
Abstract
Neurons form exuberant synapses with target cells early in development. Then, necessary synapses are selectively strengthened whereas unnecessary connections are weakened and eventually eliminated during postnatal development. This process is known as synapse elimination and is a crucial step for shaping immature neural circuits into functionally mature versions. Accumulating evidence suggests that retrograde signaling from postsynaptic cells regulates synapse elimination, but the underlying mechanisms remain unknown. Here, we show that semaphorin3A (Sema3A) and semaphorin7A (Sema7A) mediate retrograde signals for elimination of redundant climbing fiber (CF) to Purkinje cell (PC) synapses in the developing cerebellum, a representative model of synapse elimination in the central nervous system. We picked up candidate retrograde signaling molecules that are expressed in PCs during the period of CF synapse elimination and the receptors of these candidate molecules that are present in CFs. We then assessed the effects of lentivirus-mediated RNAi-knockdown of these molecules on CF synapse elimination. By this systematic screening, we found that knockdown of Sema3A in PCs or its co-receptor, plexinA4 (PlxnA4), in CFs accelerated CF synapse elimination and decreased CF-mediated synaptic inputs. Conversely, knockdown of Sema7A in PCs or either of the two receptors for Sema7A, plexinC1 (PlxnC1) and integrinB1 (ItgB1), in CFs impaired CF synapse elimination. Importantly, the effect of Sema7A involves signaling by type 1 metabotropic glutamate receptor (mGluR1), a canonical pathway in PCs for the final stage of CF synapse elimination. These results demonstrate that specific semaphorins act as retrograde signaling molecules and regulate distinct processes of CF synapse elimination during postnatal cerebellar development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Main Text
Several previous studies demonstrate that neural activity and signaling molecules in postsynaptic cells are essential for synapse elimination [1–7], a fundamental process for shaping functional neural circuits during postnatal development. Thus, retrograde signaling from postsynaptic cells is crucial for synapse elimination, since redundant and unnecessary synaptic terminals are eventually pruned. However, molecular identity of such retrograde signaling remains unknown. We searched for retrograde signaling molecules involved in the regression of redundant climbing fiber (CF) to Purkinje cell (PC) synapse during postnatal cerebellar development, a representative model of developmental synapse elimination in the central nervous system (CNS) [1, 7–9].
First, we profiled genes expressed in postsynaptic PCs during the period of CF synapse elimination. We isolated PCs from the cerebellum of L7-GFP transgenic mice, extracted mRNAs from the purified PCs, and analyzed the amount of these mRNAs using DNA microarray [10]. We picked up genes of secreted or membrane-associated molecules as possible candidates that mediate retrograde signaling. We found that several types of semaphorins are richly expressed in PCs during postnatal cerebellar development. Then, we performed loss-of-function analyses of these candidate molecules by lentivirus-mediated RNAi-knockdown in PCs in olivo-cerebellar coculture preparations [10–12]. We examined CF innervation patterns by using whole-cell recordings from knockdown and control PCs in the same slices. We found that knockdown of Sema3A caused a significant reduction in the number of CFs innervating each PC when compared to control PCs [10]. In marked contrast, knockdown of Sema7A resulted in a significant increase in the number of CFs innervating each PC [10]. These results suggest that endogenous Sema3A maintains/strengthens CF synapses, whereas Sema7A facilitates CF synapse elimination.
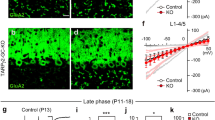
To elucidate the roles of Sema3A and Sema7A in CF synapse elimination in vivo, we injected lentiviruses expressing a miRNA against Sema3A or Sema7A (Smea3A-KD or Sema7A-KD) into the cerebellar vermis of neonatal mice and examined the CF innervation patterns in acute slices at P11–P15 or at P21–P30. We found that Sema3A knockdown caused a significant reduction in the number of CFs innervating each PC when compared to control PCs at P11–P15 [10] (Fig. 1a, c). Furthermore, the total amplitude of CF-EPSCs was significantly smaller in Sema3A-KD than in control PCs. The effect of Sema3A knockdown was rescued by co-injection of lentiviruses for the expression of a miRNA-resistant form of Sema3A (Sema3A-RES) (Fig. 1a, c). In addition, we found that VGluT2-positive CF terminals were absent around the somata of most Sema3A-KD PCs at P14–P15, whereas somatic CF terminals are abundant in control PCs [10]. However, when examined at P21–P30, CF innervation pattern of Sema3A-KD PCs was not different from that of control PCs, although the amplitude of CF-EPSCs was significantly smaller in Sema3A-KD PCs than in control PCs [10] (Table 1). Thus, Sema3A is considered to maintain or strengthen somatic CF synapses and oppose their elimination in the early phase of CF elimination [1, 7–9].

