
Abstract
Pancreatic cancer continues to be the fourth leading cause of death despite advancements in surgical and adjuvant therapeutic approaches. In the present review, the current cytotoxic therapeutic approaches and advanced targeted therapies are objectively discussed with consideration to the current literature.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer has an incidence of 10/100,000 in the general population and is the fourth leading cause of cancer-related death in males and fifth leading cause in females [1, 2]. The long-term survival rate is very low, being 14% in stage 1 disease and 1% in stage 4 disease [2]. As the majority of patients are diagnosed with either locally or systemically advanced disease stage, surgery alone will not be curative and multimodality therapy will be necessary [3]. The resectability of pancreatic cancer is around 15–25% of the cases undergoing laparotomy [4]. However, with a good preoperative work-up and accurate patient selection it can increase to 75% [4, 5]. Long-term survival following curative surgery approaches 25%, even in experienced centers [3]. These figures emphasize the importance of adjuvant therapy in providing better patient survival [6]. In the present study, we attempted to summarize current conventional and targeted adjuvant treatment protocols for pancreatic cancer.
Methods
The MeSH terms including “cytotoxic therapy and pancreatic cancer” and “targeted therapy and pancreatic cancer” were used for PubMed searches between 1985 and 2014. Suitability as a reference was decided after reading the abstract. For regularly used therapies, such as conventional cytotoxic therapy, randomized controlled trials or high-impact reviews were selected. For experimental therapies, such as gene therapy, phase 1 and 2 clinical trials were selected. The search for conventional cytotoxic therapy yielded 814 articles of which 23 were chosen as references. Searching for targeted therapy yielded 1,368 articles of which 59 were selected for this review.
Conventional Cytotoxic Therapy
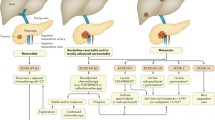
To summarize the topic properly, it would be practical to evaluate pancreatic cancer in three subclasses including resectable pancreatic cancer, locally advanced pancreatic cancer (LAPC), and metastatic pancreatic cancer. Scenarios involving resectable and metastatic (inoperable) pancreatic cancer can easily be defined for in the first scenario there is no extra-pancreatic involvement of the tumor, no major vascular involvement, and R0 resection can be obtained by pancreaticoduodenectomy. On the other hand, metastatic disease shows distant organ metastasis regardless of the condition of the primary tumor and, therefore, no definitive cure can be obtained for these patients. LAPC is usually defined as a tumor having a broad retroperitoneal margin and either requires major vascular resection to obtain negative margins or is considered unresectable intraoperatively. Nevertheless, surgery is far from providing a cure for LAPCs. The reason for this classification is based on the fact that different modalities gain importance at different stages of the disease.
The mainstay therapeutic approach in resectable disease is definitely surgery, which provides the highest chance of a successful cure [6]. The exact role for adjuvant therapy in resectable pancreatic cancer is not clear. In fact, there are various alternatives to adjuvant therapy for resectable pancreatic cancer. The Gastrointestinal Study Group (GITSG) indicated a beneficial effect of bolus 5-fluorouracil (5-FU) with radiotherapy in terms of survival [7]. Furthermore, the Radiation Therapy Oncology Group (RTOG) 9704, stated the beneficial effect of adding gemcitabine to infusion 5-FU combined with radiotherapy [8]. The Charité Onkologie Clinical-001 (CONKO-001) study randomized patients to either postoperative gemcitabine or observation and at the end of the study they reported a survival benefit of the adjuvant chemotherapeutic approach [9]. On the other hand, the European Organization of Research and Treatment of Cancer (EORTC) trial did not show any beneficial survival effects for patients treated with adjuvant chemoradiation [10]. In addition, the ESPAC-3 trial compared gemcitabine and 5-FU in an adjuvant setting and found that both therapeutic approaches provided a survival benefit but that the gemcitabine arm showed fewer adverse effects [11]. The Japanese Adjuvant Study Group of Pancreatic Cancer reported very successful results with S-1 not only in adjuvant setting but also in advanced pancreatic cancer, which was comparable to gemcitabine [12]. S-1 is an oral fluoropyrimidine that consists of tegafur, gimeracil, and oteracil. It is a prodrug form of 5-FU and an alternative to infusional 5-FU therapy [13, 14]. Nevertheless, it has been confirmed by some trials that adjuvant therapy provides a survival benefit in resectable pancreatic cancer. Most important of all is the JASPAC-01 trial comparing S-1 and gemcitabine in the patients with pathological stage II or lower, or stage III with the resection including celiac artery, macroscopically resected (R0 or R1) pancreatic cancer [15]. S-1 showed superiority to gemcitabine in overall survival with a hazard ratio of 0.56 (95% CI: 0.42–0.74; p < 0.0001 for superiority) [15]. The findings of the JASPAC-01 trial suggest that adjuvant chemotherapy with S-1 is a more effective alternative to gemcitabine in resectable pancreatic cancer [15, 16].
