
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of extreme inflammation occurring in association with genetic defects involving the granule-dependent cytotoxic pathway of CD8+ T cells or NK cells and/or a wide variety of triggers including infections, malignancies, and rheumatologic disorders. Because of its relative rarity and the non-specific nature of the individual clinical and laboratory abnormalities comprising the characteristic “pattern” of HLH, the diagnosis can be elusive. Furthermore, some of the laboratory tests included in the diagnostic criteria are time-consuming or not widely available, and since many of these patients are critically ill, HLH must be considered early on so that the diagnosis can be established and potentially life-saving treatment initiated. Since the diagnosis of HLH is truly a clinicopathologic correlation, in this article, we as a team of pediatric clinical and laboratory physicians will use exemplary cases from our own institution with a variety of clinical presentations to illustrate the many “faces” of HLH; deconstruct the diagnosis; point out some of the challenges, limitations, pearls and pitfalls; and make it easier to understand the pathophysiology in context. However, while we may see relatively more cases working in a tertiary care children’s hospital, HLH is a disease of both children and adults.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Illustrative cases
Case 1
The patient was a previously healthy black 4-month-old girl transferred from an outside hospital. Two weeks prior, she had developed low-grade fever and a rash which was initially attributed to a viral exanthem, but because of worsening fever (to 40 °C), she presented to the emergency department (ED), was found to have a platelet count of 70 × 103/mL, admitted for further evaluation including a full septic work-up, and started on empiric antibiotics. Her white blood cell count, hemoglobin, and platelet count all progressively declined (with a nadir absolute neutrophil count of 0.2 × 103/mL, hemoglobin dropping to 6.6 g/dL, and 21 × 103/mL platelets) and she developed mild hepatosplenomegaly which was confirmed by abdominal ultrasonography, prompting bone marrow examination which showed no evidence of leukemia, neuroblastoma, or hemophagocytosis. Although initially her ferritin was 17 ng/dL, at the outside hospital it ultimately reached the 500 s, while fibrinogen decreased to <100 mg/dL. Steroids were begun as treatment for HLH shortly after transfer to our institution and while an infectious etiology was not uncovered, additional testing revealed a ferritin of 4873 ng/mL, sCD25 level of 34,826 U/mL (age-specific reference range: 334–3026), absent NK cell function, and compound heterozygosity for two previously described mutations in PRF1 that are associated with familial HLH. Her initial cerebrospinal fluid (CSF) showed a minimally elevated protein without pleocytosis which normalized within 2 weeks of therapy and never exhibited neurologic symptoms. Three months later, she underwent allogeneic hematopoietic stem cell transplant. Ferritin and sCD25 were monitored on an outpatient basis until the time of transplant.
Important points are as follows:
-
1.
The patient’s clinical condition evolved as did the evidence for HLH.
-
2.
At her age, familial HLH was suspected and ultimately confirmed on mutation analysis.
-
3.
HLH-associated gene mutations vary by race/ethnicity as well as age: For example, while PRF1 mutations comprise the majority of identified genetic abnormalities observed in North American black patients like this child, they account for only about one quarter of mutations in whites.
-
4.
Hemophagocytosis was not conspicuous on review of the outside bone marrow slides or a subsequent specimen obtained in-house. Nevertheless, hemophagocytosis is neither sensitive nor specific for HLH (and its absence should not deter one from the diagnosis especially when the evidence is otherwise compelling), but bone marrow examination can be useful to evaluate for a potentially treatable condition such as a malignancy associated with HLH or altogether attributable for the presenting signs and symptoms.
Case 2
This 15-year-old previously healthy Hispanic boy was transferred to our hospital after presenting to the ED of an outside hospital with complaints of abdominal pain, vomiting, and jaundice. Laboratory findings were significant for aspartate aminotransferase of 3010 IU/L, alanine aminotransferase of 2686 IU/L, total bilirubin of 6.6 mg/dL, and an international normalized ratio (INR) of 1.4. Hepatitis viral panel was negative. Two days later, he had returned to the ED with slightly higher transaminases, total bilirubin, and INR. Albumin was low at 3.3 g/dL and acetaminophen (paracetamol) level was <2 mcg/mL.
In our hospital, the etiology of the acute hepatitis could not be determined after an extensive and unrevealing work-up for infection (viral hepatitides A, B, C, and E; Epstein-Barr virus [EBV], cytomegalovirus), ischemia, drug-induced or autoimmune hepatitis, and Wilson’s disease.
The next CBC in our laboratory (obtained by hepatology within 2 weeks of discharge) showed 31 % blasts. Subsequent flow cytometry immunophenotyping and bone marrow examination confirmed B lymphoblastic leukemia.
