
Abstract
Friedreich’s ataxia (FDRA) is the most common inherited ataxia worldwide, caused by homozygous GAA expansions in the FXN gene. Patients usually have early onset ataxia, areflexia, Babinski sign, scoliosis and pes cavus, but at least 25 % of cases have atypical phenotypes. Disease begins after the age of 25 in occasional patients (late-onset Friedreich ataxia (LOFA)). Little is known about the frequency and clinical profile of LOFA patients. One hundred six patients with molecular confirmation of FDRA and followed in three Brazilian outpatient centers were enrolled. General demographics, GAA expansion size, age at onset, cardiac, endocrine, and skeletal manifestations were evaluated and compared between LOFA and classic FDRA (cFDRA) groups. We used Mann–Whitney and Fisher tests to compare means and proportions between groups; p values <0.05 were considered significant. LOFA accounted for 17 % (18/106) and cFDRA for 83 % (88/106) of the patients. There were 13 and 48 women in each group, respectively. LOFA patients were significantly older and had smaller GAA expansions. Clinically, LOFA group had a tendency toward lower frequency of diabetes/impaired glucose tolerance (5.8 vs. 17 %, p = 0.29) and cardiomyopathy (16.6 vs. 28.4 %, p = 0.38). Skeletal abnormalities were significantly less frequent in LOFA (scoliosis 22 vs. 61 %, p = 0.003, and pes cavus 22 vs.75 %, p < 0.001) as were spasticity and sustained reflexes, found in 22 % of LOFA patients but in none of the cFDRA patients (p = 0.001). LOFA accounts for 17 % of Brazilian FDRA patients evaluated herein. Clinically, orthopedic features and spasticity with retained reflexes are helpful tips to differentiate LOFA from cFDRA patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Friedreich ataxia (FRDA) is the most common recessive ataxia worldwide with prevalence ranging from 1 to 3 for each 100,000 people [1–3]. Nikolaus Friedreich first described this condition in 1863, and the core clinical features include not only ataxia but also absent tendon reflexes, Babinski sign, scoliosis, and pes cavus [4]. It is characteristically a disease of children and adolescents, with peak incidence around puberty [5]. Several clinical criteria were proposed for the diagnosis of FDRA before the identification of the genetic basis in 1996 [1, 5], being the one proposed by Harding the most widely accepted [6]. Given this criteria required the onset of symptoms before the age of 25, a significant number of patients with FRDA phenotype and late disease onset may have been classified as “idiopathic ataxia.”
After the description of the homozygous intronic GAA repeat expansions at FXN gene as the cause of FRDA, the diagnosis of FDRA became more accurate and another issue emerged: what age groups of ataxia patients should be tested for FXN mutation? The fact that up to 25 % of all FDRA patients do not fulfill Harding’s diagnostic criteria made the answer for this question a pertinent pitfall [2, 3]. In this scenario, the patients harboring FXN mutations with onset of symptoms after the age of 25 were classified as late-onset Friedreich ataxia (LOFA) [5].
Prior studies have shown that LOFA patients present with slower disease progression and milder nonneurological symptoms [2], but there are some gaps regarding the prevalence, the spectrum of neurological presentation, and nonneurological clinical features. In the present study, we recruited a large cohort of Brazilian patients with FRDA to determine the frequency and to expand the clinical profile of LOFA patients. Our primary objective was to characterize the phenotypes of Brazilian patients with LOFA and to identify clinical clues to recognize these challenging patients. We also aimed to evaluate the frequency and the pattern of extra-neurological involvement in LOFA compared to classic FRDA.
Patients and Methods
We enrolled all patients with molecular confirmation of FRDA that were regularly followed at three Brazilian tertiary neurogenetic clinics: two in the southeast region (University of Campinas and Federal University of São Paulo) and one in the south region (Federal University of Paraná). Another center performed the molecular tests for some of the patients (Federal University of Rio Grande do Sul). This study was approved by our institution Research Ethics Committee, and a written informed consent was obtained from all participants.
We recorded information on gender, age at enrollment, age at disease onset, and disease duration for each patient. Whenever available, we also obtained data on the length of GAA repeat expansion. LOFA was defined as disease onset later than 25 years of age. Nonneurological features, including cardiac, diabetes or impaired glucose tolerance (IGT), and skeletal manifestations were assessed during routine consultation, along with the results of diagnostic tests. Cardiac involvement was assessed by echocardiograms and EKGs. IGT or diabetes were defined according to the American Diabetes Association criteria (IGT: hemoglobin A1c ≥ 5.7 and <6.5 %, fasting blood glucose ≥ 100 and < 126 mg/dL, and oral glucose tolerance test with glucose levels ranging from 140 to 199 mg/dL; diabetes: glycated hemoglobin ≥ 6.5 %, fasting glucose plasma levels ≥ 126 mg/dL, and oral glucose tolerance test with glucose levels ≥200 mg/dL). Skeletal abnormalities were assessed by clinical examination and radiographic imaging.