In the case of LAPC, the clinical scenario changes dramatically. In fact, LAPC is usually unresectable and the survival of the patients is limited to 9–10 months without any treatment [17]. Therefore, a multidisciplinary approach and potent treatment modalities are needed [18]. The currently accepted chemotherapeutic strategy for LAPC includes either capecitabine or gemcitabine, optionally combined together with radiation therapy [14]. There are some clinical trials regarding alternative treatment regimens. The Gastrointestinal Tumor Study Group has compared streptozotocin, mitomycin, and 5-FU triple therapy (SMF protocol) to the 5-FU and radiotherapy followed by the SMF protocol [19]. They concluded that multimodality combination therapy was superior to the SMF protocol alone in LAPC in terms of survival [19]. In addition, the Eastern Cooperative Oncology Group (ECOG) compared 5-FU with concurrent 5-FU with radiotherapy and maintenance and concluded that there was no difference in terms of survival [20]. Furthermore, toxicity of the combination therapy was higher than the single-agent treatment [20]. Results of the 2000-01 FFCD/SFRO study revealed that a combination of radiotherapy together with chemotherapeutics had poor prognosis when compared to gemcitabine alone, which was attributed to the toxicities of the combination therapy [21]. Although the ECOG trial found an enhanced survival benefit in the treatment arm combining gemcitabine with the radiotherapy, grade 5 toxicities were higher in the combination therapy [22].
Metastatic pancreatic cancer has an extremely dismal prognosis. Furthermore, the performance status of the patients is very poor so that the tolerability of any multimodality and highly toxic therapy becomes a big concern. Nevertheless, in 1997, gemcitabine was approved as the first-line therapy for metastatic pancreatic cancer [23]. Combining gemcitabine with any other cytotoxic agent did not show any overall survival benefits [24]. ECOG showed an enhanced overall and progression-free survival with the 5-FU, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) when compared with gemcitabine alone [25]. Nevertheless, myelosuppression was observed as a grade 3–4 adverse effect of the treatment and the toxicity of therapy was highly emphasized by the authors [25].
In conclusion, the abovementioned trials emphasize the fact that, although combination therapies provide enhanced survival or better symptom control, the side effects encountered during the treatment period limit the applicability of the treatment regimens. Therefore, more specific, personalized, and targeted therapies are needed.
Targeted Therapy for Pancreatic Cancer
Targeted therapy for cancer depends on the basic principle that the given treatment inhibits certain molecular pathways involving cell proliferation, anti-apoptosis, invasion, and metastasis that are all vital for tumorigenesis [26]. There are many strategies utilized in molecular targeted therapy for cancer. These can be classified into five groups:
-
1.
small molecule inhibitors, such as the tyrosine kinase inhibitors (erlotinib, sonic hedgehog (SHH) inhibitors);
-
2.
monoclonal antibodies, such as cetuximab and bevacizumab;
-
3.
gene therapy, including p53, p16, retinoblastoma gene, KRAS, bcl-2, etc.;
-
4.
epigenetic control of the target genes (RNA interference of certain genes such as zinc transporter and sphingosine kinase 1);
-
5.
oncolytic viruses (viral oncolysis or suicide gene therapy) [18, 27].
Monoclonal antibodies will be discussed in detail whereas the other modalities will be briefly summarized in the “Future Directions” section.
Molecular Basis of Targeted Therapy for Pancreatic Cancer
Epidermal growth factor (EGF) is a member of the erbB receptor family that are actually receptor tyrosine kinases. The erbB receptor family contains four closely related receptor kinases: erbB1, erbB2, erbB3, and erbB4. Epidermal growth factor receptor (EGFR), also known as the erbB1, has three distinct cytoplasmic domains: (1) a juxtamembrane domain which is the regulatory region; (2) a central tyrosine kinase domain which is the effector region (target for erlotinib); and (3) a non-catalyzing carboxy-terminal region. Overexpression of this receptor and the downstream signaling molecules is one of the earliest genetic aberrations during pancreatic carcinogenesis [28]. EGFR is related with molecular mechanisms governing tumor growth (uncontrollable cell division and proliferation), metastasis (loss of cell-to-cell adhesion), and epithelial-to-mesenchymal transition (EMT) [29]. Thirty to eighty percent of pancreatic ductal epithelial cancers overexpress EGFR and it is related with a poor prognosis [30, 31]. EGFR activation through the binding of the ligand activates ras- through the activation of GRB2 and Shc that activates sos-. Raf and MEK1 dual kinase are on the downstream of ras-. These complexes activate extracellular signal-regulated kinase (ERK) family. ERK translocates to the nucleus and regulates a series of transcription factors which control cell proliferation. Functional derangement in signaling causes uncontrolled cell proliferation and contributes to pancreatic carcinogenesis [32–34]. Co-expression of EGFR and its ligands in patients with pancreatic cancer has been correlated with poor prognosis and advanced-stage disease [35, 36].