Liver dysfunction, namely cholestasis, complicated treatment of the leukemia but a liver biopsy done 2 weeks after diagnosis demonstrated nodular transformation of the parenchyma with widened septa, prominent red cell extravasation, and fibrosis enveloping some of the lobules but only minimal steatosis. Mixed portal and sinusoidal infiltrates comprised an increased number of CD68-positive histiocytes and CD8-positive lymphocytes, which far outnumbered CD4-positive lymphocytes. B cells were present in the portal areas and expressed CD10, CD20, PAX-5, and TdT, supporting liver involvement by B lymphoblastic leukemia as well as features compatible with HLH in the context of elevated sCD25 and ferritin, and decreased NK cell function (Fig. 1a–c). Subsequently, a combination of flow cytometry studies (SAP and XIAP) and PRF1 sequencing to screen for genetic HLH were performed.

a Liver wedge biopsy showing nodular transformation of the hepatic parenchyma with widened septa and prominent red blood cell extravasation. Steatosis was minimal. b–d Representative portal area with predominance of CD8+ cytotoxic/suppressor T cells over CD4+ helper T cells and increased number of CD68+ histiocytes (both also present though less prominent in the sinusoids), accompanied by TdT+ leukemic infiltrate
Important points are as follows:
-
1.
HLH can be associated with a variety of malignancies as well as a hepatitis-type presentation, and certain histologic features on the liver biopsy can be suggestive of HLH.
-
2.
The underlying malignancy must be treated for the HLH to resolve.
-
3.
Even if a malignancy appears to be the trigger for HLH, the patient should be worked up to determine whether there is a genetic predisposition for HLH.
-
4.
Flow cytometry for perforin/granzyme B and CD107a degranulation (and SAP and XIAP in males) can provide a more rapid and potentially more cost-effective approach than sequencing multiple genes simultaneously.
Case 3
Patient 3 was a white boy diagnosed at the age of 3 years with ileocecal Burkitt lymphoma after suffering abdominal pain for 3 months. Conventional cytogenetic analysis of the mass revealed t(8;9)(q24.1;p13) accompanied by either +12 or a marker chromosome, with MYC rearrangement confirmed by interphase fluorescence in situ hybridization (FISH) studies. He was brought back to the operating room 2 months later for adhesive bowel obstruction but completed chemotherapy 2 months after that per POG 9317 protocol. Three years later, he had another small bowel obstruction, this time from tumor adhesion. The resection specimen again revealed Burkitt lymphoma with only two analyzable metaphases, one of which demonstrated t(8;14)(q24.1;q32) (FISH to metaphase proving MYC-IGH fusion) and der(18)t(1;18)(q21;p11.3). He subsequently attained complete remission after undergoing RICE (rituximab, ifosfamide, carboplatin, etoposide) chemotherapy followed by autologous stem cell transplant, the latter complicated by prolonged hypogammaglobulinemia. At the age of 9 years, the patient developed a rash, intermittent fevers to 39.4 °C, myalgias/arthralgias, and cervical lymphadenopathy. “Atypical lymphocytes” were seen on the peripheral blood film (Fig. 2a) and EBV viral load testing demonstrated >1,000,000 copies/mL. Anti-EBV IgM antibody was positive while IgG was negative. Because of worsening respiratory status with increased work of breathing and retractions, he was admitted directly to the pediatric intensive care unit. Pleural fluid cytology and bone marrow examination both showed no evidence of Burkitt lymphoma but the marrow was remarkable for a striking relative erythroid hyperplasia/granulocytic hyperplasia, reactive-appearing lymphocytes (predominantly CD8-positive T cells) and increased histiocytes, some performing hemophagocytosis. In situ hybridization for EBV-encoded small RNAs (EBER) showed numerous positive nuclei (Fig. 2b–e). In addition, he had significant neutropenia and thrombocytopenia, hypertriglyceridemia of 341 mg/dL (30–74), hypofibrinogenemia, and an elevated ferritin of 6975 ng/mL.

a Peripheral blood smear with reactive lymphocytes. b Bone marrow aspirate with erythroid hyperplasia/granulocytic hypoplasia, reactive lymphocytes, and no evidence of Burkitt lymphoma but increased histiocytes, some performing hemophagocytosis. c–e Hypercellular bone marrow core biopsy with predominance of CD8+ cytotoxic/suppressor T cells over CD4+ helper T cells, increased number of CD68+ histiocytes, and numerous EBV+ nuclei
He died 2 days later, and pre-mortem blood samples were sent shortly thereafter for SH2D1A and BIRC4 sequencing to evaluate for X-linked lymphoproliferative disorder (XLP). A 274C>T (Q92X) non-sense mutation was identified in SH2D1A, and a diagnosis of XLP-1 was established.
