
Abstract
SCA3 presents with a CAG expansion at 14q24.3-q32 while SCA10 shows an ATTCT expansion at 22q13-qter. SCA10 seems to be less aggressive than SCA3. For an in vivo, noninvasive approach of the correlation between central nervous system and clinical evolution, we can use optic coherence tomography (OCT) to measure retinal nerve fiber (RNFL) and ganglion cell layer (GCL) thickness. To describe OCT findings in SCA10, correlate it with expansion size and disease severity and compare with those of SCA3. We analyzed ten individuals with SCA3 and nine with SCA10 recruited from the neurology service of Hospital de Clínicas of Paraná—Brazil. They were submitted to OCT and clinical evaluation using SARA score. Expansion size, demographic data, time from disease onset, and age of onset were collected. We found no correlation between size of expansion, SARA, and RNFL or GCL thickness in SCA10. RNFL seemed to be thicker in SCA10 (p > 0.05). GCL thickness, SARA, median age, and time from disease onset did not differ between groups. SCA10 individuals had an earlier disease onset. In SCA3, there was a negative correlation between SARA and RNFL thickness in nasal area. To the best of our knowledge, this is the first paper assessing retinal changes by OCT in individuals with SCA10. The lack of correlation between disease progression, age, and time since onset supports the anatomopathological findings which suggest SCA10 is less aggressive than other SCAs. The findings in SCA3 are in accordance with the literature.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Spinocerebellar ataxias (SCAs) are a group of autosomal dominant degenerative ataxic diseases of the central nervous system that currently include up to 39 genetically distinct entities [1, 2]. SCA3, the most prevalent SCA both worldwide and in Brazil [3–6], is caused by a trinucleotide (CAG) repeat expansion on the long arm of chromosome 14 (14q24.3-q32), leading to cerebellar ataxia (primarily gait ataxia), spasticity, peripheral neuropathy, amyotrophy, and other movement disorders as well as bulging eyes (caused by lid retraction) [7–9]. SCA10 is predominantly reported in Amerindian populations [10–12] and is the second most common SCA in some regions of Mexico [13] and southern Brazil [4]. It is caused by a pentanucleotide (ATTCT) repeat expansion on chromosome 22q13-qter [14]. The clinical picture SCA10 varies from a purely cerebellar syndrome to cerebellar syndrome concomitantly with epilepsy, polyneuropathy, and hematologic heart and liver conditions [13, 15].
Optical coherence tomography (OCT) allows for quantitative objective evaluation of various structures in the eye [16], including the retinal nerve fiber layer (RNFL), which has been proposed to be used as a biomarker for degeneration of the afferent and efferent cerebellar pathways [17, 18]. We used OCT to evaluate patients with SCA3 and those with SCA10.
Materials and Methods
Patients
Demographic data including age, gender, age of onset, and disease duration were collected between 2014 and 2015 for 19 individuals (ten with SCA3 and nine with SCA10) seen at the ataxia outpatient unit in the neurology service at the Hospital de Clínicas, Federal University of Paraná (HC-UFPR). The patients signed a voluntary informed consent form, and the protocol was approved by the HC-UFPR Committee for Ethics in Human Research. All the patients underwent OCT (Spectralis OCT, software version 5.1; Heidelberg Engineering, Vista, CA, USA) of both eyes and were assessed for visual acuity with the Lea chart and color perception with Hardy-Rand-Rittler color plates. They also underwent neurological assessment with the SARA (Scale for the Assessment and Rating of Ataxia), which measures motor performance related to ataxia on a scale from 0 (absence of ataxia) to 40. The following items were measured: gait capacity (0–8), static balance (0–6), balance when sitting/posture (0–4), speech disturbance/dysarthria (0–6), upper limb coordination (finger-chase (0–4) and nose-finger tests (0–4)), diadochokinesia (fast alternating hand movement test) (0–4), and lower limb coordination (heel-shin slide test) (0–4).
SCA was diagnosed when a prior genetic study confirmed the presence of a CAG trinucleotide repeat expansion with more than 51 repeats of the ATXN3 gene in chromosome 14q24.3-q31 in the case of SCA3; or when the study confirmed the presence of an ATTCT pentanucleotide repeat expansion with more than 32 repeats in the ATXN10 gene in chromosome 22q13 in the case of SCA10; or, epidemiologically, when the patients satisfied the following three criteria: (1) having at least one relative in whom SCA3 or SCA10 had been genetically confirmed, (2) presenting with clinical signs and symptoms consistent with the ataxia in question, and (3) not having another diagnosis that could affect SARA scoring.