The K-ras gene on chromosome 12p12 encodes the p21-ras protein, which is a membrane-bound signal-transducing protein. K-ras mutation was first described in pancreatic cancer in 1988 [37]. Since then, it has been shown by many studies that a point mutation in the K-ras gene is present in 85–90% of patients [37, 38]. The mutation is almost always on codon 12 of the protein where the glycine normally present in this position is either changed to valine or aspartate. This genetic defect in the protein causes the GTP control over the protein and the mutant protein stays in the activated form amplifying extrinsic signals of cell growth and invasion [37–39]. Interestingly, Navas et al. [40] has shown that in pancreatic carcinoma K-ras-related tumorigenesis is closely related with EGFR amplification, which is not observed in colon or lung cancer. The stable mutation of the K-ras gene in the majority of the pancreatic cancers makes it an attractive target for immunotherapy and gene therapy [41].
Furthermore, many other genetic alterations are present in pancreatic cancer involving p53, c-erbB-2, and deleted in pancreatic cancer locus 4 (DPC4) which are all tumor suppressor genes. All of them play an important role in the pancreatic carcinogenesis [42]. p53 is a tumor suppressor gene that is involved in cell cycle regulation, DNA synthesis, DNA repair, apoptosis, and cell differentiation. It is generally deleted in more than 75% of patients with cancer [43]. The mutation in p53 is a loss-of-function type mutation. This loss of function leads loss of G1 arrest in case of DNA damage. This makes the cell more prone to DNA damage, and is passed on to daughter cells and is deleted in most cancer types [43, 44].
Another active area of research for a novel therapeutic target for pancreatic cancer is the tumor microenvironment (TME). Components of the TME include the extracellular matrix (ECM), angiogenesis, pancreatic stellate cells, and the tumor-associated immune cells [45]. Pancreatic cancer is rich in stromal component.
ECM is a very important part of TME that plays a critical role in carcinogenesis and tumor progression. The components of the ECM are collagen, fibronectin, proteoglycans, and hyaluronic acid. One active area of research in targeting the ECM is the SHH signaling pathway [45, 46]. The exact mechanism of the SHH pathway contributing to pancreatic carcinogenesis is not clearly known. Nevertheless, ligands for the SHH have been secreted from the ductal epithelium resulting in the downstream signaling molecules to be activated in the juxta-neoplastic stroma [47]. SHH pathway activation in the stroma leads to transcription of certain genes, such as PTCH1, SMO, HIP and GLI1, that leads to accumulation of components of ECM in the tumor known as the desmoplastic reaction [45–48]. Desmoplastic reaction causes distortions in the normal architecture of the pancreatic tissue that limits the blood supply and the lymphatic drainage. This can be a dismal factor in limiting the efficacy of cytotoxic therapy [49–51].
HER1/EGFR Tyrosine Kinase Inhibitors
Erlotinib
A key molecular event in pancreatic carcinogenesis is EGFR overexpression [32]. Erlotinib is a tyrosine kinase inhibitor that prevents the activation step of EGFR1 (erbB1) and therefore blocks the activation of the EGFR pathway (Fig. 1). It was approved by the US Food and Drug Administration in 2007 for the treatment of advanced pancreatic cancer [52]. Before the introduction of erlotinib, the major breakthrough in the treatment of pancreatic cancer was gemcitabine, which was approved in 1997 [53]. Currently, erlotinib is considered the first-line therapy in the treatment of advanced pancreatic cancer as a single agent or in combination with gemcitabine [52–55].

The mechanism of action: cetuximab and erlotinib. EGFR ligands bind to EGFR and induce dimerisation. EGFR has tyrosine kinase domain which is phosphorylated after dimerisation and then EGFR is activated. Activated EGFR stimulates the downstream signal transduction. As a result, proliferation, angiogenesis, and metastasis occur. Cetuximab is a chimeric monoclonal antibody against EGFR and inhibits the dimerisation. Erlotinib is a small molecule that inhibits phosphorylation of tyrosine domains of EGFR. EGFR Epidermal growth factor receptor, TK tyrosine kinase
Randomized controlled trials comparing erlotinib and gemcitabine to gemcitabine monotherapy showed significantly longer overall survival and median survival period [52–57]. However, the difference in the survival was only 1 month, or slightly longer, and therefore not very prominent. Although reported progression-free survival and average time to progression are longer, this does not extrapolate to a more prominent better prognosis [56, 57]. Serious adverse effects are very rare with erlotinib therapy. The most commonly reported side effects are skin rash and serious grade 3–4 adverse reactions involving fatigue, anorexia, vomiting, stomatitis, and diarrhea [58]. Stathopoulos et al. [59] reported serious hematologic complications with erlotinib monotherapy involving acute myelogenous leukemia, chronic myeloid leukemia, and myelodysplastic syndrome, but the evidence was not enough to attribute the leukemia development to erlotinib treatment. Interestingly, Moore et al. [56] reported the skin rash to be a positive prognostic marker for erlotinib therapy. Positive response predictive markers are reported to be patient age ≤65 years, ECOG performance score of 2, male gender, pain scores ≤20, presence of metastatic disease, baseline C-reactive protein >1.4 mg/L, baseline lactate dehydrogenase more than the upper limit of the normal range, and primary tumor located at the tail of the gland [56, 57]. Furthermore, distribution and level of EGFR did not predict the response to the erlotinib therapy [56].