Important points are as follows:
-
1.
EBV is the most common viral trigger for HLH.
-
2.
XLP is associated not only with fulminant, often fatal infectious mononucleosis, but also B-cell lymphomas (including Burkitt lymphoma) and HLH and should be considered in boys with “recurrent” Burkitt lymphoma or boys whose male siblings or relatives have also been diagnosed with Burkitt lymphoma. It is our current practice to consider screening male patients with Burkitt lymphoma at diagnosis for XLP.
-
3.
Genetic consultation should be offered to family members of individuals carrying a molecular diagnosis consistent with HLH, not only to determine their own personal risk of HLH but for family planning as well as identifying an appropriate (uninvolved) allogeneic hematopoietic stem cell donor.
Case 4
This previously healthy 13-year-old girl was transferred to our institution with pancytopenia and concern for rheumatologic disease after several weeks of fatigue, weight loss, occasional fevers and chills, lower back and thigh pain, and subsequent appearance of oral ulcers. At an outside ED, imaging revealed intra-abdominal lymphadenopathy and mild splenomegaly.
She continued to have pain with intermittent tactile fevers and returned to her pediatrician where laboratory testing revealed pancytopenia. She was referred to an oncologist for bone marrow examination which showed a “cellular marrow with active hematopoiesis in all cell lines,” “no leukemic infiltrate or other tumor seen” and “no excessive hemophagocytosis detected” but was subsequently admitted to another hospital for dehydration. There she was found to have worsening of her cytopenias accompanied by elevated transaminases, activated partial thromboplastin time (117.6 seconds) and erythrocyte sedimentation rate (ESR; >140 mm/h), and a positive anti-nuclear antibody. She was referred to our hospital for further work-up and management.
At our hospital, she was diagnosed with systemic lupus erythematosus and likely macrophage activation syndrome on the basis of splenomegaly, cytopenias, hemophagocytosis on our review of the marrow, absent NK cell function, and elevated ferritin, sCD25 of 2726 U/mL (45–1105) and serum neopterin. Her ESR declined with treatment (oral and intravenous corticosteroids, cyclosporin A, and hydroxychloroquine) though her hospital course was complicated by catastrophic antiphospholipid syndrome.
Important points are as follows:
-
1.
HLH can occur in the setting of collagen vascular disease and is referred to as “macrophage activation syndrome” in the rheumatology literature.
-
2.
“Hemophagocytosis” is subjective and as a criterion for HLH not clearly defined.
-
3.
It is not widely recognized that reference ranges for sCD25 vary significantly by age; note that while this patient’s sCD25 is nearly 2.5 times the upper limit of normal for her age, it would be considered normal for patient 1.
Diagnosis and pathophysiology
There are two ways to establish a diagnosis of HLH
The diagnostic criteria for HLH have evolved over the past 20+ years based on the collaborative efforts of the Histiocyte Society. The standard criteria defining HLH, formally defined in the international HLH-94 clinical trial, were revised for the HLH-2004 trial and are listed in Table 1, adapted from [1].
Jordan et al. divide these criteria into three categories to reflect causality and sequence in HLH: predisposing immunodeficiency, significant immune activation, and abnormal immunopathology [1]. Low or absent NK cell function and genetic defects involving the granule-dependent cytotoxic pathway of NK and cytotoxic CD8+ T cells underlie predisposing immunodeficiency; stated another way, intact cytotoxic function is necessary for regulating and terminating the inflammatory response. Therefore, defects in this pathway lead to prolonged activation of T cells and macrophage histiocytes and a toxic hypercytokinemia with resultant end-organ damage [2]. Immune activation is reflected by fever, splenomegaly, and elevated ferritin and sCD25 (from macrophage histiocyte and T cell activation, respectively) and permits the development of immunopathology, manifesting as cytopenias, decreased fibrinogen or increased triglycerides, and hemophagocytosis. Other features such as hepatitis and central nervous system (CNS) involvement, though not included in these criteria, are relatively common. The individual diagnostic criteria will be discussed in greater detail below as we deconstruct HLH.
