
Abstract
The CACNA1A gene encodes the alpha-1A subunit of P/Q type voltage-gated calcium channel Cav2.1, which is associated with a broad clinical spectrum and variable symptomatology. While few patients with progressive ataxia caused by CACNA1A missense variants have been reported, here we report three unrelated Chinese patients with progressive ataxia due to de novo missense variants in the CACNA1A gene, including a novel pathogenic variant (c.4999C > G) and a previously reported pathogenic variant (c.4037G > A). Our findings and a systematic literature review show the unique phenotype of progressive ataxia caused by missense variants and enlarge the genetic and clinical spectrum of CACNA1A. This suggests that in addition to routine screening for dynamic mutations, screening for CACNA1A variants is important for clinicians facing patients with progressive ataxia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Progressive ataxias are a group of complex neurological disorders that can be caused by hereditary ataxia, idiopathic cerebellar ataxia and multiple system atrophy cerebellar type [1]. The CACNA1A gene encodes the alpha-1A subunit of the P/Q type voltage-gated calcium channel Cav2.1, which is expressed especially abundantly in the cerebellar Purkinje and granule cells [2]. Pathogenic variants in this gene are associated with several dominantly inherited disorders: episodic ataxia type 2 (EA2, MIM#108,500), familial hemiplegic migraine (FHM1, MIM#141,500), spinocerebellar ataxia type 6 (SCA6, MIM#183,086), and developmental and epileptic encephalopathy 42 (DEE42, MIM#617,106) [3]. Initial reports suggested that FHM1 is caused by missense variants and EA2 by truncating variants (nonsense, frameshift, splice site) in CACNA1A, while the expanded CAG repeats cause SCA6, which is characterized by slowly progressive cerebellar ataxia with a relatively late onset [4]. However, some patients with expanded repeats presented with episodic ataxia [5], and conversely, missense variants were implicated in progressive ataxia [6]. Besides, progressive ataxia associated with missense variants differ from those caused by expanded CAG repeats in terms of early onset and distinct associated clinical characteristics [7]. The substantial phenotypic overlap among these disorders complicates the correlation between phenotypes and genotypes. To date, only a few variants associate with progressive ataxia in the CACNA1A gene have been identified worldwide, while no patients in the Chinese population have been reported.
We herein reported the genetic features and clinical findings of three Chinese families with progressive ataxia associated with de novo missense variants within CACNA1A. We subsequently summarized patients with progressive ataxia associated with CACNA1A missense variants so as to better understand this disorder.
Materials and Methods
Subjects
Participants were enrolled between February 2016 and May 2023 in the Second Affiliated Hospital of Zhejiang University. Inclusion criteria were as follows: (1) progressive ataxia; (2) negative molecular genetic tests for CAG expansions in ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, PPP2R2B, TBP, ATN1 and several ARCA genes [8]; (3) no established acquired cause of ataxia. Whole exome sequencing (WES) and bioinformatics analysis were performed in the probands. The clinical evaluations and neurological examinations were performed by at least two senior neurologists. Written informed consents were obtained from all the participants or their legal guardians. This study was approved by the Ethics Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine.
Genetic Analysis
Genomic DNA was extracted from each participant’s peripheral blood using a commercial blood genomic extraction kit (Qiagen, Hilden, Germany) and then screened by WES. Details on library preparation, sequencing protocol, bio-informatics analysis, and filtering methods of WES were conducted as described previously [9]. Sanger sequencing was performed to further validate the filtered variants in all probands and the family members. The primers for Sanger sequencing were listed in Table S1. The parenthood of patients with de novo variants were analyzed using 21 core short tandem repeat (STR) regions. The pathogenicity of variants was classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines [10]. The sequence was compared with the published human gene sequence (CACNA1A, NM_001127221) in the National Center for Biotechnology Information Database (http://www.ncbi.nlm.nih.gov).