Nevertheless, based on the available data we can say that, although erlotinib revolutionized the treatment of pancreatic cancer, it is not sufficient for a more prominent response. One explanation for this phenomenon is that all the data collected for erlotinib have been obtained from patients with metastatic disease or locally advanced unresectable disease. There are no clear data on the effect of erlotinib in localized resectable disease. Since the contribution of erlotinib in the treatment of pancreatic cancer is only marginal, more specific and powerful therapies targeting this signaling axis are needed.
Cetuximab
Cetuximab is a chimeric monoclonal antibody against EGFR that prevents the activation and downstream signaling of the receptor (Fig. 1) [60]. Preclinical models utilizing orthotropic implantation of pancreatic cancer cells yielded promising results, which was a prominent anti-tumor response when cetuximab was combined with conventional gemcitabine-based cancer therapies [61, 62]. Unfortunately, the clinical trials did not live up to the expectations raised by the preclinical data. It was first utilized in a multicenter phase II trial performed by Xiong et al. [63] where 41 patients with a locally advanced, metastatic, or recurrent pancreatic cancer were treated with cetuximab and gemcitabine. Overall 1-year survival rate was 31.7% and progression-free survival was 12%. These data seemed to be favorable when the results of the study were compared to a previous phase III trial of gemcitabine monotherapy which showed 18% overall survival and 9% progression-free survival [22]. Cascinu et al. [64] evaluated the efficacy of combining cetuximab with gemcitabine and cisplatin versus gemcitabine plus cisplatin alone. Eighty-four patients were randomized to the treatment arms containing cetuximab or non-cetuximab-based combination therapy. Objective response rates were 17.7% and 12.2% in the cetuximab and non-cetuximab treatment arms, respectively. The study concluded without any significant response in terms of objective response, progression-free survival, and overall survival. The toxicity profiles of both treatment arms were similar and the study failed to show any beneficial effect of adding cetuximab to the combination therapy [64]. Kullmann et al. [65] evaluated the efficacy of combining triple agents including cetuximab plus gemcitabine and oxaliplatin in the treatment of locally advanced or metastatic pancreatic cancer. The results of the study were compared to a pooled analysis performed by Heinmann et al. [66] in 2007. Unfortunately, the objective response rate, progression-free survival, and the overall survival were not significantly different from the gemcitabine plus oxaliplatin trials presented by Heinmann et al. [66].
While all the studies evaluated above were phase II trials with a certain level of limitation in terms of study outcome, a key randomized phase III study performed by Philip et al. [67] compared the efficacy of cetuximab plus gemcitabine to gemcitabine alone. Unfortunately this study also failed to show a significant beneficial effect of the combination therapy compared to monotherapy alone in terms of overall survival (6.3 vs. 5.9 months in the combination vs. monotherapy arms, respectively), progression-free survival, and the objective response rates. None of the studies mentioned above [60–67] could find a correlation between the response rates and the EGFR expression level. Furthermore, all of them found that skin rash due to cetuximab therapy was the most common adverse effect. In addition, as in erlotinib therapy, development of the skin rash correlated with the positive response rates to the therapy.
In conclusion cetuximab, as in erlotinib therapy, did not keep up with the expectations raised during the preclinical stage of the drug development.
Resistance to Treatment Strategies Targeting EGFR Signaling Axis
Since agents targeting the EGFR signaling axis did not meet the expectations raised in the preclinical setting, researchers became more interested in the potential mechanisms of resistance. There are many explanations for this phenomenon but most of them are at the theory level. For example, persistent activation of the tyrosine kinase is the most popular hypothesis that has not yet been tested. Goldman et al. [68] found enhanced VEGF-mediated angiogenic response in the glioblastoma multiforme models and proposed it to be a bypass pathway for EGF signaling cascade that produced resistance to EGFR antagonists.
Another resistance mechanism is a mutation that can occur in the receptor or the downstream molecule that renders the organism unable to bind and respond to monoclonal antibodies and small molecule inhibitors [69]. Therefore, a lot still needs to be done to develop a novel agent that targets the EGFR pathway.
Bevacizumab
A variety of factors are involved in the regulation of angiogenesis. Vascular endothelial growth factor (VEGF) is one of the main growth factors involved in vessel formation. VEGF, also known as vascular permeability factor (VPF), was originally described as an endothelial cell-specific mitogen [70]. VEGF is produced by many cell types including tumor cells, macrophages, platelets, keratinocytes, and renal mesangial cells. The activities of VEGF are not limited to the vascular system; VEGF plays a role in normal physiological functions such as bone formation, hematopoiesis, wound healing, and development [70]. Bevacizumab is humanized monoclonal anti-VEGF-A antibody. It blocks the VEGF-A epitope site and blocks ligand receptor interaction (Fig. 2) [71]. Since the VEGF signaling pathway provides a bypass for the EGFR signaling pathway, blocking this pathway may provide an enhanced treatment alternative. Van Cutsem et al. [57] challenged this hypothesis in a phase III trial for advanced pancreatic cancer involving 607 patients enrolled to bevacizumab plus erlotinib plus gemcitabine (n = 306) and erlotinib plus gemcitabine only (n = 301) groups. Unfortunately, the authors found no survival benefit from bevacizumab treatment [57]. A recent multicenter phase II trial published by Ko et al. [72] analyzed the effect of bevacizumab on combination therapy involving cetuximab or cetuximab plus gemcitabine. The study was terminated early due to poor results observed in the cetuximab plus bevacizumab arm [72]. A phase III trial by Kindler et al. [73] also found no survival benefit from combining bevacizumab and gemcitabine when compared to gemcitabine alone.