Postsynaptic Sema3A and Sema7A have opposite effects on CF synapse elimination in vivo. a–d Sample CF-EPSCs (Scale bars, 0.5 nA and 5 ms) and frequency distributions of the number of climbing fibers innervating each PC (c, d) for control (white columns, n = 70), Sema3A-KD (green columns, n = 79), and Sema3A-rescue (Sema3A-RES, light-green columns, n = 35) during P12-P15 (a, c), and for control (n = 93), Sema7A-KD (orange columns, n = 84) and Sema7A-rescue (Sema7A-RES, light-orange columns, n = 43,) during P21-P30 (b, d). *P < 0.05, **P < 0.005 (Mann-Whitney U test) (reproduced from Uesaka et al., 2014 [10])
In contrast to Sema3A-KD, Sema7A knockdown caused a significant increase in the number of CFs innervating each PC at P21–P30 [10] (Fig. 1b, d). We found that VGluT2-labeled CF terminals were present around the somata of most Sema7A-KD PCs at P21–P22, whereas such somatic CF synapses were hardly detectable in control PCs [10]. Sema7A knockdown did not alter the CF innervation pattern before P14 [10] (Table 1). These results indicate that Sema7A-KD specifically impairs elimination of somatic CF synapses after P15 (Table 1) and suggest that Sema7A facilitates the late phase of CF elimination [1, 7, 9].
We next tested whether the Sema7A signaling for CF synapse elimination is downstream of mGluR1, PQ-type voltage-dependent calcium channel (PQ-VDCC) or glutamate receptor δ2 (GluD2), the three molecules which act in PCs and are crucial for different aspects of CF synapse elimination [2, 13, 14]. First, we injected lentiviruses for the expression of a miRNA against mGluR1, PQ-VDCC, or GluD2 into neonatal mice and found that knockdown of one of the three molecules impaired CF synapse elimination. We then tested the effects of double knockdown of Sema7A and one of the three molecules on CF synapse elimination. We found that double knockdown of Sema7A and either PQ-VDCC or GluD2 had additive effect on CF synapse elimination when compared with single knockdown of PQ-VDCC or GluD2 [10]. In marked contrast, the effect of double knockdown of Sema7A and mGluR1 was the same as that of mGluR1 single knockdown [10]. These results indicate that mGluR1 and Sema7A are along the same signaling pathway of CF synapse elimination and suggest that Sema7A mediates synapse elimination downstream of mGluR1 signaling.
We next examined whether knockdown of Sema3A or Sema7A affects synaptic transmission from parallel fibers (PFs), the other excitatory inputs to PCs, and that from inhibitory interneurons. Sema3A KD PCs had significantly larger amplitudes of PF-EPSCs than control and Sema3A-RES PCs. The amplitude and frequency of mIPSCs was not altered in Sema3A KD. In contrast, knockdown of Sema7A had no significant effects on PF-EPSCs or mIPSCs.
Sema3A and Sema7A are known to act on Plexin A4 (PlxnA4) and Plexin C1 (PlxnC)/Integlin B1 (ItgB1), respectively [15]. If Sema3A and Sema7A retrogradely regulate CF synapse elimination, their receptors are expected to be present on CF terminals. Our in situ hybridization analyses demonstrate that mRNAs of PlxnA4, PlxnC1, and ItgB1 were expressed in inferior olivary neurons [10]. To examine the involvement of these receptors in CF synapse elimination, we knocked down respective molecules in subsets of CFs. We injected lentivirus vectors for the expression of GFP and miRNA against one of the three molecules (PlxnA4-KD, PlxnC1-KD, ItgB1-KD) into the ventral medial portion of the medulla at P0–P2. We confirmed that GFP signals were detected only in CFs at P22, suggesting that the target molecule was knocked down specifically in subsets of CFs. We found a significant reduction in the number of CFs innervating individual PCs surrounded by PlxnA4-KD CFs when compared with control PCs in the same slices that were sampled in cerebellar regions where PlxnA4-KD CFs were absent [10] (Table 1). Furthermore, the amplitude of CF-EPSCs was significantly smaller in PCs associated with PlxnA4-KD CFs than in control PCs [10]. Scrambled miRNA against PlxnA4 in CFs did not alter the CF innervation pattern. We also found that VGluT2-positive CF terminals were absent around the somata of most PCs that were associated with PlxnA4-KD CFs at P14–P15, demonstrating that the effects of PlxnA4-KD in CFs are exactly similar to those of Sema3A-KD in PCs [10]. These results indicate that Sema3A from postsynaptic PCs strengthens CF synapses and/or inhibits synapse elimination through PlxnA4 in CFs [10]. Furthermore, PF-EPSC amplitudes of PCs associated with PlxnA4-KD CFs were significantly larger than those of control PCs, which is again exactly similar to Sema3A-KD in PCs.