Literature Review
We reviewed the patients presenting with a progressive ataxia associated with CACNA1A missense variants. Primary research articles were searched in PubMed using the terms “Progressive ataxia” or “CACNA1A” until December 31, 2022. The literature search was restricted to published articles in Chinese or English. Only reports of genetically confirmed cases were considered.
Results
Pathogenic Variants Identified in CACNA1A
After genetic analysis, two variants within CACNA1A were identified in three unrelated patients. The c.4037G > A (p.R1346Q) was identified in patient 1 and patient 2, which was reported previously [11]. The novel missense variant, c.4999C > G (p.R1667G), was detected in patient 3, which was absent in the 1000 Genomes Project, ExAC and Gnome AD databases, and it has a CADD score of 23.7. Additionally, it was not found in our targeted next-generation sequencing (NGS) database covering CACNA1A, which contained 2000 unrelated Chinese individuals. According to the guidelines of ACMG, c.4999C > G (p.R1667G) was classified as pathogenic variant with PS2 (strong), PM1, PM2 and PM5 (moderate), PP3 (supporting). It is worth mentioning that both of the variants in three families were de novo. This was identified by Sanger sequencing that neither of patients’ parents carried the variant, and the kinship was verified by 21 core STR regions (Table S2).
Clinical Features of Three Patients with Progressive Ataxia and CACNA1A Missense Variants
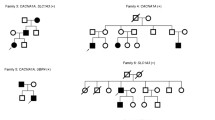
Patient 1 (Fig. 1A) carrying p.R1346Q variant is a 20-year-old male who presented with a 2-year history of slowly progressive gait disturbance, dysarthria and 3-month history of migraine episodes. Neurological examination showed wide-based gait, slurred speech, dysmetria, rotatory nystagmus, as well as positive Romberg sign. Brain MRI demonstrated cerebellar atrophy. Electromyography reveals decreased wave amplitude of sensory conduction evoked potentials in the right peroneal nerve.

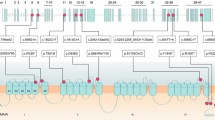
Pedigree patterns of three families with progressive ataxia and localization of CACNA1A missense variants associated with progressive ataxia. A, B, C. Family pedigree, chromatogram of the CACNA1A variants identified, and MRI images showing cerebellar atrophy in the probands. The arrow points to the proband. Black symbols represent patients while gray symbols represent preclinical individuals. T2 weighted coronal MRI showing atrophy of both cerebellar hemispheres. D. Localization of CACNA1A missense variants on the secondary structure of Cav2.1. Voltage-sensing segment S4 is shown in yellow and Ca2 + -selective pore-forming segments S5-S6 are shown in brown. Red dots indicate previously known variants, orange dots highlight variants identified in this study, and the dot featuring a yellow pentagram inside denotes the novel pathogenic variant
Patient 2 (Fig. 1B) with the same p.R1346Q variant is a 32-year-old female with a chief complaint of a 10-year history of gait disturbance and involuntary tremor of head and hands. The tremor was paroxysmal episodes, independent of body position posture. Dysarthria appeared 2 years ago, without dysphagia. On examination, she had wide-base gait, dysmetria (worse on left side), increased tendon reflexes in both lower limbs. Partial horizontal ophthalmoparesis of both eyes and horizontal nystagmus were also noted. Brain MRI showed cerebellar atrophy while electromyography was normal. Her son carries the same variant and remains asymptomatic.
Patient 3 (Fig. 1C) carrying a novel and de novo pathogenic variant c.4999C > G (p.R1667G) is a 24-year-old male. He had a history of obstructed labor and delivery with hypoxia at birth. More than 20 years ago, family members noticed that his gait was worse than that of children with similar age, subsequently, his unsteadiness of walking gradually worsened. Six years ago, he began experiencing daily involuntary head tremors lasting 10 min each without unconsciousness or involuntary movement of limbs during attacks. Fine movements could trigger the attacks, which were more frequent when the temperature was low. Neurologic examinations revealed slurred speech, dysmetria, rotatory nystagmus, as well as positive Romberg sign. Brain MRI also showed cerebellar atrophy while electromyography was normal.