The mechanism of action: bevacizumab. Bevacizumab is a humanized recombinant monoclonal antibody against to VEGF-A. It prevents the binding of VEGF-A to VEGFR1 and VEGFR2, then the receptors cannot be activated. Consequently, bevacizumab inhibits angiogenesis and tumor growth. VEGF Vascular endothelial growth factor, VEGFR vascular endothelial growth factor receptor
Future Directions
Although there have been advances in current cytotoxic therapy, we have still not reached our survival goal for pancreatic cancer in terms of adjuvant therapy. Surgery is still the only durable option providing the chance of a cure. However, the lack of neoadjuvant and adjuvant therapy severely limits the effect of surgery in prolonging the survival for patents and also preventing loco-regional recurrences. This is especially more pronounced in patients that have locally advanced tumors.
The targeted therapies have yielded only modest responses until now and there is clearly a need for more specific and potent therapies. There is clearly a need for more specific and patient-oriented approach for the treatment of pancreatic cancer. There are some experimental treatment protocols that are being tested. Some are in the preclinical setting and some have come to phase I and II clinical testing. Due to the limitations of the context of the current review, we will only define the recent most popular experimental strategies for the treatment of pancreatic cancer.
Cell-Based Vaccination Therapies
There are many strategies utilizing autologous or orthotropic-activated immune cells for the treatment of cancers. The basis of cell-based therapies is to induce an adoptive specific anti-tumor immune response in cancer patients. The main effector of this response is the cytotoxic T cell and the natural killer (NK) cells. The effector cells can be autologous or allogeneic in nature [74, 75]. Nevertheless, all of these therapies are experimental in nature and there are only a limited number of case reports regarding their utilization in advanced pancreatic cancer. Qui et al. [76] used recombinant technology to express an α1,3 galactosyl epitope in the tumor cells isolated from the lymph nodes removed from patients. The idea behind their study was that human cells did not include the α1,3 galactosyl epitope and once the engineered cells had expressed the antigen they were co-incubated with dendritic cells (DC), isolated from the peripheral blood of the patients, and cytokine-induced killer cells, isolated from the bone marrows of the patients. The study included 14 patients with advanced pancreatic cancers. After one to five cycles of DC-CIK cell-based vaccination with this protocol, six patients had stable disease, two had partial response, and six remained as progressive disease. The authors showed a positive correlation with delayed-type hypersensitivity reaction and the response rate of the patients, and concluded that the survivors showed a significant level of delayed-type hypersensitivity reaction [76]. Similarly, Long et al. [75] reported a case of advanced pancreatic cancer treated with cytokine-induced killer cell vaccination. Progression-free survival increased together with a better quality of life for the patient [75]; however, the result of the study should be evaluated with care as it is a single case report. Nevertheless, these two studies emphasize the importance of innovative therapies in pancreatic cancer.
Another strategy is to use the DC as a part of the vaccination process. DC are antigen-presenting cells which are responsible for production of long-term and sustained specific anti-tumor immunity. They can be loaded with specific tumor-associated antigens (TAA) or can be transfected with tumor-derived mRNAs to express specific TAA [77]. There are few early clinical trials utilizing DC-based vaccination therapy in advanced-stage pancreatic cancer. Lepisto et al. [78] utilized ex vivo MUC-1-derived peptide-loaded DC to treat pancreatic and biliary tract cancers in an adjuvant setting and, in a 4-year follow-up, 4 of the 12 patients survived without any evidence of the disease. Kimura et al. [79] recently published a phase I/II clinical trial using a DC-based vaccine in combination with lymphokine-activated killer cells (LAKC) and standard gemcitabine and or S-1 chemotherapy in advanced-stage pancreatic cancer. The authors concluded that multimodality combination yielded superior overall survival and that DC vaccination was effective only when combined with LAKC [79].
The main barrier underlying the utilization of various immunocytes in adoptive immune therapy of cancers, and especially pancreatic cancer, is that there is always an ex vivo component which is in great need for utilizing the right TAA to target the immunocytes for the tumor. In the case of pancreatic cancer, it not always easy to find that specific TAA. Furthermore, the tumor tissue develops certain ways to circumvent the immune surveillance and generally the TME is immunosuppressive [73–76]. Therefore, there is need to develop new strategies for stimulating the immune system of the patient against the hosted tumor. For this reason, whole cancer cell-based vaccines are being developed for human use which eliminates the need for developing epitopes for specific TAA and ex vivo stimulation of the immunocytes because it utilizes the irradiated/homogenized tumor cells itself to stimulate the host immune system. They can be considered as autologous vaccines that stimulate lymphocytes and patients’ sera against the TAA [80, 81]. This strategy was further improved by engineering the tumor cells to express certain cytokines such as granulocyte–macrophage colony-stimulating factor (GM-CSF), i.e., GVAX as trademark) for enhanced immune response [80]. Murine α-1,3-galactosyl transferase for α-galactosylated epitope expression in the human cancer cells lines (algenpantucel-L), which is foreign to the human immune system and evokes a potent anti-cancer immune response, might also be used to improve this strategy [81]. Furthermore, experimentally oncolytic viruses seem to be effective tools to evoke a sustained and potent anti-tumor immune response in individuals with a very good safety profile due to the inability to infect normal tissues but producing a lytic infection in the tumor tissues. It can also remedy the resistance mechanisms of the tumor tissue against the host immune surveillance [82, 83].
