversity and gene flow among the giant mud crabs (Scylla serrata) in anthropogenic-polluted mangroves of mainland Tanzania: implications for conservation. Fish Res 205:96–104. https://doi.org/10.1016/j.fishres.2018.04.015
Russell B, Houston W (1989) Offshore fishes of the Arafura Sea. Beagle Rec North Territ Museum Arts Sci 6:69–84
Russo CAM, Selvatti AP (2018) Bootstrap and rogue identification tests for phylogenetic analyses. Mol Biol Evol 35:2327–2333
Sahyoun R, Guidetti P, Di Franco A, Planes S (2016) Patterns of fish connectivity between a marine protected area and surrounding fished areas. PLoS One 11:e0167441. https://doi.org/10.1371/JOURNAL.PONE.0167441
Saleh MF, Arshaad WM, Hassan RB, Masaya K, Latun AR, Fatah NN, Jaafar K (2020) Towards the sustainable management of purse seine fisheries in Southeast Asia. In: Fish for the People. SEAFDEC, Bangkok, pp 20–26
Semba M, Lumpkin R, Kimirei I, Shaghude Y, Nyandwi N (2019) Seasonal and spatial variation of surface current in the Pemba Channel, Tanzania. PLoS One 14:e0210303. https://doi.org/10.1371/journal.pone.0210303
Silva SE, Silva IC, Madeira C, Sallema R, Paulo OS, Paula J (2013) Genetic and morphological variation in two littorinid gastropods: evidence for recent population expansions along the East African coast. Biol J Linn Soc 108:494–508. https://doi.org/10.1111/J.1095-8312.2012.02041.X
Sululu JS, Kamukuru AT, Sekadende BC, Mahongo SB, Igulu MM (2020) Reproductive biology of the anchovy (Stolephorus commersonnii, Lacepède, 1803) and spotted sardine (Amblygaster sirm, Walbaum, 1792) from Tanga Region, Tanzania. West Indian Ocean J Mar Sci:81–94. https://doi.org/10.4314/wiojms.si2020.1.8
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. https://doi.org/10.1093/genetics/123.3.585
Tamura K, Stecher G, Kumar S (2021) MEGA11: Molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. https://doi.org/10.1093/NAR/22.22.4673
Van Nieuwenhove AHM, Ratsimbazafy HA, Kochzius M (2019) Cryptic diversity and limited connectivity in octopuses: recommendations for fisheries management. PLoS One 14:e0214748. https://doi.org/10.1371/journal.pone.0214748
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Mol Ecol Notes 7:965–968. https://doi.org/10.1111/J.1471-8286.2007.01821.X
Whitehead PJP (1988) FAO species catalogue: an annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, shads, anchovies and wolf-herrings. Pt 2 – Engraulididae. FAO Fisheries Synopsis, Rome
Zemeckis DR, Martins D, Kerr LA, Cadrin SX (2014) Stock identification of Atlantic cod (Gadus morhua) in US waters: an interdisciplinary approach. ICES J Mar Sci 71:1490–1506. https://doi.org/10.4135/9781412953924.n678
Zhang DX, Hewitt GM (1996) Nuclear integrations: challenges for mitochondrial DNA markers. Trends Ecol Evol 11:247–251. https://doi.org/10.1016/0169-5347(96)10031-8
Zink RM, Barrowclough GF (2008) Mitochondrial DNA under siege in avian phylogeography. Mol Ecol 17:2107–2121. https://doi.org/10.1111/J.1365-294X.2008.03737.X
Acknowledgements
The authors would like to thank the Western Indian Ocean Marine Sciences Association (WIOMSA) through the MARG I 2019 program for financial support to conduct this study. The authors also would like to thank the Tanzania Fisheries Research Institute (TAFIRI) and the Institute of Marine Sciences (IMS) for hosting and providing logistical support. Thanks to the College of Natural and Applied Sciences (CoNAS) of the Sokoine University of Agriculture (SUA) for providing space for molecular laboratory analysis. The authors appreciate the assistance of Asha Said and Alex Zuberi at the Department of Biosciences, SUA, during sample analysis in the lab. Also, the authors would like to thank all anonymous reviewers whose comments and advice helped improve this manuscript.
Funding
This study was funded by the Western Indian Ocean Marine Sciences Association (WIOMSA) through the MARG I 2019 program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
All applicable international guidelines for animal testing and the use of animals were followed by the authors.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities and are mentioned in the acknowledgements. The study is compliant with CBD and Nagoya protocols.
Data availability
The dataset of all COI sequences are available in the GenBank repository with the accession numbers ON631654 - ON631731.
Authors’ contribution
The study was conceptualized, planned, and carried out by Fabiani G and Chauka JL. Fabiani G and Rumisha C analyzed laboratory molecular samples. Fabiani G and Mtonga JC conducted a statistical analysis of the data. Mtonga JC and Fabiani G wrote the manuscript. Muhando AC, Chauka JL, and Rumisha C revised the manuscript. The final paper was read and approved by all authors.
Additional information
Communicated by R. Thiel
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fabiani, G., Mtonga, C.J., Chauka, L.J. et al. Mitochondrial DNA revealed a single stock structure of the Spotted sardinella Amblygaster sirm (Walbaum, 1792) (Teleostei; Clupeidae) in Tanzanian coastal waters. Mar. Biodivers. 53, 40 (2023). https://doi.org/10.1007/s12526-023-01351-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12526-023-01351-2