OCT
In the frequency-domain OCT (FD-OCT), the thickness of the RNFL was measured overall and in six different regions of the retina of each eye (temporal, temporal superior, temporal inferior, nasal, nasal superior, and nasal inferior). Total retinal thickness and ganglion cell layer (GCL) thickness were measured at the thickest points above and below the macula. In one individual with SCA10, the retinal and GCL thickness measurements were not used because of technical problems during image acquisition. The data used for the analysis are the means of the values for both eyes.
Statistics
The nonparametric Mann-Whitney test was used to compare the results for the two patient groups (SCA3 and SCA10), and Spearman’s correlation coefficient was used to assess the correlation between two variables. All the statistical analysis was performed with Excel and Statistica (version 7) using a significance level of 5%.
Results
There was no difference between the two groups in terms of gender, age, or disease duration. However, the group with SCA10 had a lower age of onset of disease (SCA3, 42.4 ± 8.2 years; SCA10, 29.3 ± 10.9 years; p = 0.011) (Table 1). The mean ± standard deviation, minimum, maximum, and median SARA scores in the SCA3 group were 16.2 ± 8.5, 1, 25, and 15.8, respectively. The corresponding figures for the SCA10 group were 12.6 ± 6.9, 4, 26, and 9.5. There were no statistically significant differences between the two groups.
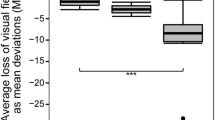
Color vision was normal in all the individuals, although subjects with SCA3 made more errors in the screening stage (plates 7 and 10) in the HRR test. Visual acuity measured with the Lea chart was the same in both groups. There was no correlation between the size of the repeat expansion and the thickness of the RNFL in SCA10 patients. The RNFL tended to be thicker in these patients than in SCA3 patients, although this difference was not statistically significant (Fig. 1). Changes in RNFL thickness were observed in at least one region in six of the ten individuals with SCA3 and in only two of the nine with SCA10.

RNFL thickness in patients with SCA3 and SCA10. RNFL retinal nerve fiber layer, TS temporal superior, NS nasal superior, T temporal, G overall, N nasal, TI temporal inferior, NI nasal inferior. p > 0.05
In the SCA3 patients, RNFL thickness in the nasal region was inversely related to SARA score (Fig. 2). When the items in the SARA test were assessed individually, only dysarthria, limb coordination, tremor, and diadochokinesia showed this correlation. There was no correlation with total SARA score in SCA10 patients, although there was a negative correlation between gait capacity and RNFL thickness in the temporal superior region (r = −0.670, p = 0.048) and between dysarthria and RNFL thickness both overall and in the nasal inferior region (r = −0.710, p = 0.032). While in patients with SCA3 there was a negative correlation between RNFL thickness in the nasal superior region and disease duration, this correlation was not observed in SCA10 patients. No statistically significant difference in either total retinal thickness or GCL thickness was observed between the two groups (Table 2).

RNFL thickness vs. SARA score. There is a negative correlation between SARA score and RNFL thickness in the nasal region in SCA3 patients (r = −0.695, P = 0.025). RNFL retinal nerve fiber layer, SARA scale for assessment and rating of ataxia
Retinal thickness in the upper and lower regions tended to decrease with increasing age (r = −0.514, p = 0.029, and r = 0.563, p = 0.015, respectively), but this trend was only observed when both groups were assessed together. Although most of the correlations indicated that the longer the disease duration, the thinner the RNFL, these findings were not statistically significant. When disease progression was assessed on the SARA scale, there was no direct correlation with GCL thickness in either group. However, total retinal thickness in the upper and lower regions was inversely correlated with SARA score (r = −0.517, p = 0.028, and r = −0.698, p = 0.001, respectively). This correlation remained statistically significant for the SCA10 group on its own (superior-retinal region, r = −0.781, p = 0.022; inferior-retinal region, r = −0.732, p = 0.39) but not for the SCA3 group. The only items on the SARA scale for which there was a correlation with reduced retinal thickness were static balance and dysarthria.
Discussion
Although in the classical description of spinocerebellar ataxias cerebellar or olivopontocerebellar atrophy is considered the most prominent pathological change [19], there is increasing evidence that many SCAs also affect other regions of the central and peripheral nervous systems as well as the retinal nervous system, which can be explained by the fact that they have the same embryological (neuroectodermal) origin.
To date, there are no studies assessing the role of OCT as a diagnostic tool or of RNFL thickness as a biomarker for SCA10, whether in populations with a pure cerebellar ataxia phenotype, like those in Brazil, or in populations with a phenotype associated with epilepsy, like those in Mexico and Argentina. Our findings show that RNFL thickness tends to be closer to normal in SCA10 patients than in individuals with SCA3. While there is no correlation between total SARA score and RNFL thickness, a weak negative correlation was observed between the SARA gait subscore and RNFL thickness in the temporal superior region and SARA speech subscore and RNFL thickness both overall and in the nasal inferior regions.