Results were expressed with descriptive statistics. We used Mann–Whitney and Fisher tests to compare means and proportions between LOFA and classical FRDA with disease onset earlier than 25 years old (cFDRA) groups. P values <0.05 were considered significant. All statistical analyses were performed using the SYSTAT v13.0 software.
Results
One hundred six consecutive FDRA patients were enrolled from all three centers. LOFA accounted for 17 % (18/106) and cFDRA for 83 % (88/106) of the patients. Demographic, genetic, and clinical characteristics of LOFA and cFDRA patients are summarized in Table 1.
Although all patients had molecular confirmation, GAA repeat length quantification was available only for 63 patients. Patients with LOFA had shorter GAA repeat expansion than patients with cFRDA in both alleles. Mean GAA1 length was 328.6 ± 180 repeats for LOFA and 492.8 ± 263.7 repeats for cFDRA (p < 0.001) whereas mean GAA2 expansion length was 761.8 ± 198.8 repeats and 923.5 ± 83.1 repeats (p < 0.001), respectively.
The assessment of neurological and nonneurological features revealed that LOFA patients had a tendency toward lower frequency of diabetes/IGT and heart disease (5.8 vs 17 %, p = 0.29, and 16.6 vs 28.4 %, p = 0.38, respectively). Compared to cFRDA, LOFA patients had a significantly lower frequency of scoliosis (4/18 vs 54/88, p = 0.003) and pes cavus (4/18 vs 66/88, p < 0.001). Another relevant clinical difference was spasticity and sustained reflexes, which were found in 4/18 (22 %) of LOFA patients but in none of the cFDRA patients (p < 0.001).
Discussion
The identification of the genetic basis of FRDA broke a paradigm in the ataxia clinics, particularly for those patients with late-onset autosomal recessive ataxia and idiopathic sporadic late-onset cerebellar ataxia (ILOCA) [1, 2]. The underlying pathological homozygous GAA repeat expansion in chromosome 9q13-q21.1 leads to frataxin deficiency in different tissues, which results in a multisystemic disease not restricted to neurological impairment. The ubiquitous distribution of this protein throughout the body is related to the overt nonneurological findings present in FDRA [7]. In this context, a significant number of patients on movement disorders clinics were finally diagnosed with FDRA thanks to the inclusion of the molecular testing for FDRA in the workup of late-onset ataxias [8].
In the present study, LOFA accounted for 17 % of the FDRA patients. Two previously published series involving FDRA patients, with different genetic background from those herein described, demonstrated the very same 17 % of LOFA prevalence [9, 10]. However, there are other studies in which the prevalence of LOFA patients ranged from 13.5 to 25 % [2, 11]. Also in line with previous reports, these patients had shorter GAA repeats [2, 8–10]. This shorter expansion plays a major role in the later onset of symptoms and absent/milder nonneurological features seen in LOFA. As previously demonstrated, a possible reason is that smaller GAA expansions enable residual frataxin expression, modifying the classical FDRA phenotype [7].
The disease manifestations have been described with GAA expansions as small as 67 [12], and the onset of ataxia seems to be related to the dysfunction of the dentate nuclei of cerebellum, which is a delayed event in LOFA [8].
LOFA is an important diagnostic pitfall for general neurologists as it contradicts several of the classical clinical features of FDRA, especially those stated on traditional diagnostic criteria: age of onset, loss of reflexes, pes cavus, scoliosis, cardiomyopathy, and diabetes [6]. Even the autosomal recessive inheritance pattern may be challenging as it was recently described in a Brazilian family with LOFA and a pseudo-dominant inheritance pattern [13].
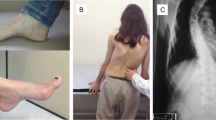
Regarding the clinical picture, skeletal abnormalities are more prominent in cFRDA as the deleterious effects of the absence of frataxin take place in the actively growing and maturing bones. This is in contrast with LOFA, where the disease begins later in life when the skeletal development has been completed. Another possible explanation for that finding may lay in the paraspinal muscle weakness that may be milder in LOFA patients, although Labelle and cols in the premolecular diagnosis era, had demonstrated that the scoliosis degree itself was not related to the axial/limb muscle weakness [14]. Those aspects place scoliosis and pes cavus as secondary clinical features in LOFA.