Molecular diagnosis of HLH
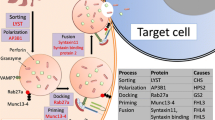
As above, defects involving the granule-dependent cytotoxic pathway of NK and cytotoxic CD8+ T cells characterize genetically determined HLH, which has an autosomal recessive or hemizygous pattern of inheritance. Normally, these effector cells are able to mediate immune contraction via granule-dependent cytotoxicity to target cells. This occurs through a complex interplay of proteins involved in the trafficking, docking, priming for exocytosis or membrane fusion of granules which diffuse across the immunologic synapse to access the cytoplasm of the target cell (perforin) and activate a cascade of caspases (granzyme B), ultimately leading to its death through apoptosis. While the genetic defects identified to date compromise the cytotoxic function of NK and CD8+ T cells, neutrophil and platelet degranulation can also be affected, which explains an association in some cases with inflammatory bowel disease and bruising/bleeding. Table 2 summarizes the genes, types of mutations, proteins they encode and associated features in genetically determined HLH [1, 3–13].
The general NK cell function test (discussed below) is important, but one of its limitations is that it does not enable one to reliably distinguish between primary and secondary HLH. Nowadays, as illustrated by case 2, more specific flow cytometry assays are available to screen for some of the genetic abnormalities described in HLH, whose turnaround time is measured in days rather than the weeks sequencing typically requires. Initially, girls and boys should be evaluated for perforin/granzyme B protein expression to screen for PRF1 abnormalities and if perforin is decreased, PRF1 sequencing should be performed. If perforin is normal or increased, girls should reflexively undergo flow cytometry for CD107a mobilization [14]. Boys should be tested for signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) and X-linked inhibitor of apoptosis protein (XIAP) encoded by SH2D1A and BIRC4 and mutated in XLP-1 and XLP-2, respectively [15, 16], and if SAP and XIAP are normal, CD107a mobilization should be performed next. CD107a (also known as lysosomal-associated membrane protein 1, or LAMP1) colocalizes with perforin in the lytic granules of both NK and CD8+ T cells, with engagement of activating receptors in the case of the former and the T cell receptor on the latter, leading to surface expression of CD107a, which reflects exocytosis of the granules. As one might predict, abnormally low-resting NK cell degranulation can be used to screen for abnormalities in UNC13D, STXBP2, RAB27A, and STX11 [14] but will be normal in HLH associated with mutations in PRF1, SH2D1A, BIRC4, and “secondary” HLH (HLH triggered by infection, rheumatologic disease, or malignancy—though it must be remembered that infection sometimes unmasks genetic HLH as in case 3 above). However, leukopenia/lymphopenia may preclude an accurate flow cytometric evaluation, in which case sequencing should be pursued.
Clinical and laboratory criteria
-
1.
Fever: This is one of the most consistent features in HLH, seen in about 91 % of patients [17]. Worthy of additional comment is the observation that fever is uncommon in HLH during the neonatal period (particularly in preterm infants) when HLH can be mistaken for sepsis [2].
-
2.
Splenomegaly: Approximately 84 % of patients with HLH are found to have splenomegaly. Although not currently one of the diagnostic criteria, hepatomegaly is even more frequent, present in 90–95 % [2].
-
3.
Cytopenias: Greater than 80 % of patients with HLH have anemia and thrombocytopenia [1]. It is likely that the cytokine milieu, rich in tumor necrosis factor (TNF) alpha and interferon (IFN) gamma, plays a prominent role in the suppression and inducing apoptosis of blood elements, instead of being attributable solely to hemophagocytosis [18–20].
-
4.
Hypertriglyceridemia and/or hypofibrinogenemia: In a review of familial and secondary HLH, hypertriglyceridemia/hypofibrinogenemia was only seen in 60–70 and 40 % of cases, respectively [4]. Elevated triglycerides are the result of decreased lipoprotein lipase activity due to elevated levels of TNF alpha and IFN gamma [19, 20]. Fibrinogen is decreased in HLH because of high plasmin levels and hyperfibrinolysis as a result of activated macrophages secreting plasminogen activator [20].
-
5.