In contrast to PlxnA4, knockdown of PlxnC1 in CFs caused a significant increase in the number of CFs innervating individual PCs associated with PlxnC1-KD CFs at P21–P30 [10] (Table 1). Knockdown of ItgB1 also significantly impaired CF synapse elimination at P21–P30 [10] (Table 1). Moreover, VGluT2-labeled CF terminals were present around the somata of most PCs associated with PlxnC1-KD or ItgB1-KD CFs at P21–P22, indicating that elimination of somatic CF synapses was impaired in such PCs [10]. These results indicate that the effects of PlxnC1-KD or ItgB1-KD in CFs are exactly similar to those of Sema7A-KD in PCs. From these results, we conclude that Sema7A facilitates elimination of CF synapses from PC somata through acting on PlxnC1 and ItgB1 in CFs [10].
While the importance of semaphorins as axon guidance molecules has been well established, their roles in refinement of neural circuitry during postnatal development have been unclear. Here, we have disclosed a novel role of Sema3A and Sema7A as retrograde signaling molecules that regulate developmental synapse elimination. Our results suggest that Sema3A and Sema7A have opposite effects and are involved in different stages of synapse elimination. Since Sema7A is activated downstream of mGluR1 signaling [10], Sema7A action becomes obvious after P15 when mGluR1 signaling plays a crucial role in elimination of redundant somatic CF synapses. Sema3A may presumably be regulated by cellular processes that are active earlier than mGluR1 signaling in PCs. It remains to be investigated how Sema3A signaling is regulated and whether Sema3A and Sema7A interact to coordinate CF synapse elimination during postnatal development.
References
Hashimoto K, Kano M. Synapse elimination in the developing cerebellum. Cell Mol Life Sci. 2013;70:4667–80.
Hashimoto K, Tsujita M, Miyazaki T, Kitamura K, Yamazaki M, Shin HS, et al. Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc Natl Acad Sci U S A. 2011;108:9987–92.
Kawamura Y, Nakayama H, Hashimoto K, Sakimura K, Kitamura K, Kano M. Spike timing-dependent selective strengthening of single climbing fibre inputs to Purkinje cells during cerebellar development. Nat Commun. 2013;4:2732.
Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. 2000;25:269–78.
Lorenzetto E, Caselli L, Feng G, Yuan W, Nerbonne JM, Sanes JR, et al. Genetic perturbation of postsynaptic activity regulates synapse elimination in developing cerebellum. Proc Natl Acad Sci U S A. 2009;106:16475–80.
Nakayama H, Miyazaki T, Kitamura K, Hashimoto K, Yanagawa Y, Obata K, et al. GABAergic inhibition regulates developmental synapse elimination in the cerebellum. Neuron. 2012;74:384–96.
Watanabe M, Kano M. Climbing fiber synapse elimination in cerebellar Purkinje cells. Eur J Neurosci. 2011;34:1697–710.
Crepel F. Regression of functional synapses in the immature mammalian cerebellum. Trends Neurosci. 1982;5:266–9.
Kano M, Hashimoto K. Synapse elimination in the central nervous system. Curr Opin Neurobiol. 2009;19:154–61.
Uesaka N, Uchigashima M, Mikuni T, Nakazawa T, Nakao H, Hirai H, et al. Retrograde semaphorin signaling regulates synapse elimination in the developing mouse brain. Science. 2014;344:1020–3.
Mikuni T, Uesaka N, Okuno H, Hirai H, Deisseroth K, Bito H, et al. Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron. 2013;78:1024–35.
Uesaka N, Mikuni T, Hashimoto K, Hirai H, Sakimura K, Kano M. Organotypic coculture preparation for the study of developmental synapse elimination in mammalian brain. J Neurosci. 2012;32:11657–70.
Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, et al. Roles of glutamate receptor δ2 subunit (GluRδ2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci. 2001;21:9701–12.
Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, et al. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18:71–9.
Pasterkamp RJ, Giger RJ. Semaphorin function in neural plasticity and disease. Curr Opin Neurobiol. 2009;19:263–74.
Acknowledgments
The authors thank A. Nienhuis, St. Jude Children’s Research Hospital and George Washington University for the gifts of the lentiviral backbone vector and the packaging plasmid; K. Kitamura and K. Hashimoto for helpful discussions; and K. Matsuyama, M. Sekiguchi, S. Tanaka, and A. Koseki for technical assistance. This work was supported by Grants-in-Aid for Scientific Research (19100005 to M.W. 21220006 and 25000015 to M.K. and 23650160 to N.U.), the Funding Program for Next Generation World-Leading Researchers (LS021) to H.H., the Strategic Research Program for Brain Sciences (Development of biomarker candidates for social behavior), and the Global COE Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms) from MEXT, Japan.
Conflict of Interest
The authors declare that there are no conflicts of interest in the submission of this manuscript to The Cerebellum.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Uesaka, N., Uchigashima, M., Mikuni, T. et al. Retrograde Signaling for Climbing Fiber Synapse Elimination. Cerebellum 14, 4–7 (2015). https://doi.org/10.1007/s12311-014-0615-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-014-0615-y