Other peculiar features seen in this series were spasticity and retained reflexes, present exclusively in LOFA patients with a frequency of 22 % (Table 1). Such distinct profile raises attention not only because it contradicts the archetypal finding of areflexia in FDRA but also because ataxia may not be the main clinical feature in some patients, expanding the differential diagnoses to nonataxic disorders such as complicated forms of hereditary spastic paraplegia [12].
As seen in our series, LOFA patients had the trend toward less glucose intolerance/diabetes and cardiomyopathies. Given the complete absence of frataxin results in hypersensitivity to oxygen radicals and tissue damage, tissues with high-energy demand, such as cardiac muscle, are particularly susceptible to disease-related damage. Residual levels of frataxin may endorse minor cardiovascular impairment in LOFA patients [7].
An interesting point not evaluated by this study, but largely explored in the field of neurodegenerative disorders, is the impact of nonmotor features, such as fatigue, depression, and daytime somnolence. These aspects were already evaluated in cFDRA demonstrating that fatigue is a frequent feature and has a direct relation to depressive symptoms [15].
Finally, it is important for general neurologists and movement disorders specialists to keep low thresholds for testing late-onset ataxia patients for FDRA. An important point is to consider FRDA molecular testing for patients with late-onset cerebellar ataxia or spastic paraplegia, even if they have absent or minor cardiac, glycemic, or skeletal abnormalities, regardless of the family history of similar cases or consanguinity. This is particularly relevant in the care of patients presenting with ILOCA. Given that LOFA is a rare condition, collaborative studies are very important in order to gather information to delineate as close as possible the whole clinical picture and nuances of this condition.
References
Campuzano V, Montermini L, Molto MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–7.
Bhidayasiri R, Perlman SL, Pulst SM, Geschwind DH. Late-onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol. 2005;629:1865–9.
Abrahão A, Pedroso JL, Braga-Neto P, Bor-Seng-Shu E, Aguiar PC, Barsottini OG. Milestones in Friedreich ataxia: more than a century and still learning. Neurogenetics. 2015;16:151–60.
Friedreich N. Über degenerative Atrophie der spinalen Hinterstränge (On degenerative atrophy of the spinal dorsal columns), Virchows Arch. Pathol Anat Physiol Klin Med. 1863;26:391–419.
de Michele G, Filla A, Cavalcanti F, Di Maio L, Pianese L, Castaldo I, et al. Late onset Friedreich’s disease: clinical features and mapping of mutation to the FRDA locus. J Neurol Neurosurg Psychiatry. 1994;57:977–9.
Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620.
Bayot A, Santos R, Camadro JM, Rustin P. Friedreich’s ataxia: the vicious circle hypothesis revisited. BMC Med. 2011;9:112.
Koeppen AH, Morral JA, McComb RD, Feustel PJ. The neuropathology of late-onset Friedreich’s ataxia. Cerebellum. 2011;10:96–103.
Reetz K, Dogan I, Costa AS, Dafotakis M, Fedosov K, Giunti P, et al. Biological and clinical characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS) cohort: a cross-sectional analysis of baseline data. Lancet Neurol. 2015;14:174–82.
Lecocq C, Charles P, Azulay JP, Meissner W, Rai M, N’Guyen K, et al. Delayed-onset Friedreich’s ataxia revisited. Mov Disord. 2015. doi:10.1002/mds.26382.
Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335:1169–75.
Berciano J, Mateo I, De Pablos C, Polo JM, Combarros O. Friedreich ataxia with minimal GAA expansion presenting as adult-onset spastic ataxia. J Neurol Sci. 2002;194:75–82.
Moro A, Martinez ARM, Karuta CVS, Munhoz RP, Moscovich M, Germiniani FMB, et al. Pseudo-Dominant’ inheritance in Friedreich’s ataxia: clinical and genetic study of a Brazilian family. Movmnt Disords Clncl Pract. 2014;1:361–3.
Labelle L, Tohmé S, Duhaime M, Allard P. Natural history of scoliosis in Friedreich’s ataxia. J Bone Joint Surg Am. 1986;68:564–72.
da Silva CB, Chevis CF, D’Abreu A, Lopes-Cendes I, França Jr MC. Fatigue is frequent and multifactorial in Friedreich’s ataxia. Parkinsonism Relat Disord. 2013;19:766–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Martinez, A.R.M., Moro, A., Abrahao, A. et al. Nonneurological Involvement in Late-Onset Friedreich Ataxia (LOFA): Exploring the Phenotypes. Cerebellum 16, 253–256 (2017). https://doi.org/10.1007/s12311-015-0755-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-015-0755-8