Disease duration does not appear to affect RNFL thickness, GCL thickness, or total retinal thickness in individuals with SCA10. This may be because SCA10 has a less aggressive pathology than SCA3. In the former, the cerebral cortex, hippocampus, midbrain, and pons are normally preserved [20], while in SCA3, there is severe atrophy of the cerebellum, brainstem, and spinocerebellar tracts and moderate atrophy of the internal globus pallidus, subthalamic nucleus, substantia nigra, pontine nuclei, and dentate nucleus [19]. This would suggest why the RNFL is slightly thicker in individuals with SCA10 than in those with SCA3. However, this finding contradicts the reduction in total retinal thickness observed in SCA10.
Two studies investigated retinal changes in SCA3 patients assessed with OCT. In the first, RNFL thickness was measured in nine individuals (18 eyes) and was found to be reduced in the upper region in ten eyes, in the lower region in eight, and in the nasal region in four. The temporal region was not affected. The same study also found a negative correlation between mean RNFL thickness and SARA score [21]. The second study assessed five individuals and found, in addition to the same negative correlation between thickness and SARA score, a more marked reduction in RNFL thickness in the nasal and lower regions and in the macular region, 3 mm from the fovea [22]. In the present study, there was also a significant reduction in RNFL thickness in the nasal region, and this was inversely proportional to SARA score. We also found that the thickness in the nasal superior region was associated with disease duration and that the GCL and total retinal thickness did not vary in the same way as the RNFL thickness.
These findings suggest that the papillomacular bundle is preserved in SCA10 and that RNFL thinning is not caused by a loss of cells in the GCL, corroborating the conclusions reported by Alvarez et al. [21]. They also suggest that retinal degeneration in SCA3 is similar to the degeneration that occurs in multiple sclerosis [23] but different from the degeneration in idiopathic Parkinson’s disease, in which, although the RNFL becomes thinner as the patient’s clinical condition worsens, it is accompanied by neuron loss in the GCL [24, 25], and also different from the degeneration in Alzheimer’s disease, where there is mainly loss of neurons in the GCL [26, 27]. It is also different from the pattern found in SCA1, where there is more neuronal loss in the temporal region, suggesting that the papillomacular bundle is affected [28].
Conclusions
This is the first study to use OCT to assess changes in the thickness of the RNFL and GCL in individuals with SCA10. Degeneration in SCA10 is apparently less severe than in SCA3 and is unrelated to disease duration. Our findings corroborate those reported in other studies of SCA3 to date.
Author’s Roles
Fernando Spina Tensini: conception, organization, execution of clinical evaluations, interpretation of results, and manuscript
Mario Teruo Sato: conception and organization
Naoye Shiokawa: execution of OCT
Tetsuo Ashizawa: execution of genetic analysis, review, and critique
Helio Afonso Ghizoni Teive: conception, organization, review, and critique
References
Teive HAG, Ashizawa T. Primary and secondary ataxias. Curr Opin Neurol. 2015;28:413–22.
Pestronk A. Hereditary ataxies: dominant. Washington University in Saint Louis (2016). at <http://neuromuscular.wustl.edu/ataxia/domatax.html>
Cintra VP, et al. Mutational screening of 320 Brazilian patients with autosomal dominant spinocerebellar ataxia. J Neurol Sci. 2014;347:375–9.
Teive HAG, et al. Spinocerebellar ataxias—genotype-phenotype correlations in 104 Brazilian families. Clinics. 2012;67:443–9.
Freund AA, et al. Spinocerebellar ataxias: microsatellite and allele frequency in unaffected and affected individuals. Arq Neuropsiquiatr. 2009;67:1124–32.
Silveira I, et al. Frequency of spinocerebellar ataxia type 1, dentatorubropallidoluysian atrophy, and Machado-Joseph disease mutations in a large group of spinocerebellar ataxia patients. Neurology. 1996;46:214–8.
Woods BT, Schaumburg HH. Nigro-spino-dentatal degeneration with nuclear ophthalmoplegia. A unique and partially treatable clinico-pathological entity. J Neurol Sci. 1972;17:149–66.
Nakano KK, Dawson DM, Spence A. Machado disease. A hereditary ataxia in Portuguese emigrants to Massachusetts. Neurology. 1972;22:49–55.
Stevanin G, et al. The gene for spinal cerebellar ataxia 3 (SCA3) is located in a region of approximately 3 cM on chromosome 14q24.3-q32.2. Am J Hum Genet. 1995;56:193–201.