Hemophagocytosis: As illustrated in the first two cases above, hemophagocytosis is neither a necessary nor sufficient criterion for the diagnosis of HLH. More common causes of hemophagocytosis include blood transfusion, infection, and autoimmune disease among others [1]. In a study of 21 pediatric patients with HLH, the sensitivity of hemophagocytosis (defined as 1 or more hemophagocytic cell among 500 nucleated cells examined) was only 58 %. Furthermore, all of these patients were diagnosed with HLH regardless of the presence of absence of hemophagocytosis [21]. Goel et al. subsequently compared six marrow aspirates from three HLH patients to 20 random control marrows and found that rare hemophagocytosis can be seen outside of HLH and also, although statistically significantly differences in hemophagocytosis are observed in HLH (0–0.31 %; mean, 0.082 %) versus controls (0–0.04 %; mean, 0.009 %), “overlap…precludes using hemophagocytosis as a reliable indicator of HLH” [22]. More recently, Ho and colleagues found no correlation between hemophagocytosis on aspirate or core biopsy and disease probability [23]. Some investigators have proposed that CD68 or CD163 immunohistochemistry be performed on bone marrow core biopsies in HLH but we are not aware of any classical studies detailing the normal quantity and distribution of CD68- and/or CD163-positive cells in the bone marrow. Hemophagocytosis can also be identified in other reticuloendothelial organs including the liver. Chen et al. reviewed 25 liver specimens from 19 pediatric HLH patients and described four histopathologic patterns: chronic hepatitis-like, leukemia-like, histiocytic storage disorder-like, and neonatal giant cell hepatitis-like. Common to all cases were portal and sinusoidal infiltrates of CD3+, CD8+ granzyme B+ lymphocytes and CD68+, CD1a-histiocytes performing hemophagocytosis. Additionally, endothelialitis of portal and central veins and lymphocyte-mediated bile duct injury were present [24].
-
6.
Low or absent NK cell activity: This is an important test though not widely available and labor-intensive, often involving radioactivity. It is not simply a quantitative assay to determine the number of NK cells present but rather their function. However, limitations include the fact that many HLH patients have few circulating NK cells and the inability of this assay to distinguish between primary and secondary HLH [14]. Nevertheless, in a study of familial HLH patients as well as their asymptomatic parents and siblings, impaired NK-function was observed in all of the patients and some of their relatives, but not in >100 adult and >50 pediatric controls [25]. Therefore, while useful in the diagnosis of HLH, results must be interpreted with caution in siblings of familial HLH patients if being used to predict their risk of developing the disease.
-
7.
Hyperferritinemia: Ferritin has two primary roles in the body. The first is as an intracellular store of iron, and the second is as serum ferritin, an acute phase reactant released from dying cells or from breakdown of red blood cells [26]. The current cutoff for ferritin under the HLH-2004 criteria is 500 ng/dL, which provides a sensitivity of 84 % [27]. This number was derived from 31 children with familial disease on whom ferritin levels were available. A subsequent study of 330 patients at Texas Children’s Hospital whose ferritin was >500 ng/dL revealed that a ferritin level greater than 10,000 ng/dL was 90 % sensitive and 96 % specific for HLH, though only 10 patients had HLH [28]. More recently, 2000 ng/dL was proposed as a compromise with a sensitivity of 70 % and specificity of 68 % and similar results between familial and virus-associated acquired HLH. With a much larger cohort of patients with HLH (123) versus other hyperferritinemic conditions (320), a cutoff of 10,000 ng/dL would have a sensitivity of only 29 % [29]. However, in adults, marked hyperferritinemia (exceeding 50,000 ng/dL) is associated more frequently with renal failure, hepatocellular injury, infection, hematologic malignancy, and rheumatologic/inflammatory conditions than with HLH; therefore, ferritin appears to be a more specific marker of HLH in children than in adults [30].
-
8.
Elevated sCD25: CD25 is the alpha chain of interleukin (IL)-2 receptor. Elevated soluble CD25 reflects T cell activation and in the HLH-2004 review by Henter et al. was stated to have a sensitivity of 93 % [27].
Other diagnostic tests and scenarios
HLH is characterized by a systemic cytokine storm which underlies organ dysfunction. These cytokines include IL-1 beta, TNF alpha, IL-6, IL-8, IFN gamma, IL-18, and IL-10. As above, elevated levels of sCD25 and IFN gamma reflect T cell activation, while macrophage histiocyte activation results in high ferritin, CD163, and neopterin. Xu found that a specific cytokine profile with markedly elevated IFN gamma and IL-10 and modestly elevated IL-6 has high diagnostic accuracy for HLH and can be used to differentiate it from infection in febrile children [31].
At our institution, serum neopterin is also ordered frequently in the work-up of HLH. Ibarra and colleagues, in a study comparing 21 HLH patients (3 with familial and 18 with secondary HLH) with 50 active juvenile dermatomyositis patients, found that a cutoff of 38.9 nmol/L was 70 % sensitive and 90 % specific for HLH and that neopterin levels correlated significantly with ferritin [32].
Just as flow cytometry plays an important role in the diagnosis of (and screening for genetic defects in) HLH, several other quantitative and qualitative immunophenotypic abnormalities have been described in HLH. These include a relative decrease in the marrow myeloid component (gain of HLA-DR and decreased CD10+ cells is common in cases associated with EBV) accompanied by a relative increase in the lymphoid component, particularly T cells. HLA-DR expression is usually seen on CD8+ cytotoxic T cells reflecting their activated state, and downregulation of pan-T cell antigens such as CD7 and CD5 and an inverted CD4:CD8 ratio (both fairly frequent in EBV-associated HLH) should not be overinterpreted as evidence of leukemia or lymphoma [33].