In addition to using cell-based vaccines or oncolytic viruses, there are some experimental protocols utilizing that target the TME and modify it to enhance anti-tumor immune response. These are all yet experimental approaches; however, animal models have yielded promising results. The studies mostly utilized standard cytotoxic chemotherapy to circumvent myeloid-derived suppressor cells to reduce tumor-associated immunosuppression. Many preclinical studies have utilized gemcitabine at clinically relevant dose and have combined it with modified tumor vaccines or IFN gamma gene bearing adenovirus vaccines [84, 85]. These studies have shown abundant CD8 cytotoxic lymphocyte-mediated specific anti-tumor immune responses that were coupled to reduced Gr-1+/CD11b+ myeloid-derived suppressor cells [84, 85]. These two studies mainly emphasize the importance of targeting the TME.
Conclusions
The diagnosis and treatment of pancreatic cancer has improved in the last decade and there is now more hope for enhancing the survival of patients. There are more specific therapies being developed and being tested for human use. Nevertheless, there are still limitations to be overcome for the success of specific therapies. We believe the answer lies in the TME and modulation of the host immune system against tumor cells. More experimental work and clinical correlations are needed but the future seems to be the biological therapies and adaptive immunotherapy adjunct to the routine targeted and cytotoxic therapies for pancreatic cancer.
This review is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
References
Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics, 1997. CA Cancer J Clin. 1997;47(1):5–27.
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29.
Gaedcke J, Gunawan B, Grade M, et al. The mesopancreas is the primary site for R1 resection in pancreatic head cancer: relevance for clinical trials. Langenbecks Arch Surg. 2010;395(4):451–8.
Balcom JH, Castillo CF. Can we predict resectability in pancreatic cancer? Ann Gastroenterol. 2000;13(3):201–6.
Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363(9414):1049–57.
Hackert T, Buchler M, Werner J. Current state of the surgical management of pancreatic cancer. Cancers. 2011;3:1253–73.
Kalser MH, Ellenberg SS. Pancreatic cancer. Adjuvant combined radiation and chemotherapy following curative resection. Arch Surg. 1985;120(8):899–903. Erratum in: Arch Surg 1986 Sep;121(9):1045.
Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350(12):1200–10.
Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297(3):267–77.
Klinkenbijl JH, Jeekel J, Sahmoud T, et al. Adjuvant radiotherapy and 5-fluorouracil after curative resection of cancer of the pancreas and periampullary region: phase III trial of the EORTC gastrointestinal tract cancer cooperative group. Ann Surg. 1999;230(6):776–82.
Neoptolemos JP, Stocken DD, Bassi C, et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304(10):1073–81.
Maeda A, Boku N, Fukutomi A, et al. Randomized phase III trial of adjuvant chemotherapy with gemcitabine versus S-1 in patients with resected pancreatic cancer: Japan Adjuvant Study Group of Pancreatic Cancer (JASPAC-01). Jpn J Clin Oncol. 2008;38(3):227–9.
Saif MW, Syrigos KN, Katirtzoglou NA. S-1: a promising new oral fluoropyrimidine derivative. Expert Opin Investig Drugs. 2009;18(3):335–48.
Saif MW. Advancements in the management of pancreatic cancer: 2013. JOP. 2013;14(2):112–8.
Uesaka K, Fukutomi A, Boku N, et al. Randomized phase III trial of adjuvant chemotherapy with gemcitabine versus S-1 for patients with resected pancreatic cancer (JASPAC-01 study). J Clin Oncol 30. 2012;(suppl 34; abstr 145).
Fukutomi A, Uesaka K, Boku N, et al. JASPAC 01: Randomized phase III trial of adjuvant chemotherapy with gemcitabine versus S-1 for patients with resected pancreatic cancer. J Clin Oncol 31. 2013;(suppl; abstr 4008).
Zhu CP, Shi J, Chen YX, Xie WF, Lin Y. Gemcitabine in the chemoradiotherapy for locally advanced pancreatic cancer: a meta-analysis. Radiother Oncol. 2011;99(2):108–13.
Zhang Y, Satoh K, Li M. Novel therapeutic modalities and drug delivery in pancreatic cancer – an ongoing search for improved efficacy. Drugs Context. 2012;2012:212244. doi:10.7573/dic.212244
Gastrointestinal Tumor Study Group. Treatment of locally unresectable carcinoma of the pancreas: comparison of combined-modality therapy (chemotherapy plus radiotherapy) to chemotherapy alone. J Natl Cancer Inst. 1988;80(10):751–5.