Clinical Phenotypes of Patients with Progressive Ataxia and CACNA1A Missense Variants Worldwide
We reviewed all of the previously reported patients with progressive ataxia associated with CACNA1A missense variants. In addition to the three patients reported here (Table 1), we found 18 affected individuals from 14 families, published in 9 articles. Their detailed characteristics are summarized in Table 2 and the variants distribution in the CaV2.1 channel are illustrated in Fig. 1D.
Combined with our study and previous studies, individuals with progressive ataxia associated with CACNA1A missense variants all have typical manifestations of cerebellar ataxia and cerebellar oculomotor disturbances. Basal ganglia sign (44.4%) and headache (28.6%) are also common. The mean age onset was 26.2 years, and more than 50% of patients had their onset before the age of 26 years, which increases to 80% while extending the onset to 32 years.
Discussion
In our study, we reported three Chinese families with CACNA1A missense variants, including a novel pathogenic variant (c.4999C > G) and a previously reported pathogenic variant (c.4037G > A). They all had a clinical characteristic of progressive ataxia which is typically seen in spinocerebellar ataxia. The literature review revealed progressive ataxia associated with CACNA1A missense variants differ from those caused by expanded CAG repeats, the individuals manifested basal ganglia signs and headache more often and did not exhibit any pyramid signs. Moreover, the onset of progressive ataxia was also earlier similar to the prior study [7]. These missense variants seem to result in a heterogeneous ataxia disorder with clinical phenotypes between SCA6, FHM1 and EA2.
The CACNA1A gene encodes the alpha-1A subunit of the P/Q type voltage-gated calcium channel Cav2.1, which consists of four homologous regions, each containing six transmembrane segments [2]. Pathogenic variants in this gene cause various alterations in calcium channel kinetics and loss or gain of P/Q-type channel function and have been associated with EA2 and FMH1, respectively. Expanded CAG repeats found in SCA6 do not directly affect the P/Q-type channel but may be mediated by a transcription factor, the C-terminus of the alpha-1A subunit, which is expressed through the internal ribosomal entry site (IRES) located in CACNA1A [12]. Three variants identified from patients with progressive ataxia were located in the S5-S6 connecting section of domain I (p.G293R, p.D302N) and domain II (p.E668K). The S5-S6 connecting section forms a channel hole, which selectively allows the passage of ions. And three variants were located in the loop between I-II (p.A405T, p.A454T) and II- III (p.R803S). The previous report presumed that these variants might affect the interaction between the loop and the β subunit leading to dysregulation of the inactivation kinetics [18]. The two pathogenic variants reported in this study, combined with four previously reported variants, were located in the S4 transmembrane segments of domain II (p.R583Q), domain III (p.R1346Q), and domain IV (p.R1664Q, p.R1667G, p.R1668W). The S4 segment is attached to a positive amino acid, forming the S4 transmembrane alpha-helix, which acts as a “voltage sensor”. These variants replace a polar positively charged highly conserved arginine by neutral amino acid (glutamine) or non-polar amino acids (tryptophane and glycine), which may cause an excess of intracellular calcium and thus death of neurons. However, no correlation seems to exist between specific variants and phenotypic presentations, even the same variant can present diverse clinical phenotypes. The p.R1346Q missense variant has also been described in association with EA2 and FHM1 [11]. Furthermore, a different amino acid exchange at the codon 1667 (p.R1667P) has been identified in a patient with fatal brain edema [20].
When clinicians facing patients presenting with progressive ataxia always focus on searching for CAG expansions and fails to identify missense variants in CACNA1A. We establish here that screening for CACNA1A missense variants is of interest to clinicians facing patients presenting with progressive ataxia, especially when combined with headache as well as basal ganglia signs. Additionally, the variants identified here were de novo, indicating that CACNA1A missense variant warrant attention in sporadic cases with undiagnosed progressive ataxia.