Although not part of the diagnostic criteria, HLH patients frequently present with liver dysfunction [1]. Our case 2 above had acute hepatitis with markedly elevated transaminases indicating hepatocellular damage, the etiology of which could not be initially be determined even after an extensive work-up. Similarly, additional case 2 below had acute liver failure, anemia, and neutropenia. We have also seen liver failure in neonates with HLH.
Neurologic findings are also relatively common, at least in children. In a study of 193 pediatric HLH patients on whom CSF data at diagnosis were available, 63 % had either neurological symptoms (seizures, meningismus, or irritability being the most frequent) and/or abnormal CSF findings (elevated protein and/or cell count). Hemophagocytosis was not assessed in this study, although frequently clinically queried in our practice. Nevertheless, the authors found that HLH patients with abnormal CSF at diagnosis faced significantly higher mortality [34].
Finally, case 4 above is an example of HLH in the setting of rheumatologic disease, so-called macrophage activation syndrome (MAS). The diagnostic criteria for MAS are not as clearly defined as the HLH-2004 criteria, but do show some overlap. They were developed by circulating a questionnaire listing 28 clinical, laboratory, and histopathologic features of MAS to 505 pediatric rheumatologists worldwide, who were asked to choose and rank the 10 most important features of MAS in their opinion. Interestingly, the features chosen by more than 50 % of the respondents were the following: falling platelet count, hyperferritinemia, evidence of macrophage hemophagocytosis in the bone marrow, increased liver enzymes, falling leukocyte count, persistent continuous fever of ≥38 °C, falling erythrocyte sedimentation rate, hypofibrinogenemia, and hypertriglyceridemia [35]. Note that almost all of these are laboratory values, underscoring the importance of the clinical laboratory in establishing the diagnosis of MAS.
Additional cases
Three additional cases and accompanying figures are included to further illustrate presenting features of HLH (Figs. 3, 4, and 5).

Additional case 1: 3-year-old boy with skin nodules clinically suspected to represent deep fungal, mycobacterial, Nocardia, or Actinomyces infection and found to be anaplastic large cell lymphoma (ALCL), ALK+, associated with HLH. As one might expect, HLH not uncommonly complicates T and NK cell malignancies. a–b Punch biopsy of representative skin lesion showing deep dermal and subcutaneous infiltrate of ALCL. c–e The bone marrow was not involved by ALCL but hemophagocytosis was quite conspicuous

Additional case 2: This was a 10-year-old boy with acute liver failure of unknown etiology, anemia, and neutropenia. There were no circulating blasts. He underwent concurrent liver and bone marrow biopsies. a Bone marrow examination revealed 85–90 % blasts with an early T cell precursor acute lymphoblastic leukemia (ETP-ALL) phenotype: CD1a−, CD2+, cytoplasmic CD3+ but surface CD3−, CD4−, CD5−, CD7+, CD8−, CD13+, CD33−, CD34+, CD38+, CD45+ (dim), HLA-DR-, MPO-, TdT+. b The liver biopsy showed many swollen hepatocytes, scattered apoptotic bodies, and a moderately dense lymphohistiocytic infiltrate. c–e ETP-ALL involved the liver, tumor cells being positive for CD3 (c) and CD99 (d) but negative for CD5 (e) by immunohistochemistry. f–h In addition, CD8+ cytotoxic/suppressor T cells (f) predominated over CD4+ helper T cells (g). An increased number of histiocytes was present throughout, staining weakly for CD4 and strongly positive for CD68 (h). Additional laboratory testing revealed decreased NK cell function, markedly elevated ferritin (21,357 ng/mL), and high sCD25 (7258 U/mL (45–1105]), clinching the diagnosis of HLH. This is the first case to our knowledge associating ETP-ALL and HLH

Additional case 3: A 19-year-old female with an history of acute myelomonocytic leukemia who had relapsed approximately 2 months prior to this bone marrow examination, which was performed immediately following magnetic resonance imaging to evaluate for cerebrovascular accident after new onset of hemiparesis. Since relapse, she had received a 5-day course of chemotherapy, followed by granulocyte-colony stimulating factor for approximately 6 weeks during which she was persistently febrile and pancytopenic. New pulmonary and pleural-based infiltrates were thought to have represented an infectious (fungal or atypical) etiology. In addition, she had hypertriglyceridemia of 551 mg/dL (which might have been attributable to being on total parenteral nutrition) and a ferritin of 12,358 ng/mL (11–320). a Peripheral blood smear from the time of relapse with circulating blasts. b Peripheral blood smear from the day of bone marrow examination revealing pancytopenia (the white blood cell count was <0.1 K/uL, hemoglobin was 8.7 g/dL, and platelet count was 35 K/uL). c–d. Bone marrow aspirate smear showing about 80 % histiocytes with readily identifiable hemosiderin pigment and some erythrophagocytosis. e. The majority of cells present in the bone marrow core biopsy were histiocytes. Histochemical stains were negative for fungal organisms and acid-fast bacilli. HLH has been described as a terminal complication of numerous malignancies (probably related to infection) but also following administration of colony-stimulating factor or interleukin
Conclusions
In this review, we have deconstructed HLH into its clinical, laboratory, and molecular diagnostic criteria. While the individual features are non-specific, when taken together, they form a unique pattern reflecting a combination of immunodeficiency, immune activation, and immunopathology. By keeping in mind these criteria and the pathophysiology they represent, laboratory and clinical hematologists are well-prepared to work together and promptly recognize HLH.