Klaassen DJ, MacIntyre JM, Catton GE, Engstrom PF, Moertel CG. Treatment of locally unresectable cancer of the stomach and pancreas: a randomized comparison of 5-fluorouracil alone with radiation plus concurrent and maintenance 5-fluorouracil—an Eastern Cooperative Oncology Group study. J Clin Oncol. 1985;3(3):373–8.
Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2000-01 FFCD/SFRO study. Ann Oncol. 2008;19(9):1592–9.
Loehrer PJ Sr, Feng Y, Cardenes H, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol. 2011;29(31):4105–12.
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–13.
Saif MW, Kim R. Role of platinum agents in the management of advanced pancreatic cancer. Expert Opin Pharmacother. 2007;8(16):2719–27.
Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25.
Dienstmann R, Braña I, Rodon J, Tabernero J. Toxicity as a biomarker of efficacy of molecular targeted therapies: focus on EGFR and VEGF inhibiting anticancer drugs. Oncologist. 2011;16(12):1729–40.
Yu X, Zhang Y, Chen C, Yao Q, Li M. Targeted drug delivery in pancreatic cancer. Biochim Biophys Acta. 2010;1805(1):97–104.
Berlanga-Acosta J, Gavilondo-Cowley J, Barco-Herrera DG, Martin-Machado J, Guillen-Nieto G. Epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) as tissue healing agents: clarifying concerns about their possible role in malignant transformation and tumor progression. J Carcinog Mutagen. 2011;2:1.
Einama T, Ueda S, Tsuda H, et al. Membranous and cytoplasmic expression of epidermal growth factor receptor in metastatic pancreatic ductal adenocarcinoma. Exp Ther Med. 2012;3(6):931–6.
Normanno N, Maiello MR, De Luca A. Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs): simple drugs with a complex mechanism of action? J Cell Physiol. 2003;194(1):13–9.
Mendelsohn J. Blockade of receptors for growth factors: an anticancer therapy—the fourth annual Joseph H. Burchenal American Association of Cancer Research Clinical Research Award Lecture. Clin Cancer Res. 2000;6(3):747–53.
Xiong HQ. Molecular targeting therapy for pancreatic cancer. Cancer Chemother Pharmacol. 2004;54(Suppl 1):S69–77.
Alroy I, Yarden Y. The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand–receptor interactions. FEBS Lett. 1997;410(1):83–6.
Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61(2):203–12.
Dong M, Nio Y, Guo KJ, Tamura K, Tian YL, Dong YT. Epidermal growth factor and its receptor as prognostic indicators in Chinese patients with pancreatic cancer. Anticancer Res. 1998;18(6B):4613–9.
Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, Korc M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993;13(3):565–9.
Mu DQ, Peng YS, Xu QJ. Values of mutations of K-ras oncogene at codon 12 in detection of pancreatic cancer: 15-year experience. World J Gastroenterol. 2004;10(4):471–5.
Zhu H, Liang ZY, Ren XY, Liu TH. Small interfering RNAs targeting mutant K-ras inhibit human pancreatic carcinoma cells growth in vitro and in vivo. Cancer Biol Ther. 2006;5(12):1693–8.
Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53(4):549–54.
Navas C, Hernández-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–30.
Peace DJ, Chen W, Nelson H, Cheever MA. T cell recognition of transforming proteins encoded by mutated ras proto-oncogenes. J Immunol. 1991;146(6):2059–65.
Shin SH, Kim SC, Hong SM, et al. Genetic alterations of K-ras, p53, c-erbB-2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas. 2013;42(2):216–22.
Sirivatanauksorn V, Sirivatanauksorn Y, Lemoine NR. Molecular pattern of ductal pancreatic cancer. Langenbecks Arch Surg. 1998;383(2):105–15.
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76.
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61.
Tian H, Callahan CA, DuPree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci USA. 2009;106(11):4254–9.
Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–6.
Wehr AY, Furth EE, Sangar V, Blair IA, Yu KH. Analysis of the human pancreatic stellate cell secreted proteome. Pancreas. 2011;40(4):557–66.
Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60(6):861–8.
Mahadevan D, Von Hoff DD. Tumor–stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–97.
Le A, Rajeshkumar NV, Maitra A, Dang CV. Conceptual framework for cutting the pancreatic cancer fuel supply. Clin Cancer Res. 2012;18(16):4285–90.
Yang ZY, Yuan JQ, Di MY, et al. Gemcitabine plus erlotinib for advanced pancreatic cancer: a systematic review with meta-analysis. PLoS One. 2013;8(3):e57528.
Li J, Saif MW. Any progress in the management of advanced pancreatic cancer? Highlights from the 45th ASCO annual meeting. Orlando, FL, USA. May 29–June 2, 2009. JOP. 2009;10(4):361–5.
Squadroni M, Fazio N. Chemotherapy in pancreatic adenocarcinoma. Eur Rev Med Pharmacol Sci. 2010;14(4):386–94.