Conclusion
In summary, we reported three Chinese patients with progressive ataxia associated with de novo missense variants within CACNA1A. The variants including a novel pathogenic variant (c.4999C > G) and a previously reported pathogenic variant (c.4037G > A). These cases and the pertinent literature review also suggest that in addition to routine screening for dynamic CAG variants, screening for CACNA1A missense mutations is of importance to clinicians facing patients presenting with progressive ataxia, especially when combined with basal ganglia signs as well as headache.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Giordano I, Harmuth F, Jacobi H, Paap B, Vielhaber S, Machts J, Schöls L, Synofzik M, Sturm M, Tallaksen C, Wedding IM, Boesch S, Eigentler A, van de Warrenburg B, van Gaalen J, Kamm C, Dudesek A, Kang JS, Timmann D, Silvestri G, Masciullo M, Klopstock T, Neuhofer C, Ganos C, Filla A, Bauer P, Tezenas du Montcel S, Klockgether T. Clinical and genetic characteristics of sporadic adult-onset degenerative ataxia. Neurology. 2017;89:1043–9. https://doi.org/10.1212/WNL.0000000000004311.
Lübbert M, Goral RO, Keine C, Thomas C, Guerrero-Given D, Putzke T, Satterfield R, Kamasawa N. Young SM Jr. CaV2.1 α1 Subunit Expression Regulates Presynaptic CaV2.1 Abundance and Synaptic Strength at a Central Synapse. Neuron. 2019;101(2):260-273.e6. https://doi.org/10.1016/j.neuron.2018.11.028.
Hommersom MP, van Prooije TH, Pennings M, Schouten MI, van Bokhoven H, Kamsteeg EJ, van de Warrenburg BPC. The complexities of CACNA1A in clinical neurogenetics. J Neurol. 2022;269(6):3094–108. https://doi.org/10.1007/s00415-021-10897-9.
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15(1):62–9. https://doi.org/10.1038/ng0197-62.
Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, Francia A, Spadaro M, Pierelli F, Salvi F, Ophoff RA, Frants RR, Frontali M. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6(11):1973–8. https://doi.org/10.1093/hmg/6.11.1973.
Yue Q, Jen JC, Nelson SF, Baloh RW. Progressive ataxia due to a missense mutation in a calcium-channel gene. Am J Hum Genet. 1997;61(5):1078–87. https://doi.org/10.1086/301613.
Coutelier M, Coarelli G, Monin ML, Konop J, Davoine CS, Tesson C, Valter R, Anheim M, Behin A, Castelnovo G, Charles P, David A, Ewenczyk C, Fradin M, Goizet C, Hannequin D, Labauge P, Riant F, Sarda P, Sznajer Y, Tison F, Ullmann U, Van Maldergem L, Mochel F, Brice A, Stevanin G, Durr A. SPATAX network. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain. 2017;140(6):1579–94. https://doi.org/10.1093/brain/awx081.
Cheng HL, Shao YR, Dong Y, Dong HL, Yang L, Ma Y, Shen Y, Wu ZY. Genetic spectrum and clinical features in a cohort of Chinese patients with autosomal recessive cerebellar ataxias. Transl Neurodegener. 2021;10(1):40. https://doi.org/10.1186/s40035-021-00264-z.
Xie JJ, Ni W, Wei Q, Ma H, Bai G, Shen Y, Wu ZY. New clinical characteristics and novel pathogenic variants of patients with hereditary leukodystrophies. CNS Neurosci Ther. 2020;26(5):567–75. https://doi.org/10.1111/cns.13284.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.
Alonso I, Barros J, Tuna A, Seixas A, Coutinho P, Sequeiros J, Silveira I. A novel R1347Q mutation in the predicted voltage sensor segment of the P/Q-type calcium-channel alpha-subunit in a family with progressive cerebellar ataxia and hemiplegic migraine. Clin Genet. 2004;65(1):70–2. https://doi.org/10.1111/j.2004.00187.x.
Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, Hansel C, Gomez CM. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell. 2013;154(1):118–33. https://doi.org/10.1016/j.cell.2013.05.059.
Balck A, Hanssen H, Hellenbroich Y, Lohmann K, Münchau A. Adult-onset ataxia or developmental disorder with seizures: two sides of missense changes in CACNA1A. J Neurol. 2017;264(7):1520–2. https://doi.org/10.1007/s00415-017-8494-z.
Bürk K, Kaiser FJ, Tennstedt S, Schöls L, Kreuz FR, Wieland T, Strom TM, Büttner T, Hollstein R, Braunholz D, Plaschke J, Gillessen-Kaesbach G, Zühlke C. A novel missense mutation in CACNA1A evaluated by in silico protein modeling is associated with non-episodic spinocerebellar ataxia with slow progression. Eur J Med Genet. 2014;57(5):207–11. https://doi.org/10.1016/j.ejmg.2014.01.005.
Duque KR, Marsili L, Sturchio A, Mahajan A, Merola A, Espay AJ, Kauffman MA. Progressive Ataxia with Hemiplegic Migraines: a Phenotype of CACNA1A Missense Mutations Not CAG Repeat Expansions. Cerebellum. 2021;20(1):134–9. https://doi.org/10.1007/s12311-020-01185-9.
Marti S, Baloh RW, Jen JC, Straumann D, Jung HH. Progressive cerebellar ataxia with variable episodic symptoms–phenotypic diversity of R1668W CACNA1A mutation. Eur Neurol. 2008;60(1):16–20. https://doi.org/10.1159/000127974.
Romaniello R, Zucca C, Tonelli A, Bonato S, Baschirotto C, Zanotta N, Epifanio R, Righini A, Bresolin N, Bassi MT, Borgatti R. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J Neurol Neurosurg Psychiatry. 2010;81(8):840–3. https://doi.org/10.1136/jnnp.2008.163402.
Cricchi F, Di Lorenzo C, Grieco GS, Rengo C, Cardinale A, Racaniello M, Santorelli FM, Nappi G, Pierelli F, Casali C. Early-onset progressive ataxia associated with the first CACNA1A mutation identified within the I-II loop. J Neurol Sci. 2007;254(1–2):69–71. https://doi.org/10.1016/j.jns.2007.01.008.
Tonelli A, D’Angelo MG, Salati R, Villa L, Germinasi C, Frattini T, Meola G, Turconi AC, Bresolin N, Bassi MT. Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J Neurol Sci. 2006;241(1–2):13–7. https://doi.org/10.1016/j.jns.2005.10.007.
Gauquelin L, Hawkins C, Tam EWY, Miller SP, Yoon G. Pearls & Oy-sters: Fatal brain edema is a rare complication of severe CACNA1A-related disorder. Neurology. 2020;94(14):631–4. https://doi.org/10.1212/WNL.0000000000009223.
Funding
This work was supported by the grants from the National Natural Science Foundation of China to Zhi-Ying Wu (82071260, 82230062).
Author information
Authors and Affiliations
Contributions
Chen-Hao Zhu: data collection, analysis and interpretation, draft of manuscript. Jin-Yang Yu: data collection, analysis and interpretation. Yin Ma: technical support. Yi Dong: analysis and interpretation. Zhi-Ying Wu: study concept and design, critical revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study was approved by the ethics committee of Second Affiliated Hospital of Zhejiang University (Hangzhou).
Consent to Participate
Written informed consent was obtained from each participant.
Consent to Publish
Not applicable.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhu, CH., Yu, JY., Ma, Y. et al. Progressive Ataxia due to de novo Missense Variants in the CACNA1A Gene. Cerebellum (2024). https://doi.org/10.1007/s12311-024-01710-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s12311-024-01710-0