The history and physical examination and readily available laboratory testing provide most of the data needed to render a diagnosis of HLH but a couple of the tests require additional time and since many of these patients have life-threatening disease, once the disease is being considered, more esoteric testing like NK cell activity and sCD25 should be ordered promptly; sometimes therapy has to be initiated prior to having a complete data set. Bone marrow examination probably has greater utility in evaluating for a treatable cause of HLH like malignancy or infection than it does for establishing the diagnosis, though the procedure is frequently indicated/performed to investigate the cytopenias. In many children’s hospitals, patients are sedated for procedures, which require scheduling operating room time but also allows for coordination with lumbar puncture. Additional testing not represented in the diagnostic criteria but useful include hepatic panel, PT, aPTT, and D-dimer. Lymph node biopsy is only performed in our institution to evaluate lymphadenopathy, not to rule-in HLH. Genetic testing may not be critical for acute management of HLH but is essential for definitive management and determining the risk of recurrence and need for stem cell transplantation.
Important take home points include the following:
-
Clinical condition and evidence for HLH are not static and can evolve over time.
-
A genetic basis should be suspected in infants with HLH, but finding an infectious (commonly, EBV), malignant, or rheumatologic “trigger” should not preclude work-up for a genetic defect in granule-dependent cytotoxicity given obvious implications on recurrence risk, likelihood in future pregnancies, and choice of stem cell donor.
-
That stated, genetically determined HLH occurs in older children as well as adults, who in most if not all cases should undergo such a work-up, which can be accomplished by flow cytometry (assuming adequate white blood cells/lymphocytes) and/or gene sequencing.
-
Hemophagocytosis, though part of the disease name, is subjective, neither sensitive nor specific for, and by no means required to establish a diagnosis of HLH.
-
In general, higher ferritin levels are more suspicious for HLH, but in adults there are other more common causes for marked hyperferritinemia.
-
sCD25 reference ranges vary by and are inversely related to age.
-
Testing for XLP can be considered in boys diagnosed with non-endemic Burkitt lymphoma.
References
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL (2011) How I treat hemophagocytic lymphohistiocytosis. Blood 118:4041–4052
Chandrakasan S, Filipovich AH (2013) Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr 163:1253–1259
Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, Pende D, Griffiths G, Aricò M (2014) Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol 16:167
George MR (2014) Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med 12:69–86
Sieni E, Cetica V, Mastrodicasa E, Pende D, Moretta L, Griffiths G, Aricò M (2012) Familial hemophagocytic lymphohistiocytosis: a model for understanding the human machinery of cellular cytotoxicity. Cell Mol Life Sci 69:29–40
Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, Ciambotti B, Brugnolo F, zur Stadt U, Pende D, Moretta L, Griffiths GM, Henter JI, Janka G, Aricò M (2011) Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet 48:343–352
An O, Gursoy A, Gurgey A, Keskin O (2013) Structural and functional analysis of perforin mutations in association with clinical data of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) patients. Protein Sci 22:823–839
Ueda I, Ishii E, Morimoto A, Ohga S, Sako M, Imashuku S (2006) Correlation between phenotypic heterogeneity and gene mutational characteristics in familial hemophagocytic lymphohistiocytosis (FHL). Pediatr Blood Cancer 46:482–488
Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, Villanueva J, Risma KA, Wei Q, Klein PS, Filipovich AH (2011) Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood 118:5794–5798
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachée-Chardin M, Chedeville G, Tamary H, Minard-Colin V, Vilmer E, Blanche S, Le Diest F, Fischer A, de Saint BG (2003) Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 115:461–473
Horne A, Ramme KG, Rudd E, Zheng C, Wali Y, al-Lamki Z, Gürgey A, Yalman N, Nordenskjöld M, Henter JI (2008) Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol 143:75–83