Páez D, Labonte MJ, Lenz HJ. Pancreatic cancer: medical management (novel chemotherapeutics). Gastroenterol Clin North Am. 2012;41(1):189–209.
Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6.
Van Cutsem E, Vervenne WL, Bennouna J, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27(13):2231–7.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32.
Stathopoulos GP, Trafalis D, Athanasiou A, Bardi G, Chandrinou H. Serious hematologic complications following erlotinib treatment. Anticancer Res. 2010;30(3):973–6.
Faloppi L, Andrikou K, Cascinu S. Cetuximab: still an option in the treatment of pancreatic cancer? Expert Opin Biol Ther. 2013;13(5):791–801.
Bruns CJ, Harbison MT, Davis DW, et al. Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms. Clin Cancer Res. 2000;6(5):1936–48.
Cohenuram M, Saif MW. Epidermal growth factor receptor inhibition strategies in pancreatic cancer: past, present and the future. JOP. 2007;8(1):4–15.
Xiong HQ, Rosenberg A, LoBuglio A, et al. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: a multicenter phase II Trial. J Clin Oncol. 2004;22(13):2610–6.
Cascinu S, Berardi R, Labianca R, et al. Cetuximab plus gemcitabine and cisplatin compared with gemcitabine and cisplatin alone in patients with advanced pancreatic cancer: a randomised, multicentre, phase II trial. Lancet Oncol. 2008;9(1):39–44.
Kullmann F, Hollerbach S, Dollinger MM, et al. Cetuximab plus gemcitabine/oxaliplatin (GEMOXCET) in first-line metastatic pancreatic cancer: a multicentre phase II study. Br J Cancer. 2009;100(7):1032–6.
Heinemann V, Labianca R, Hinke A, Louvet C. Increased survival using platinum analog combined with gemcitabine as compared to single-agent gemcitabine in advanced pancreatic cancer: pooled analysis of two randomized trials, the GERCOR/GISCAD intergroup study and a German multicenter study. Ann Oncol. 2007;18(10):1652–9.
Philip PA, Benedetti J, Corless CL, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol. 2010;28(22):3605–10.
Goldman CK, Kim J, Wong WL, King V, Brock T, Gillespie GY. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiforme pathophysiology. Mol Biol Cell. 1993;4(1):121–33.
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74.
Pavlidis ET, Pavlidis TE. Role of bevacizumab in colorectal cancer growth and its adverse effects: a review. World J Gastroenterol. 2013;19(31):5051–60.
Welch S, Spithoff K, Rumble RB, Maroun J, Gastrointestinal Cancer Disease Site Group. Bevacizumab combined with chemotherapy for patients with advanced colorectal cancer: a systematic review. Ann Oncol. 2010;21(6):1152–62.
Ko AH, Youssoufian H, Gurtler J, et al. A phase II randomized study of cetuximab and bevacizumab alone or in combination with gemcitabine as first-line therapy for metastatic pancreatic adenocarcinoma. Invest New Drugs. 2012;30(4):1597–606.
Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–22.
Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3(9):666–75.
Long Y, Sun Q, Wu J, Wang Y, Jiao S. Allogeneic cell-based immunotherapy combined with chemotherapy and targeted therapy in advanced pancreatic cancer with metastases: a case report. Oncol Lett. 2014;7(5):1594–8.
Qiu Y, Yun MM, Xu MB, Wang YZ, Yun S. Pancreatic carcinoma-specific immunotherapy using synthesised alpha-galactosyl epitope-activated immune responders: findings from a pilot study. Int J Clin Oncol. 2013;18(4):657–65.
Salman B, Zhou D, Jaffee EM, Edil BH, Zheng L. Vaccine therapy for pancreatic cancer. Oncoimmunology. 2013;2(12):e26662.
Lepisto AJ, Moser AJ, Zeh H, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008;6(B):955–64.
Kimura Y, Tsukada J, Tomoda T, et al. Clinical and immunologic evaluation of dendritic cell-based immunotherapy in combination with gemcitabine and/or S-1 in patients with advanced pancreatic carcinoma. Pancreas. 2012;41:195–205.
Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte–macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90(8):3539–43.
Hardacre JM, Mulcahy M, Small W, et al. Addition of algenpantucel- L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17:94–100.
Gujar SA, Lee PW. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front Oncol. 2014;4:77.
Nakao A, Kasuya H, Sahin TT, et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011;18(3):167–75.
Ishizaki H, Manuel ER, Song GY, et al. Modified vaccinia Ankara expressing survivin combined with gemcitabine generates specific antitumor effects in a murine pancreatic carcinoma model. Cancer Immunol Immunother. 2011;60(1):99–109.
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–21.
Acknowledgments
No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Conflict of interest
Fusun Ozmen, Tevfik Tolga Şahin, and M. Mahir Ozmen declare no conflict of interest.
Compliance with ethics guidelines
This review is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ozmen, F., Şahin, T.T. & Ozmen, M.M. Current Adjuvant Therapeutic Approaches for Pancreatic Cancer. Adv Ther 32, 42–56 (2015). https://doi.org/10.1007/s12325-015-0177-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-015-0177-5