Leonardi L, et al. Spinocerebellar ataxia type 10 in Peru: the missing link in the Amerindian origin of the disease. J Neurol. 2014;261:1691–4.
Matsuura T, et al. Spinocerebellar ataxia type 10 is rare in populations other than Mexicans. Neurology. 2002;58:983.
Fujigasaki H, et al. Spinocerebellar ataxia type 10 in the French population. Ann Neurol. 2002;51:408.
Grewal RP, et al. Clinical and genetic analysis of a distinct autosomal dominant spinocerebellar ataxia. Neurology. 1998;51:1423–6.
Matsuura T, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–4.
Teive HAG, et al. Spinocerebellar ataxia type 10—a review. Park. Relat. Disord. 2011;17:655–61.
Huang D, et al. Optical coherence tomography. Science. 1991;254:1178–81.
Aleman TS, et al. Relationship between optic nerve appearance and retinal nerve fiber layer thickness as explored with spectral domain optical coherence tomography. Transl Vis Sci Technol. 2014;3:4.
Zagozewski JL, Zhang Q, Eisenstat DD. Genetic regulation of vertebrate eye development. Clin Genet. 2014;86:453–60.
Seidel K, et al. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1–21.
Xia G, et al. Purkinje cell loss is the major brain pathology of spinocerebellar ataxia type 10. J Neurol Neurosurg Psychiatry. 2013;84:1409–11.
Alvarez G, et al. Optical coherence tomography findings in spinocerebellar ataxia-3. Eye. 2013;27:1376–81.
Pula JH, et al. Retinal nerve fibre layer and macular thinning in spinocerebellar ataxia and cerebellar multisystem atrophy. Neuro-Ophthalmology. 2011;35:108–14.
Xu LT, Bermel RA, Nowacki AS, Kaiser PK. Optical coherence tomography for the detection of remote optic neuritis in multiple sclerosis. J Neuroimaging. 2016;26:283–8.
Bayhan HA, Aslan Bayhan S, Tanık N, Gürdal C. The association of spectral-domain optical coherence tomography determined ganglion cell complex parameters and disease severity in Parkinson’s disease. Curr Eye Res. 2014;3683:1–6.
Mailankody P, et al. Optical coherence tomography as a tool to evaluate retinal changes in Parkinson’s disease. Park Relat Disord. 2015;21:1164–9.
Ong YT, et al. Retinal neurodegeneration on optical coherence tomography and cerebral atrophy. Neurosci Lett. 2015;584:12–6.
Garcia-Martin E, et al. Ganglion cell layer measurements correlate with disease severity in patients with Alzheimer’s disease. Acta Ophthalmol. 2016; doi:10.1111/aos.12977.
Stricker S, et al. Temporal retinal nerve fiber loss in patients with spinocerebellar ataxia type 1. PLoS One. 2011;6:1–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The patients signed a voluntary informed consent form, and the protocol was approved by the HC-UFPR Committee for Ethics in Human Research.
Conflict of Interest
Fernando Spina Tensini has received no relevant financial or nonfinancial supports including, but not limited to, grants or funding, employment, affiliations, patents, inventions, honoraria, consultancies, royalties, stock options/ownership, or expert testimony, in the past 3 years to disclose.
Mario Teruo Sato has received no relevant financial or nonfinancial supports including, but not limited to, grants or funding, employment, affiliations, patents, inventions, honoraria, consultancies, royalties, stock options/ownership, or expert testimony, in the past 3 years to disclose.
Naoye Shiokawa has received no relevant financial or nonfinancial supports including, but not limited to, grants or funding, employment, affiliations, patents, inventions, honoraria, consultancies, royalties, stock options/ownership, or expert testimony, in the past 3 years to disclose.
Tetsuo Ashizawa receives research/grant support as a principal investigator of studies for Ionis Pharmaceuticals, Inc. (1515598769-CS2); the Myotonic Dystrophy Foundation; and the National Institutes of Health (NS083564).
Helio Afonso Ghizoni Teive has received no relevant financial or nonfinancial supports including, but not limited to, grants or funding, employment, affiliations, patents, inventions, honoraria, consultancies, royalties, stock options/ownership, or expert testimony, in the past 3 years to disclose.
Rights and permissions
About this article

Cite this article
Spina Tensini, F., Sato, M.T., Shiokawa, N. et al. A Comparative Optical Coherence Tomography Study of Spinocerebellar Ataxia Types 3 and 10. Cerebellum 16, 797–801 (2017). https://doi.org/10.1007/s12311-017-0856-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-017-0856-7