Meeths M, Entesarian M, Al-Herz W, Chiang SC, Wood SM, Al-Ateeqi W, Almazan F, Boelens JJ, Hasle H, Ifversen M, Lund B, van den Berg JM, Gustafsson B, Hjelmqvist H, Nordenskjöld M, Bryceson YT, Henter JI (2010) Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood 116:2635–2643
Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, Kanegane H, Lopez-Granados E, Mejstrikova E, Pellier I, Galicier L, Galambrun C, Barlogis V, Bordigoni P, Fourmaintraux A, Hamidou M, Dabadie A, Le Diest F, Haerynck F, Ouachée-Chardin M, Rohrlich P, Stephan JL, Lenoir C, Rigaud S, Lambert N, Milili M, Schiff C, Chapel H, Picard C, de Saint BG, Blanche S, Fischer A, Latour S (2011) Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 117:1522–1529
Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, Chiang SC, Marcenaro S, Meazza R, Bondzio I, Walshe D, Janka G, Lehmberg K, Beutel K, zur Stadt U, Binder N, Arico M, Moretta, Henter JI, Ehl S (2012) A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood 119:2754–2763
Gifford CE, Weingartner E, Villanueva J, Johnson J, Zhang K, Filipovich AH, Bleesing JJ, Marsh RA (2014) Clinical flow cytometric screening of SAP and XIAP expression accurately identifies patients with SH2D1A and XIAP/BIRC4 mutations. Cytometry B Clin Cytom 86:263–271
Marsh RA, Bleesing JJ, Filipovich AH (2010) Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods 362:1–9
Mehta RS, Smith RE (2013) Hemophagocytic lymphohistiocytosis (HLH): a review of literature. Med Oncol 30:740
Larroche C, Mouthon L (2004) Pathogenesis of hemophagocytic syndrome (HPS). Autoimmun Rev 3:69–75
Jordan MB, Hildeman D, Kappler J, Marrack P (2004) An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 104:735–743
Rosado FGN, Kim AS (2013) Hemophagocytic lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol 139:713–727
Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem (2008) The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 51:402–404
Goel S, Polski JM, Imran H (2012) Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann Clin Lab Sci 42:21–25
Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML (2014) Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol 141:62–71
Chen JH, Fleming MD, Pinkus GS, Pinkus JL, Nichols KE, Mo JQ, Perez-Atayde AR (2010) Pathology of the liver in familial hemophagocytic lymphohistiocytosis. Am J Surg Pathol 34:852–867
Sullivan KE, Delaat CA, Douglas SD, Filipovich AH (1998) Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res 44:465–468
Kell DB, Pretorius (2014) Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 6:748–773
Henter JI, Horne A, Aricò M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48:124–131
Allen CE, Yu X, Kozinetz CA, McClain KL (2008) Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 50:1227–1235
Lehmberg K, McClain KL, Janka GE, Allen CE (2014) Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 61:2101–2103
Schram AM, Campigotto F, Mullally A, Fogerty A, Massarotti E, Neuberg D, Berliner N (2015) Marked hyperferritinemia does not predict for HLH in the adult population. Blood 125:1548–1552
Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, Shi SW, Shen HP, Mao JQ, Zhang LY, Pan BH (2012) Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr 160:984–990
Ibarra MF, Klein-Gitelman M, Morgan E, Proytcheva M, Sullivan C, Morgan G, Pachman LM, O’Gorman MR (2011) Serum neopterin levels as a diagnostic marker of hemophagocytic lymphohistiocytosis syndrome. Clin Vaccine Immunol 18:609–614
McCall CM, Mudali S, Arceci RJ, Small D, Fuller S, Gocke CD, Vuica-Ross M, Burns KH, Borowitz MJ, Duffield AS (2012) Flow cytometric findings in hemophagocytic lymphohistiocytosis. Am J Clin Pathol 137:786–794
Horne A, Trottestam H, Aricò M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI, Histiocyte Society (2008) Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol 140:327–335
Davì S, Consolaro A, Guseinova D, Pistorio A, Ruperto N, Martini A, Cron RQ, Ravelli A, MAS Study Group (2011) An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol 38:764–768
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Weinstein, J.L., Badawy, S.M., Bush, J.W. et al. Deconstructing the diagnosis of hemophagocytic lymphohistiocytosis using illustrative cases. J Hematopathol 8, 113–125 (2015). https://doi.org/10.1007/s12308-015-0254-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-015-0254-3